- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

A Bioreactor Method to Generate High-titer, Genetically Stable, Clinical-isolate Human Cytomegalovirus
Published: Vol 7, Iss 21, Nov 5, 2017 DOI: 10.21769/BioProtoc.2589 Views: 9736
Reviewed by: Modesto Redrejo-RodriguezMarielle CavroisAnonymous reviewer(s)
Original research article
The authors used this protocol in:
Nov 2017
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics








Abstract
Human cytomegalovirus (HCMV) infection is a major cause of morbidity and mortality in transplant patients and a leading cause of congenital birth defects (Saint Louis, 2016). Vaccination and therapeutic studies often require scalable cell culture production of wild type virus, represented by clinical isolates. Obtaining sufficient stocks of wild-type clinical HCMV is often labor intensive and inefficient due to low yield and genetic loss, presenting a barrier to studies of clinical isolates. Here we report a bioreactor method based on continuous infection, where retinal pigment epithelial (ARPE-19) cells adhered to microcarrier beads are infected in a bioreactor and used to produce high-titers of clinical isolate HCMV that maintain genetic integrity of key viral tropism factors and the viral genome. In this bioreactor, an end-stage infection can be maintained by regular addition of uninfected ARPE-19 cells, providing convenient preparation of 107-108 pfu/ml of concentrated TB40/E IE2-EYFP stocks without daily cell passaging or trypsinization. Overall, this represents a 100-fold increase in gain of virus production of 100-times compared to conventional static-culture plates, while requiring 90% less handling time. Moreover, this continuous infection environment has the potential to monitor infection dynamics with applications for real-time tracking of viral evolution.
Keywords: Human cytomegalovirusBackground
Congenital CMV infection is a leading cause of birth defects with an annual direct cost of one billion dollars in the US alone, and represents a global unmet medical need (Bristow et al., 2011; Griffiths et al., 2013; Manicklal et al., 2013; Saint Louis, 2016). This would be preventable with an effective vaccine or therapeutic targeting women in their reproductive years. Human cytomegalovirus (HCMV) can infect a wide range of cell types, but a major barrier in the field is that extended passage of clinically derived HCMV strains in fibroblast cells leads to a loss of viral tropism for other cell types (Waldman et al., 1991; Sinzger et al., 1999). In the late 1960s, several laboratory-adapted strains of CMV were serially passaged in fibroblasts–including the HCMV AD169, Towne, and Davis strains, as well as the Smith strain of murine CMV–and became some of the first tools used to study the molecular biology of CMV (Plotkin et al., 1975). These laboratory-adapted strains–often created during unsuccessful attempts to generate a live attenuated vaccine–were found to have a number of mutations affecting (i) their ability to infect different cell types, (ii) rate of viral replication, and (iii) altering latency phenotypes (Albrecht and Weller, 1980; Yamane et al., 1983; Waldman et al., 1989; Kahl et al., 2000). Specifically, the HCMV open reading frames (ORFs) UL128, UL130, UL131, which comprise a viral glycoprotein entry complex, were found to accumulate mutations during passage in fibroblasts, leading to the loss of viral tropism for infection of epithelial cells, endothelial cells, macrophages, and dendritic cells (Sinzger et al., 1999, Hahn et al., 2004; Wang and Shenk, 2005; Adler et al., 2006). These lab-adapted HCMV strains were also shown to have lost several genes in the UL/b’ region of the viral genome, a region that confers immune evasion functions and replication dissemination in vivo (Cha et al., 1996). It is now known that sustained viral growth on fibroblast cultures removes the selection pressure to retain these sequences, resulting in genetic loss or rearrangement of sequences essential for replication and dissemination in other host cells and tissues. However, passage of HCMV clinical isolates (e.g., TB40/E and VR1814) in epithelial and endothelial cell settings maintains selection pressure to prevent loss of tropism for non-fibroblast cell types (Waldman et al., 1991; Hahn et al., 2002; Sinzger et al., 2008).
Differences between established laboratory-adapted strains of HCMV and clinical isolates of HCMV are important considerations when planning experiments, as the choice of virus strain used may influence results. Clinical isolates are much more similar to the viruses that replicate within patients, making them preferable for understanding clinical symptoms, as well as natural and drug-selected genetic variability of human CMV. These clinical isolates also maintain productive infection and latency phenotypes that best represent the wild-type virus population (Lee et al., 2015). Because clinical isolates spread through a cell-associated manner, yields from clinical isolates are significantly lower than those collected from laboratory-adapted virus strains, due in part to clinical strains being more limited to cell-associated spread. This contributes to time consuming and labor-intensive aspects of clinical virus stock preparation.
Here, we report a new, more efficient method for generating stocks of clinically derived HCMV isolates, represented by TB40/E IE2-EYEFP. This virus is genetically tagged with an EYEFP fusion to enable convenient monitoring of continuous infection in the bioreactor environment. Using a two-stage bioreactor system (Figure 1) with microcarrier beads and the advantage of the prolonged period of virus production characteristic of HCMV, we are able to maintain an end-stage infection and generate high titer virus stocks on primary adherent cell cultures that preserve genetic integrity of key viral tropism factors and the viral genome. We have used the TB40/E-IE2 EYFP tagged virus in development and characterization of the bioreactor infection. The fluorescent tag enables convenient monitoring of the virus in the bioreactor culture, by surveying aliquots of the infected samples with fluorescent microscopy.
Materials and Reagents
- 15 cm tissue culture dish (Corning, Falcon®, catalog number: 353025 )
- Vented screw caps (Corning, Falcon®, catalog number: 354639 ; Corning, catalog number: 3968 )
- 1.5 ml polypropylene microcentrifuge tubes (Corning, Axygen®, catalog number: MCT-150-C )
- Conical sterile polypropylene centrifuge tubes, 50 ml (Fisher Scientific, catalog number: 05-539-12 )
- 96-well flat-bottom cell culture plates (Corning, catalog number: 3596 )
- 12-well reagent reservoirs (Corning, Costar®, catalog number: 4877 )
- 40 μm cell strainers (Corning, catalog number: 431750 )
- Conical sterile polypropylene centrifuge tubes, 15 ml (Fisher Scientific, catalog number: 06-443-18 )
- 0.45 μm sterile syringe filters (EMD Millipore, catalog number: SLHV033RS )
- Disposable sterile syringes with Luer-Lock tips (BD, catalog number: 309646 )
- Serological pipettes (50 ml, 25 ml, 10 ml, 5 ml, 2 ml)
- 0.2 or 0.1 μm filter system
- ARPE-19 cells (ATCC, catalog number: CRL-2302 )
- MRC-5 cells (ATCC, catalog number: CCL-171 )
- TB40/E IE2-EYFP bacmid cloned virus
TB40/E IE2-EYFP is derived from the bacmid clone of TB40/E, a clinically derived HCMV isolate (Sinzger et al., 2008). The viral double stranded DNA genome has been cloned as a bacmid and can produce infectious virus encoding the tropism factors required for replication in multiple host cell types. This virus contains an EYFP fluorescent tag fused to the carboxyl terminus of a key viral transactivator, the IE2 gene, as previously reported (Teng et al., 2012). This bacmid clone was generated using the galK selection and counterselection recombination protocol described previously for bacmid cloning with HCMV (Murphy et al., 2008, Warming et al., 2005). Expression of this fluorescent tag occurs throughout the virus lytic infection cycle and enables convenient real time monitoring of continuous infection in the bioreactor environment. - DPBS without calcium and magnesium, DPBS-CMF (Mediatech, catalog number: 21-031-CV )
- Trypsin-EDTA, 0.25% (Mediatech, catalog number: 25-053-CI )
- Trypsin-EDTA, 0.05% (Mediatech, catalog number: 25-052-CI )
- Dry ice pellets
- Isopropanol (Fisher Scientific, catalog number: A451-4 )
- DMEM with L-glutamine and sodium pyruvate (Mediatech, catalog number: 10-013-CV )
- DMEM/F-12 with L-glutamine and 15 mM HEPES (Mediatech, catalog number: 10-092-CV )
- Fetal bovine serum (FBS) (Mediatech, catalog number: 35-011-CV )
- Penicillin-streptomycin solution (Pen/Strep) (Mediatech, catalog number: 30-002-CI )
- Sigmacote (Sigma-Aldrich, catalog number: SL2-25ML )
- 1 M Tris, pH 7.5
- Magnesium chloride (MgCl2)
- Hydrochloric acid (HCl)
- Cytodex 1 microcarrier beads, 60-87 μm (Sigma-Aldrich, catalog number: C0646-5G )
- Culture media for titration assays (TCID50) (see Recipes)
- Culture media for bioreactor (see Recipes)
- Siliconizing glassware with SigmaCote (see Recipes)
- 20% sorbitol (see Recipes)
- Microcarrier beads ratio and preparation (see Recipes)
- Adherent cell ratio (see Recipes)
Equipment
- Personal protective equipment, PPE, required for working at BSL-2: Laboratory coats or gowns, eye and face protection, gloves
- Wheaton Celstir spinner flasks, 150 ml, 250 ml, and 500 ml (WHEATON, catalog numbers: 356879 and 356882 )
- Tissue culture incubator: 37 °C, 5% CO2
- 4 position slow-speed stir plate (Corning, catalog number: 440814 )
- Class II biosafety cabinets (The Baker)
- Vortex mixer
- Micropipette (1-1,000 μl capacity), multiple channel pipettes (1-200 μl capacity) (P1000 pipette and P200 pipette [Pipetteman])
- Hemocytometer (Hausser Scientific, catalog number: 1475 )
- Cell culture inverted epi-fluorescent microscope
- Tabletop centrifuge
- Water bath, 37 °C
- 100 ml autoclavable glass bottle (WHEATON, catalog number: W818012406 )
- Approved BSL-2 laboratory facilities
- Autoclave
- Pipette controller (Pipette-aid, Drummond Scientific, model: Pipet-Aid® XP2, catalog number: 4-000-501 )
- Freezers: -80 °C, -20 °C, 4 °C
Procedure
TIMING
Expanding ARPE-19 cells (step A1: 2 weeks)
Preparing the culture feeder flask and beads (steps A2-A6: 3 h to be performed the day before seeding)
Seeding ARPE-19 cells in feeder flask (step A7-A11: 6.5 h)
Replacing media in feeder bioreactor (step A12: 15 min every 3-4 days)
Preparing the samples for cell count (steps B1-B8: 25 min)
Incubating cells in the bioreactor until confluent (step C1: 1-2 weeks)
Infecting cells in bioreactor (steps C2-B4: 10 min)
Replacing media and cells in infected bioreactor (steps D1-D7: 25 min)
Preparing the 96-well plate for TCID50 (step E1: 15 min)
Setting up TCID50 infections (steps E2-E11: 25 min)
Determining titer from TCID50 (steps E12-E13: 10 min)
Preparation of virus collected from bioreactor (steps F1-F6: 25 min)
Concentrating virus stocks (step F7: 3 h)
See Figure 1 for bioreactor system setup for the experiment.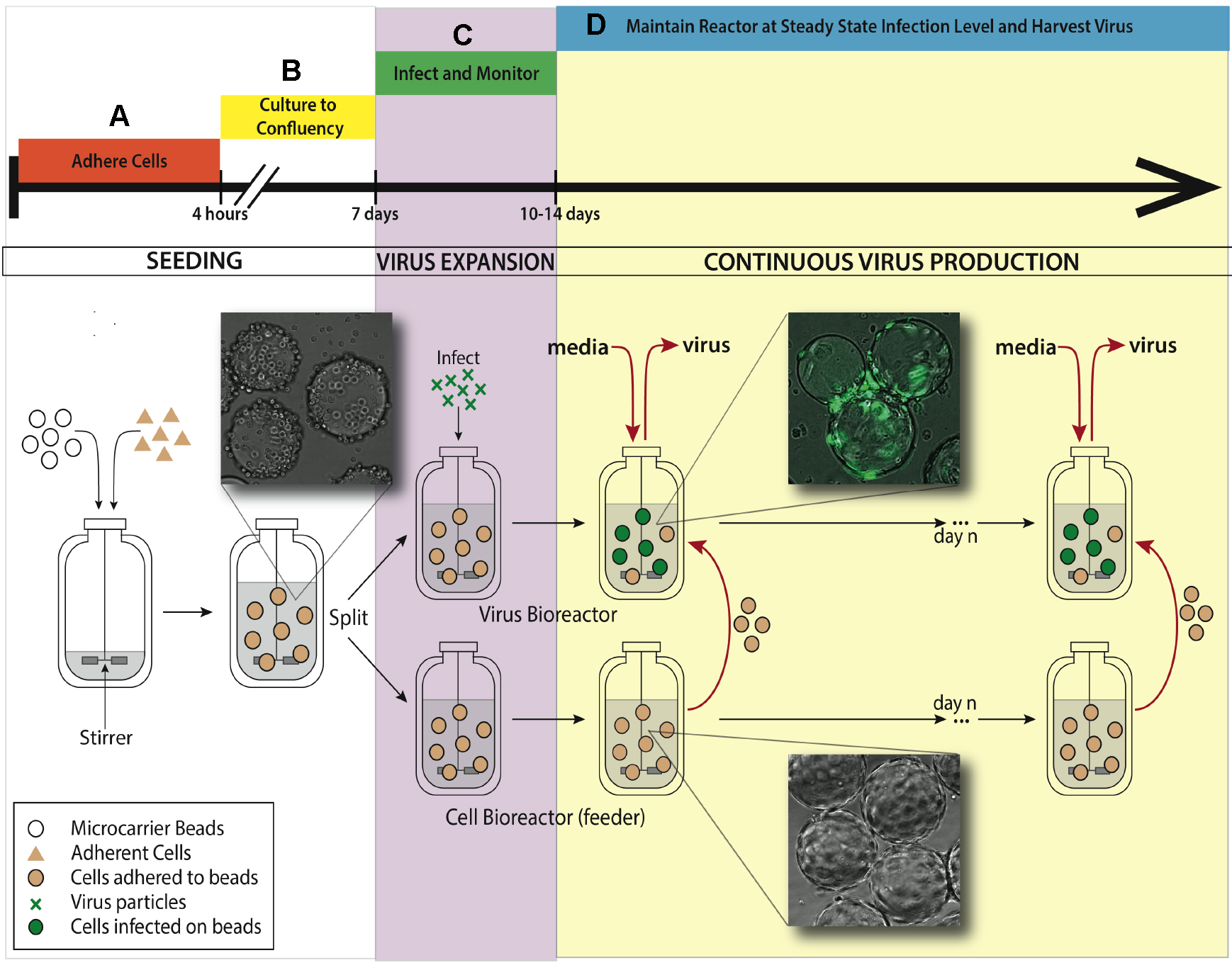
Figure 1. Bioreactor system setup for maintaining continuous virus production. A. An adherent cell line (ARPE-19) is added to a 500 ml silicon coated glass feeder flask containing 2.5 g hydrated microcarrier beads in 150 ml DMEM-F12 media. This feeder flask is placed in a 37 °C incubator for 4 h to allow ARPE-19 cells to adhere to microcarrier beads, mixing every half an hour. B. After 4 h, the volume of media is brought up to working concentration of 500 ml and put on a stirring plate set at 60 rpm back in the same incubator. Allow a week for the cells to divide on the microcarrier beads and reach confluence in the feeder flask. C. When confluence is reached, 250 ml of stirred microcarrier beads from the feeder flask are transferred to a smaller, 250 ml bioreactor. This smaller bioreactor is then infected with TB40/E IE2-EYFP at an MOI of 0.01. Both the small and the large bioreactors are placed back in the incubator on the stir-plate. D. When virus expansion reaches desired steady state, slurry (microcarrier beads/media, taken when spinning) and supernatant (media collected when stir plate is off and microcarrier beads settle) are collected from the infected bioreactor. Uninfected ARPE-19 cells are taken from the feeder flask and added into the infected bioreactor, along with fresh media. The rate of microcarrier bead and media replacement maintains end stage viral production of HCMV. The volume of the infected bioreactor and the microcarrier bead concentration remains constant. This induced steady state can be continued for as long as desired, and collected samples from infected reactor are concentrated to create virus stocks.
- Seeding Cytodex 1 microcarrier beads with ARPE-19 cells (Figure 1A)
- Culture sufficient ARPE-19 cells on 15 cm cell culture dishes such that you have 1 x 106 cells/ml final volume. For a 500 ml Celstir spinner flask (feeder flask), you need 5 x 108 cells.
- The day before seeding, siliconize 500 ml feeder flask and an autoclavable 100 ml glass container with Sigmacote according to manufacturer’s instructions (http://www.sigmaaldrich.com/content/dam/sigma-aldrich/docs/Sigma/Product_Information_Sheet/1/sl2pis.pdf).
Note: This procedure has been optimized for greatest adherence of ARPE-19 cells to the prepared Cytodex 1 microcarrier beads during seeding. Sigmacote is used to prevent cells from adhering to the walls of the spinner flasks. Given that it can be toxic to cells, rigorous rinsing is needed after siliconizing. - Rinse feeder flask and glass container thoroughly with MilliQ water, at least 10 times. Place a new vented screw-on caps onto each arm of the flask.
- Weigh the appropriate amount of dry Cytodex 1 microcarrier beads to reach 0.5 g/100 ml bioreactor final volume. For 500 ml feeder flask, measure out 2.5 g microcarrier beads.
- Pour dry Cytodex 1 microcarrier beads into a siliconized 100 ml glass container, and add 100 ml DPBS-CMF (DPBS without calcium and magnesium). Resuspend beads thoroughly. For more information refer to GE manual: http://www.gelifesciences.com/file_source/GELS/Service%20and%20Support/Documents%20and%20Downloads/Handbooks/pdfs/Microcarrier%20Cell%20Culture.pdf.
- Autoclave feeder flask and resuspended microcarrier beads.
- Remove by pipetting 50 ml DPBS-CMF from settled beads in a 100 ml glass container, replace with 50 ml DMEM-F12 with 10% FBS. Resuspend by vortexing. Incubate the glass container with the beads at 37 °C until beginning step A9.
- Trypsinize 5 x 108 ARPE-19 cells, resuspend in 90 ml DMEM-F12 with 10% FBS, and place in feeder flask.
- Take the glass container with settled beads from the 37 °C incubator and remove by pipetting 60 ml (i.e., most of the liquid). Resuspend these microcarrier beads in the remaining 40 ml of DMEM-F12 with 10% FBS left in the glass container and transfer them to the feeder flask. Add another 20 ml of 100% FBS to enhance cell adhesion. Final volume in the feeder flask should be about 150 ml and the final FBS concentration should roughly be 25%.
- Place feeder flask in the 37 °C incubator to allow cells to adhere to beads for 4 h. Every 30 min, place the feeder flask on a slow-speed stir-plate set at 60 rpm within the incubator for 30 sec to gently agitate bioreactor and ensure even seeding of cells.
- After 4 h, bring the feeder flask to full 500 ml volume by adding 350 ml DMEM-F12 with 10% FBS.
- Place the feeder flask on 4 position slow-speed stir-plate set to 60 rpm in the incubator.
- Replace media within the feeder flask every 2-3 days to maintain cell density. To do this, let beads settle for 10 min, then remove 50-75% of the total media volume (250-375 ml), and replace it with the same amount of fresh media (DMEM-F12 with 5% FBS). Let cells expand across the microcarrier beads for a week, or until maximum cell density (step C1) is reached (monitor through cell counts in Procedure B and by eye).
Note: If seeding is too low, the maximum cell density in the spinner flask will also remain low over time. Once seeding is completed, if there are any microcarrier beads without cells, they will always remain uncoated. After the spinner flask is placed permanently on the stir-plate, cells are unable to bind to new cells, and the only method of cell spreading is through cell division.
- Culture sufficient ARPE-19 cells on 15 cm cell culture dishes such that you have 1 x 106 cells/ml final volume. For a 500 ml Celstir spinner flask (feeder flask), you need 5 x 108 cells.
- Monitoring ARPE-19 cell density on microcarrier beads (Figure 1B)
Note: In the procedure, we are obtaining accurate cell counts for the feeder reactor. To do this, we take a sample of the slurry, perform a wash to remove debris, and then trypsinize the cells off of the microcarrier beads. Because the microcarrier beads settle much more quickly than the cells, we are able to easily separate the two without centrifuging.- Remove the feeder flask from the incubator and place on a stir-plate set at 60 rpm inside a class II biosafety cabinet.
- Pipette 1 ml slurry (mixed cells/microcarrier beads/media) from the feeder flask while it stirs on stir-plate and dispense into a 1.5 ml microcentrifuge tube. Put the feeder flask back into the incubator on stir-plate, you will not need to use it for the remainder of this procedure.
- Let microcarrier beads in the microcentrifuge tube settle for 2 min. The microcarrier beads can be observed visually settling to the bottom of the microcentrifuge tube by gravity, no centrifugation is necessary.
- Wash by pipetting out 0.8 ml of media from the microcentrifuge tube, and resuspending with 0.8 ml of DPBS-CMF. Resuspend beads by gently vortexing on vortex mixer or by pipetting up and down, then let microcarrier beads settle again.
- Remove 0.8 ml of DPBS-CMF from the microcentrifuge tube and replace with 0.8 ml of 0.25% trypsin. Resuspend cells by gently vortexing.
- Place microcentrifuge tube in the 37 °C incubator and let cells trypsinize for 15-20 min, resuspending every 5 min by gently vortexing or flicking to ensure even trypsinization.
- Place microcentrifuge tube back in the biosafety cabinet and pipette vigorously to resuspend using P1000 pipette. This step also helps to remove all of the cells from the microcarrier beads and break up aggregates. Let beads settle for 30 sec by gravity.
- Pipette 10 μl media containing the detached cells from the microcentrifuge tube, avoiding taking up any microcarrier beads. Transfer this 10 μl to a hemocytometer, and determine the cell concentration.
Note: Cell counts should be performed regularly (every 2 or 3 days) to ensure that cells are healthy. Cell density in the feeder flask should increase until max density (1 x 106 cells/ml) is reached and then remains constant. It is also possible to view the cells adhered to the beads under a microscope. If the cells appear to be dissociating from the beads or cell counts are suddenly dropping, it is probably because the media is not being replaced frequently enough.
- Remove the feeder flask from the incubator and place on a stir-plate set at 60 rpm inside a class II biosafety cabinet.
- Infection of ARPE-19 cells on microcarrier beads in the bioreactor with TB40/E-IE2-EYFP (Figure 1C)
Note: Our EYFP fusion tagged IE2 virus was used to track viral dissemination in epithelial cells within the bioreactor setting by eye using an inverted epi-fluorescent microscope.- Once the cell concentration in the feeder flask reaches roughly 1 x 106 cells/ml, as measured through cell counts (Procedure B), transfer 250 ml of slurry to a smaller, siliconized, rinsed, and autoclaved 250 ml WHEATON spinner flask (referred herein as bioreactor). Continue maintaining cell growth in the feeder flask by replacing media (DMEM-F12 with 5% FBS) regularly (see step A13).
- Once roughly 1 x 106 cells/ml is reached, as measured through cell counts, transfer 250 ml of slurry to a smaller, siliconized and autoclaved 250 ml WHEATON spinner flask (bioreactor). Continue maintaining what remains in the feeder flask by replacing media regularly (refer to step C1).
- Inoculate the cells in the new bioreactor with TB40/E-IE2-EYFP at a multiplicity of infection, MOI, of 0.01 based on the viable cell counts and TCID50 of the virus stock.
- Place the bioreactor (250 ml) and the feeder flask (500 ml) on the same 4 position stir-plate set to 60 rpm within the 37 °C incubator.
- Monitor virus expansion by taking 200 μl aliquots from the stirred bioreactor, and visualizing EYFP spread through a fluorescent microscope. Expansion should also be monitored through regular TCID50.
Note: The infected cells will fall off of the beads.
- Once the cell concentration in the feeder flask reaches roughly 1 x 106 cells/ml, as measured through cell counts (Procedure B), transfer 250 ml of slurry to a smaller, siliconized, rinsed, and autoclaved 250 ml WHEATON spinner flask (referred herein as bioreactor). Continue maintaining cell growth in the feeder flask by replacing media (DMEM-F12 with 5% FBS) regularly (see step A13).
- Continuous high-titer viral production by maintenance of steady-state TB40/E IE2-EYFP infection (Figure 1D)
Note: In order to maintain an ongoing CMV infection and avoid a crash of the infection, we need to regularly provide uninfected cells from the feeder flask, which newly produced viruses will then infect. Fresh media is also added to prevent cell death. The rate at which media and cells need to be replaced depends on the particular virus’s kinetics. For TB40/E IE2-EYFP, we determined that replacing media and cells every 48 h resulted in highest viral production over time. Maintaining as regular of a feeding schedule as possible is ideal for sustaining steady viral production. Additionally, maintaining healthy naïve cells in the feeder flask is essential for continued viral production. Media should be replaced, and the cell density should remain constant in this feeder flask (see step A13).- Regular visual monitoring can follow the health of the infected culture in the bioreactor. When 80% of the cells are killed (and detach from the beads), which usually occurs at 5-7 days post infection, start regular feeding with cells and media as followed.
- Every 48 h, remove 37.5 ml (15% of total bioreactor volume) slurry from the infected bioreactor as it is stirred on a stir-plate in the biosafety cabinet. Transfer this slurry into a 50 ml conical tube, and centrifuge for 5 min at 300 x g (to removed non-adhered cells). Aliquot 3 supernatant samples of 500 μl (to be used for TCID50). Freeze the three aliquots and the 50 ml conical tubes of media at -80 °C to be used later (for step E2).
- Set aside an aliquot of 500 μl for TCID50 determination (for step E2) and freeze the rest at -80 °C for later.
- Removed the bioreactor from the stir-plate, and let beads settle for 5-10 min. Remove another 25 ml (10% of total bioreactor volume) of supernatant from the top of the infected bioreactor. Do not pipette the settled beads.
- From the uninfected feeder flask, pipette 37.5 ml of the slurry while spinning and transfer it to the infected bioreactor. Be sure not to cross contaminate your feeder flask.
- Pipette 25 ml DMEM-F12 with 5% FBS into the infected bioreactor. This media should have been warmed to 37 °C.
- Place both spinner flasks back onto the stir-plate in the incubator.
- Regular visual monitoring can follow the health of the infected culture in the bioreactor. When 80% of the cells are killed (and detach from the beads), which usually occurs at 5-7 days post infection, start regular feeding with cells and media as followed.
- Monitoring viral production in TB40/E IE2-EYFP infected bioreactor using TCID50 (Figure 2)
- Seed MRC-5 fibroblast cells in a 96-well flat-bottom plate by pipetting 100 μl per well of a cell suspension at 6,000 cells/ml. Incubate overnight at 37 °C to allow cell adhesion and growth.
- Take one aliquot of 500 μl of supernatant (prepared in step D2), and freeze-thaw 3 times by alternating incubation in a dry ice/isopropanol bath and, once frozen, transfer the tube to a 37 °C water bath to thaw. Make sure to vortex between each freeze-thaw. This step releases the virions that remain within the infected cells.
- Centrifuge 500 μl aliquot for 3 min at 300 x g.
- Add 1 ml DMEM with 10% FBS to 6 wells of a 12-well reagent reservoir.
- To the first well, pipette 110 μl of prepared viral supernatant into media. Using P1000, mix thoroughly while avoiding bubbles. The viral dilution in the first well is 1:10.
- Pipette 110 μl media from well #1 to well #2 to obtain 1:100 dilution of the viral preparation. Mix thoroughly using a P1000 Pipetman and avoid bubbles.
- Repeat serial dilutions of 1:10 through the remaining wells to reach in the last well a dilution of 1:1,000,000.
- Take prepared 96-well plate with adhered MRC-5 cells out of the incubator and aspirate media.
- Using a multichannel P200 pipette, transfer 100 μl of each serial dilution onto the MRC-% fibroblast plate. Repeat the transfer 12 times into successive rows to make sure that 8 wells of infected MRC-5 are being infected with each viral dilution.
- Place plate in a 37 °C incubator and monitor using fluorescent microscope over the next week.
- At day 7, score the eight wells within each dilution as either positive or negative for cytopathic effect.
- Calculate TCID50 using score (Reed Muench, 1938). TCID50 is the infectious virus titer at which pathological changes in 50% of inoculated cell cultures occur. This is determined by inoculating the series of diluted virus to susceptible cells. An Excel file from ResearchGate is a simple way to complete this calculation: https://www.researchgate.net/file.PostFileLoader.html?id=58dad730f7b67ea37125593f&assetKey=AS%3A476999471898624%401490736944531.
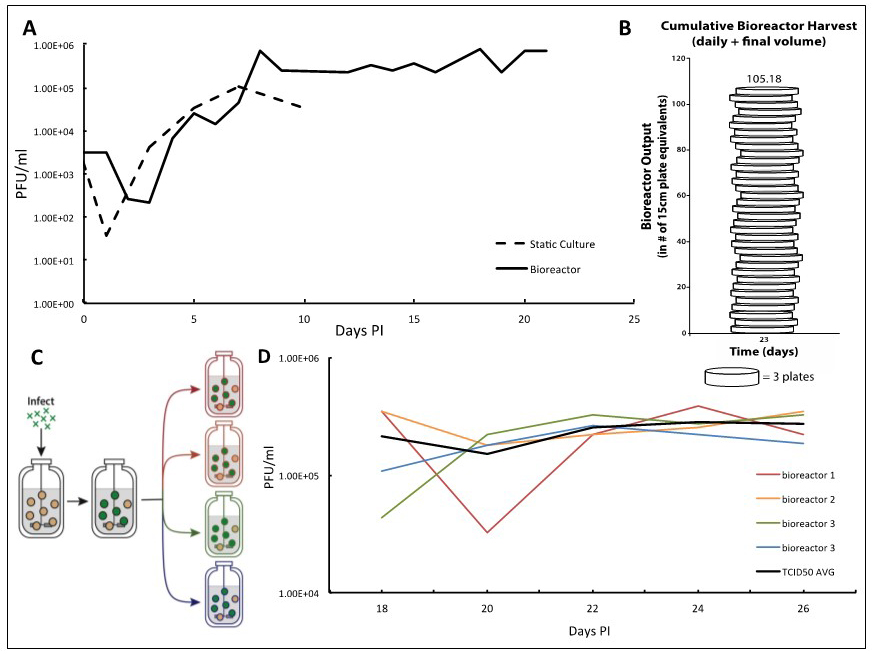
Figure 2. TB40/E IE2-EYFP virus infection can be reproducibly held at end-stage for long periods of time enabling high-titer TB40/E IE2-EYFP production. A. Adherent ARPE-19 cells infected at an MOI of 0.01 show a characteristic eclipse phase one day after infection, where the concentration of virus drops to low levels. By day two, the viral concentration begins to increase until reaching its peak end stage viral production by day 7. After this point, viral concentrations begin to decline when using static culturing methods, as there are no more cells to infect. This same viral kinetics curve is observed in the bioreactor setting. With this system, however, it is possible to maintain peak viral production stage by regular replacements of microcarrier beads coated with naïve ARPE-19 cells. B. We demonstrate that 100-times more TB40/E IE2-EYFP virus can be generated in comparison to the static culture plates in a three-week timeframe. Virus stocks taken at each time-point are cumulated, and compared with the average titration from collecting virus from one 15 cm cell culture dish, estimated at 7.5E6 total pfu per plate. This estimates how many 15 cm plates infected with TB40/E IE2-EYFP it would take to obtain the same concentration of virus as the cumulative samples collected from the bioreactor. Once steady state viral production is reached, and regular feedings begin, the number of plates it would take to reach the same total pfu increases quickly. On day 23 it would take 105 plates to have the same pfu/ml as the reactor system. C. To show reproducibility of the maintained end-stage infection, a bioreactor containing microcarrier beads coated with ARPE-19 was infected with TB40/E IE2-EYFP at an MOI of 0.01. Once peak viral production was reached, steady state was established and maintained. After a period of time, the infected bioreactor was split equally into 4 smaller 100 ml bioreactors, in which the steady states were maintained equivalently by regularly changing the same percentage of the slurry (15%) and of the supernatant (10%) as before the split. D. TCID50 titrations were completed using the supernatant samples from each 48 h time point of all four bioreactors. Each day’s results for the four bioreactors were averaged and standard deviations were determined. The results show that the maintained viral production state is reproducible. The longer the bioreactors are carried, the more consistent their viral production becomes.
- Seed MRC-5 fibroblast cells in a 96-well flat-bottom plate by pipetting 100 μl per well of a cell suspension at 6,000 cells/ml. Incubate overnight at 37 °C to allow cell adhesion and growth.
- Preparing stocks of virus for use in other experiments
Note: Depending on your TCID50 results (determined from Procedure E), virus collected from the bioreactor during the regular feedings may be at a high enough concentration for certain experiments. If you require a higher concentration, however, standard ultracentrifugation using a sorbitol underlay can be performed. Supernatant and slurry samples need to be processed differently before use. Frozen supernatant samples (no microcarrier beads) simply need to be thawed and filtered. Slurry samples (frozen samples with microcarrier beads) require multiple freeze thaws to isolate the virus from within the cells.- Supernatant samples
- Thaw frozen conical tube samples of virus from days that have been titered with TCID50 in a water bath.
- Supernatant samples can be used directly (prepared in step D7). Before using, however, filter media using a cell strainer to remove any remaining microcarrier beads. After, media should also be filtered using a 40 μm filter.
- Thaw frozen conical tube samples of virus from days that have been titered with TCID50 in a water bath.
- Slurry samples
- Thaw frozen conical tube samples of virus from days that have been titered with TCID50 in a water bath.
- For slurry samples, centrifuge thawed conical tubes for 5 min and pipet off all media except for 5 ml that contain pelleted microcarrier beads. Media removed can be filtered and used as explained in step F2.
- Freeze thaw the remaining 5 ml of slurry in the conical tube 3 times by placing it in a dry ice/isopropanol bath and then transferring to a water bath once frozen. In between each freeze-thaw, vortex all samples. This step releases the virions that remain within the infected cells.
- Centrifuge 5 ml for 3 min at 300 x g.
- Remove remaining media from pellet, and pipette through cell strainer. This can now be used for infections or frozen for later.
- Thaw frozen conical tube samples of virus from days that have been titered with TCID50 in a water bath.
- Optional: To concentrate the virus stock further, the protocol developed by Stinski et al. (1976) should be used. Briefly, on 23 ml viral preparation, underlay 7 ml of 20% sorbitol. After 1.5 h of centrifuge at 7,200 x g (20,000 rpm) at 18 °C, remove carefully the supernatant and resuspend the viral pellet in 2 ml of DMEM with no FBS.
- Supernatant samples
Data analysis
We report a bioreactor system (Figure 1) for producing continuous high-titer stocks of clinically derived isolates of CMV that also maintains the integrity of the virus genome and tropism factors. Infection in the bioreactor system exhibits kinetics similar to infections in static culture, but maintains a reproducible production of virus for extended periods of time (Figure 2A) at the steady state that is reproducible (Figures 2C-2D). This bioreactor approach enables preparation of clinical isolate TB40/E IE2-EYFP HCMV virus at 107-108 pfu/ml over a 23-day period without requiring daily cell passaging or trypsinization, and the virus maintains cell-tropism factors. Overall, this system increased TB40/E IE2-EYFP virus production by more than 100 times in comparison to conventional static-culture plates (Figure 2B), while requiring substantially less time and manpower. Although we have used TB40/E IE2-EYFP to clearly portray the procedure and benefits of this system, this bioreactor method can be used to generate stocks of any clinically derived CMV strain. The end-stage steady-state infection exhibits minimal variation (Figure 2C) indicating that this system may potentially be useful for other assays such as real-time tracking of viral evolution.
Expected results
- Given that an average of 7.5 x 106 pfu can be collected from a single 15 cm dish of TB40/E IE2-EYFP-infected cells, we can relate the amount of HCMV viruses produced in our bioreactor to the number of plates that would have been needed. In our bioreactor, once steady state viral production is reached, and regular feedings begin, the virus production increases very rapidly. By day 8, it would take 8 plates to obtain the same amount of total viral particles, and by day 22, it would take 32.5 plates. On day 23, when the full bioreactor volume of 250 ml was collected, it would take 105 plates to obtain the amount of virus reached in our bioreactor system (Figure 2B).
- The sample collected at each time point, can be frozen, pooled and concentrated to achieve high concentrated HCMV stocks (i.e., obtaining up to 3 ml of a viral preparation at more than 2 x 107pfu/ml).
Notes
Troubleshooting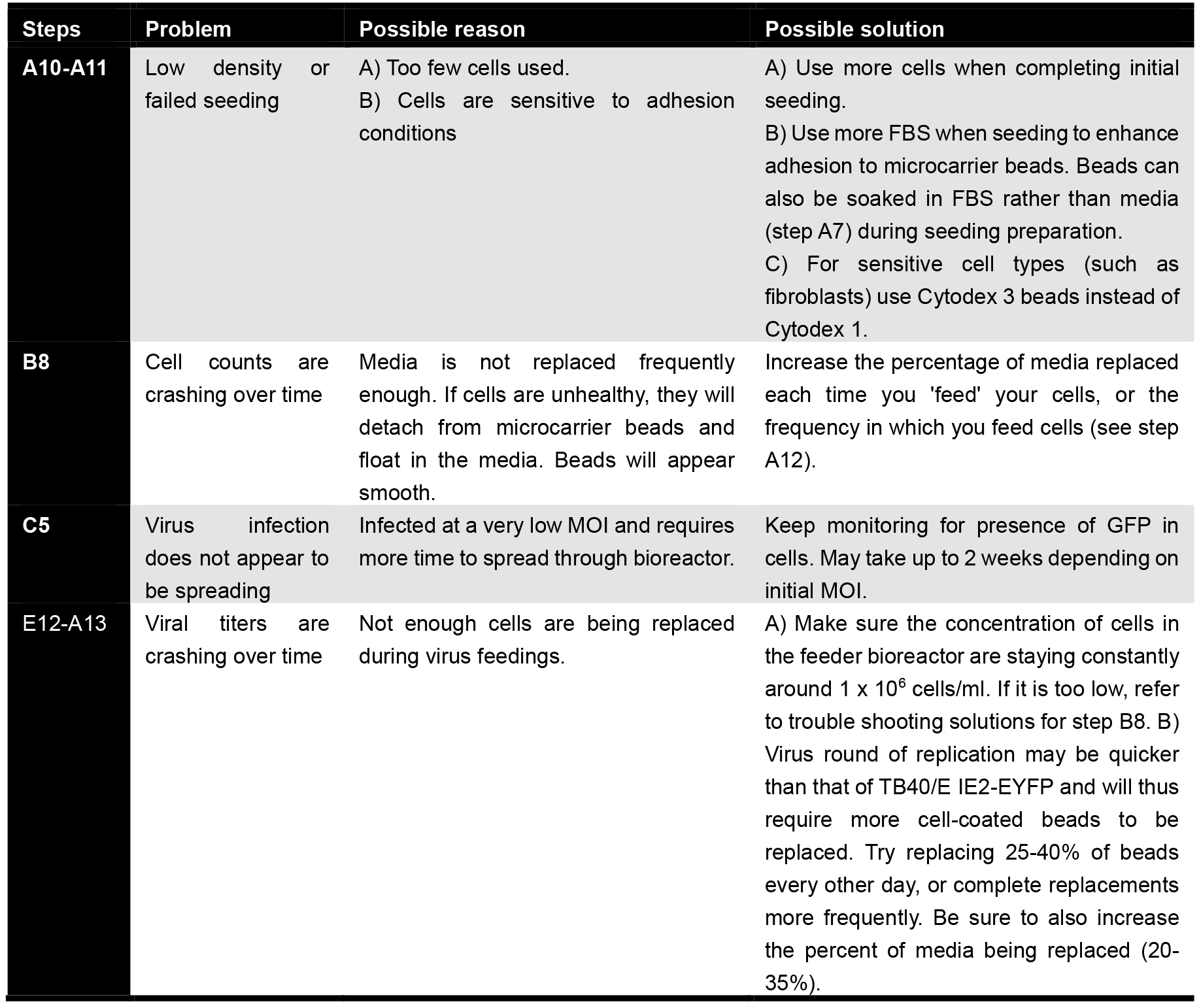
Recipes
- Culture media for titration assays (TCID50)
Dulbecco’s modified eagle medium (DMEM/F-12) with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin
Store at 4 °C
Bring to 37 °C before use - Culture media for bioreactor
DMEM/F12 with 5% FBS and 1% penicillin-streptomycin
Store at 4 °C
Bring to 37 °C before use - Siliconizing glassware with Sigmacote
Clean glassware (stir flask, 100 ml containers) with deionized water and dry
Apply 3 ml of Sigmacote in the glass flask and swirl such that liquid covers all of the inside surface of the glassware for 10 sec. Remove excess solution, and save for reuse
Rinse the glass flask with deionized water thoroughly to ensure that excess Sigmacote is removed
Note: Store Sigmacote at 4 °C. - 20% sorbitol
- Combine:
100 g sorbitol
25 ml 1 M Tris, pH 7.5
0.5 ml 1 M MgCl2 (30.5 g MgCl2 + 150 ml MilliQ water) - Add enough water to bring to 350 ml
- Add HCl dropwise to bring to pH 7.2
- Add enough MilliQ water to bring to 500 ml
- Filter stock with 0.2 or 0.1 μm filter system
- Combine:
- Microcarrier beads ratio and preparation
0.5 g Cytodex 1 microcarrier beads per 100 ml final bioreactor volume (dry weight)
Store dry microcarrier beads at room temperature - Adherent cell ratio
1 x 105 cells per 100 ml final bioreactor volume (cells should be low-passage)
Acknowledgments
This work was supported by funding from NIH grants, DP1DE024408 and DP2OD006677 (to L. Weinberger).
Conflict of Interest Disclosure: L. Weinberger is a co-founder of Autonomous Therapeutics Inc. Other authors declare no competing financial interest.
References
- Adler, B., Scrivano, L., Ruzcics, Z., Rupp, B., Sinzger, C. and Koszinowski, U. (2006). Role of human cytomegalovirus UL131A in cell type-specific virus entry and release. J Gen Virol 87(Pt 9): 2451-2460.
- Albrecht, T. and Weller, T. H. (1980). Heterogeneous morphologic features of plaques induced by five strains of human cytomegalovirus. Am J Clin Pathol 73(5): 648-654.
- Bristow, B. N., O'Keefe, K. A., Shafir, S. C. and Sorvillo, F. J. (2011). Congenital cytomegalovirus mortality in the United States, 1990-2006. PLoS Negl Trop Dis 5(4): e1140.
- Cha, T. A., Tom, E., Kemble, G. W., Duke, G. M., Mocarski, E. S. and Spaete, R. R. (1996). Human cytomegalovirus clinical isolates carry at least 19 genes not found in laboratory strains. J Virol 70(1): 78-83.
- Griffiths, P., Plotkin, S., Mocarski, E., Pass, R., Schleiss, M., Krause, P. and Bialek, S. (2013). Desirability and feasibility of a vaccine against cytomegalovirus. Vaccine 31 Suppl 2: B197-203.
- Hahn, G., Khan, H., Baldanti, F., Koszinowski, U. H., Revello, M. G. and Gerna, G. (2002). The human cytomegalovirus ribonucleotide reductase homolog UL45 is dispensable for growth in endothelial cells, as determined by a BAC-cloned clinical isolate of human cytomegalovirus with preserved wild-type characteristics. J Virol 76(18): 9551-9555.
- Hahn, G., Revello, M. G., Patrone, M., Percivalle, E., Campanini, G., Sarasini, A., Wagner, M., Gallina, A., Milanesi, G., Koszinowski, U., Baldanti, F. and Gerna, G. (2004). Human cytomegalovirus UL131-128 genes are indispensable for virus growth in endothelial cells and virus transfer to leukocytes. J Virol 78(18): 10023-10033.
- Kahl, M., Siegel-Axel, D., Stenglein, S., Jahn, G. and Sinzger, C. (2000). Efficient lytic infection of human arterial endothelial cells by human cytomegalovirus strains. J Virol 74(16): 7628-7635.
- Lee, S. H., Albright, E. R., Lee, J. H., Jacobs, D. and Kalejta, R. F. (2015). Cellular defense against latent colonization foiled by human cytomegalovirus UL138 protein. Sci Adv 1(10): e1501164.
- Manicklal, S., Emery, V. C., Lazzarotto, T., Boppana, S. B. and Gupta, R. K. (2013). The “silent” global burden of congenital cytomegalovirus. Clin Microbiol Rev 26(1): 86-102.
- Murphy, E., Vanicek, J., Robins, H., Shenk, T., Levine, A. (2008). Supression of immegiate-early viral gene expression by herpesvirus-coded micoRNAs: Implications for latency. PNAS 105(14): 5453-5458.
- Plotkin, S. A., Furukawa, T., Zygraich, N. and Huygelen, C. (1975). Candidate cytomegalovirus strain for human vaccination. Infect Immun 12(3): 521-527.
- Reed, L. J. Muench, H. (1938). A simple method of estimating fifty percent endpoints. J Hygiene 27(3): 493-497.
- Saint Louis, C. (2016). CMV is a greater threat to infants than Zika, but far less often discussed. The New York Times.
- Sinzger, C., Hahn, G., Digel, M., Katona, R., Sampaio, K. L., Messerle, M., Hengel, H., Koszinowski, U., Brune, W. and Adler, B. (2008). Cloning and sequencing of a highly productive, endotheliotropic virus strain derived from human cytomegalovirus TB40/E. J Gen Virol 89(Pt 2): 359-368.
- Sinzger, C., Schmidt, K., Knapp, J., Kahl, M., Beck, R., Waldman, J., Hebart, H., Einsele, H. and Jahn, G. (1999). Modification of human cytomegalovirus tropism through propagation in vitro is associated with changes in the viral genome. J Gen Virol 80 (Pt 11): 2867-2877.
- Stinski, M. F. (1976). Human cytomegalovirus: glycoproteins associated with virions and dense bodies. J Virol 19(2): 594-609.
- Teng, M. W., Bolovan-Fritts, C., Dar, R. D., Womack, A., Simpson, M. L., Shenk, T. and Weinberger, L. S. (2012). An endogenous accelerator for viral gene expression confers a fitness advantage. Cell 151(7): 1569-1580.
- Waldman, W. J., Roberts, W. H., Davis, D. H., Williams, M. V., Sedmak, D. D. and Stephens, R. E. (1991). Preservation of natural endothelial cytopathogenicity of cytomegalovirus by propagation in endothelial cells. Arch Virol 117(3-4): 143-164.
- Waldman, W. J., Sneddon, J. M., Stephens, R. E. and Roberts, W. H. (1989). Enhanced endothelial cytopathogenicity induced by a cytomegalovirus strain propagated in endothelial cells. J Med Virol 28(4): 223-230.
- Wang, D. and Shenk, T. (2005). Human cytomegalovirus UL 131 open reading frame is required for epithelial cell tropism. J Virol 7910330-10338.
- Warming, S., Constantino, N., Court, D.L., Jenkins, N.A., Copeland, N.G. (2005). Simple and high efficient BAC recombineering using galK selection. Nucleic Acid Res 33(4): e36.
- Yamane, Y., Furukawa, T. and Plotkin, S. A. (1983). Supernatant virus release as a differentiating marker between low passage and vaccine strains of human cytomegalovirus. Vaccine 1(1): 23-25.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Saykally, V. R., Rast, L. I., Sasaki, J., Jung, S., Bolovan-Fritts, C. and Weinberger, L. S. (2017). A Bioreactor Method to Generate High-titer, Genetically Stable, Clinical-isolate Human Cytomegalovirus. Bio-protocol 7(21): e2589. DOI: 10.21769/BioProtoc.2589.
Category
Cell Biology > Cell isolation and culture > Cell growth
Microbiology > Microbial cell biology > Cell isolation and culture
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.