- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Articles In Press
Articles In Press are peer reviewed and have been accepted for publication. Please note that these versions may be subject to further edits before their final online publication. Nevertheless, Articles In Press are citable using the DOI. Upon the formal online publication, the article will no longer be listed here, but existing links will automatically redirect to the final version in the corresponding issue.
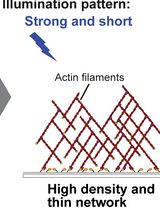
Optical Control of Actin Network Assembly on the Supported Lipid Bilayer
The spatiotemporal dynamics and density of actin networks are key determinants of actin cytoskeleton–mediated cellular functions. In vitro reconstitution systems have been widely used to study actin cytoskeletal dynamics; however, many existing approaches offer limited flexibility in controlling the geometry, thickness, and density of the assembled actin networks. Here, we present an in vitro optogenetic protocol that enables precise control of actin network assembly on supported lipid bilayers using an improved light-induced dimer (iLID)-SspB-based light-inducible dimerization system. In this system, His-mEGFP-iLID is anchored to a Ni-NTA-containing lipid bilayer, while SspB-mScarlet-I-VCA, a nucleation-promoting factor fused with SspB, together with other actin cytoskeletal proteins, is supplied in bulk solution. Upon blue light illumination, SspB-mScarlet-I-VCA is recruited to the membrane in a spatially and temporally defined manner, inducing localized actin polymerization. By tuning illumination patterns and duration, actin networks with defined density, thickness, and geometry can be generated, and polymerization can be rapidly halted by stopping illumination. This protocol provides a versatile platform for reconstructing actin networks with controlled spatial organization and density, enabling quantitative analysis of density-dependent interactions between actin networks and actin-binding proteins.
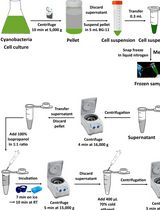
A Simple and Easy Method for RNA Extraction from the Cyanobacterium Synechocystis sp. PCC 6803
Cyanobacteria have been widely used as model organisms in photobiochemical research and have recently been exploited as hosts in numerous pilot studies to produce valuable biochemicals via genetic and metabolic modifications. Analyzing cellular RNA is a suitable method for studying genetic changes in cells. Several methods have previously been reported for cyanobacterial RNA extraction. However, the majority of these methods rely heavily on phenol and chloroform, which are hazardous. Additionally, these methods are time-consuming and difficult to perform. Using Synechocystis sp. PCC 6803 as a model, this study developed a novel method for extracting total ribonucleic acid (RNA) using standard centrifugation techniques and laboratory chemicals such as citric acid, ethylenediaminetetraacetic acid, sodium dodecyl sulfate, sodium chloride, and tri-sodium citrate dihydrate to extract RNA from cyanobacterial cells. This method does not necessitate the use of hazardous chemicals, especially phenol and chloroform. Furthermore, it is cost-effective since it does not require expensive chemicals. The results of the quantification, purity, and integrity checks show the effectiveness of this method for extracting good-quality RNA. Furthermore, RT-qPCR results demonstrate that the quality of the extracted RNA is suitable for downstream applications.
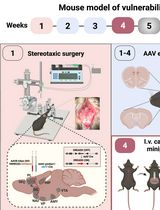
A Male Mouse Model of WIN 55,212–2 Self-Administration to Study Cannabinoid Addiction
Despite substantial progress in preclinical cannabinoid research, translational studies on cannabis use disorders (CUD) are still insufficient due to the absence of robust, validated animal models that fully recapitulate the multifactorial clinical phenotype of human CUD. The complex nature of CUD and the incomplete understanding of its underlying neurobiological mechanisms contribute to the limited availability of effective treatments. To address this gap, we developed an operant conditioning–based mouse model that enables the identification of individual vulnerability or resilience to CUD development. This highly translational model is based on the Diagnostic and Statistical Manual of Mental Disorders, 5th Edition (DSM-5) criteria for substance use disorders. The model allows the assessment of addiction-like behaviors by evaluating three behavioral domains: 1) persistence of responding during periods of cannabinoid unavailability, 2) motivation for cannabinoid seeking measured using a progressive ratio schedule, and 3) compulsivity, assessed when cannabinoid reward is paired with an aversive consequence such as a mild electric foot shock. A major strength of this paradigm is its ability to quantify two phenotypic traits proposed as predisposing factors for addiction vulnerability and two parameters related to craving. In addition, the model is specifically designed to evaluate genetic and circuit-level manipulations using chemogenetic approaches, with minor modifications required by surgical viral-vector delivery. Using this protocol, we can determine whether altering the excitability of specific neural networks promotes resilience or vulnerability to developing cannabinoid addiction. Elucidating these mechanisms is expected to facilitate the identification of novel and more effective therapeutic interventions for CUD.
Workflow for Crystallographic Fragment Screening by Crystal Soaking for Protein Targets: A Case Study on Thioredoxin Glutathione Reductase from Schistosoma mansoni
Among the biophysical techniques used in fragment-based drug discovery (FBDD) campaigns, crystallography is the most sensitive, allowing for the identification of low-affinity ligands and the characterization of protein–ligand complexes at atomic resolution. Although powerful, the proper application of this technique depends on obtaining crystals capable of diffracting X-rays at high resolution. Additionally, in crystallographic compound screening, the crystals must be resistant to multiple organic solvents used in chemical libraries, such as DMSO. In this protocol, we describe recombinant protein production suitable for crystallization and procedures for X-ray crystallographic screening of a library of 768 fragments. As a case study, we used the Schistosoma mansoni thioredoxin glutathione reductase (SmTGR), a redox enzyme with a key role in controlling oxidative stress in parasites of the Schistosoma genus, which causes schistosomiasis. As a validated pharmacological target, SmTGR is used in the development of new schistosomicidal drugs. The experimental pipeline includes SmTGR expression, purification, and crystallization, crystal soaking, diffraction data collection, and refinement. The 768 fragments from the Diamond-SGC Poised Library (DSPL) were individually soaked onto the crystals, and diffraction data were collected and processed at the I04-1 beamline of the Diamond Light Source synchrotron. Diffraction data were subsequently analyzed using PanDDA to identify fragment-binding events and to enable reliable detection of low-occupancy ligands within the protein crystal structures. In addition to the core experimental steps, this protocol incorporates systematic approaches to overcome limitations frequently encountered in crystallographic screening campaigns, including assessment of crystal solvent tolerance, acceleration of crystal mounting through the use of auxiliary devices, acoustic dispensing–based soaking of hundreds of fragments for low material consumption and high throughput, automated data collection, and efficient data analysis pipeline for the detection of weakly bound ligand. This protocol can be broadly applied to screen diverse compound sets against multiple targets amenable to crystallization.
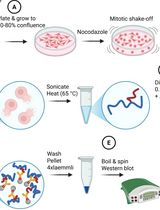
Denaturing SUMO Immunoprecipitation From Mitotic Cells
Small ubiquitin-related modifiers (SUMOs) are covalently conjugated onto the proteome and serve as signaling molecules in many aspects of eukaryotic cell biology, from S. cerevisiae and C. elegans to H. sapiens. The conjugatable SUMO variants, SUMO1 and the almost identical SUMO2 and SUMO3 (designated SUMO2/3), are processed by an E1(SAE1:SAE2)-E2(UBC9)-E3 enzyme cascade to produce SUMO-modified proteins. The prerogative of the SUMO biology field is to identify and study the specific proteins undergoing SUMOylation, which grants us insights into the biological pathway of interest. This protocol was developed using the human osteosarcoma cell line U2OS to enable the investigation of SUMO conjugates in mitosis, the cell division phase of the cell cycle. We enrich the cell population for mitotic cells, which are isolated and subjected to stringent lysis conditions involving a high concentration of SDS and DTT in RIPA buffer, to promote complete protein denaturation. The lysates in high SDS RIPA buffer are diluted to reduce the overall SDS concentration and undergo conventional immunoprecipitation using SUMO1- or SUMO2/3-specific antibodies bound to protein A/G agarose beads. The samples are then compatible with downstream readouts such as western blots and mass spectrometry. This protocol detects endogenous SUMOylated proteins and avoids exogenous SUMO overexpression, which can alter SUMO conjugate formation. Furthermore, this denaturing protocol ensures only SUMOylated proteins are immunoprecipitated, and not their interactors.

A Simple Method for Estimating the Spatiotemporal Distribution of Phenoloxidase Proteins in Insect Tissues
Laccase2 (Lac2), a member of the phenoloxidase (PO) family, is an essential oxidase for melanin pigmentation in insects. The identification of the in vivo spatial distribution of Lac2 is crucial for understanding the molecular mechanisms underlying color pattern formation. However, it is technically difficult to determine the distribution because Lac2 expression peaks at late pupal stages, when adult cuticle sclerotization has already begun. Here, we report a simple and rapid protocol for estimating the distribution of endogenous PO proteins, prophenoloxidases (proPOs) and phenoloxidases (POs), in insect tissues. In this method, the spatial distribution of endogenous PO proteins is estimated based on staining patterns formed by dopamine melanin synthesis in tissues incubated in a solution containing isopropanol and dopamine. We validated that tissues collected at approximately 80% of the total pupal duration yielded staining patterns corresponding to adult melanin-forming regions in three insect species. By comparing staining patterns across developmental stages, this protocol enables estimation of the timing of color pattern formation. Furthermore, the contrast between stained and unstained regions within the same tissue allows region-specific sampling, thereby facilitating an investigation of the underlying molecular mechanisms regulating spatial PO distribution. Taken together, this method facilitates the study of melanin biosynthesis and enables the identification of the genes involved in regulating color pattern formation. This protocol does not require antibodies, transgenic lines, or specialized equipment and can be completed within a short time frame. Its effectiveness has been validated in multiple coleopteran and lepidopteran species, demonstrating its broad applicability as a versatile tool for studying insect pigmentation and color pattern formation.

MDISCO: A High-Throughput Tissue-Clearing Protocol for Preservation of Endogenous Fluorescence in Whole Mouse Brains

Organic solvent–based tissue clearing methods are widely used for whole-brain imaging but often compromise endogenous fluorescence. Existing protocols, such as iDISCO and fluorescence-preserving variants, have improved optical transparency but still present trade-offs between fluorescence retention, tissue stability, and workflow complexity. Here, we present MDISCO, a modified iDISCO-based clearing protocol designed to enhance preservation of endogenous fluorescence while maintaining high transparency and stable tissue morphology. MDISCO is directly compared with FDISCO+, an established fluorescence-preserving protocol, for the preservation of endogenous tdTomato and YFP. Performance across clearing steps is evaluated by measuring brain weight, anteroposterior and mediolateral dimensions, and optical transparency before and after solvent clearing and refractive index matching. Fluorescence preservation is assessed using whole-brain light-sheet microscopy with standardized imaging parameters to enable direct comparison. This protocol provides an accessible and high-throughput, reproducible workflow for solvent-based clearing with robust endogenous fluorescence preservation, offering clear advantages for whole-brain 3D imaging of genetically encoded fluorescent reporters.
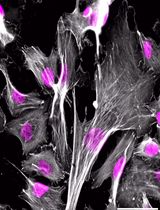
Isolation, Culture, and Differentiation of Bovine Muscle Resident Stem Cells
Bovine muscle satellite cells (MuSC) and fibro-adipogenic progenitor cells (FAP) are muscle resident stem cells that are responsible for postnatal muscle growth, intramuscular fat deposition, and extracellular matrix generation. These cells are of increasing interest for the cultivated meat community due to their ability to generate all the major components of meat; additionally, these cells are of interest to conventional animal science research to elucidate mechanisms to improve meat quality. To use these cells for these goals, efficient and accurate cell isolation, culture, and differentiation are essential to evaluate their cell fate decisions and behaviors. In this protocol, we detail a simultaneous isolation of both MuSCs and FAPs with multiple intermediate stopping points, allowing for flexibility for day-of time constraints. We also detail improved growth conditions to maximize cell expansion and procedures to assess cell differentiation. This protocol provides a flexible isolation procedure that is compatible with sampling in modern slaughterhouses or from biopsies. Additionally, the differentiation procedures provide improved differentiation but still allow in vitro treatment and assessment.

3D STED Super-Resolution Imaging Strategy for Visualizing Synaptic Nano-architecture in Brain Cryosections
Super-resolution imaging of synapses in intact brain tissue remains challenging because light scattering, photobleaching, and limited probe penetration, along with antigen accessibility within the densely packed postsynaptic densities (PSDs), constrain resolution and labeling efficiency. Here, we present a protocol utilizing thin brain cryosections and tau-stimulated emission depletion (STED) nanoscopy to visualize the intricate nano-architecture of excitatory synapses in situ. Slicing the brain into 6 μm sections allows for highly efficient and even penetration of probes throughout sections while ensuring that the resolution is not significantly impacted by the imaging depth of the tissue. We outline step-by-step instructions for labeling pre- and postsynaptic nano-architecture using antibodies and nanobodies, highlighting how fixative choice influences the labeling efficiency of synaptic proteins. While this protocol is compatible with both confocal and super-resolution imaging, when combined with rapid image acquisition times of tau-STED, it enables clear separation of key synaptic features in three dimensions with minimal photobleaching. Thus, this approach enables robust multiplex imaging of fluorescently labeled synaptic proteins in the brain, providing exceptional spatial resolution for visualization and quantification of synaptic nanoarchitecture in its native environment.
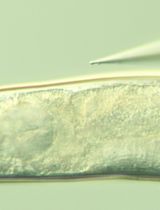
Microinjection of Synthetic Peptides Into Caenorhabditis elegans
The genome of the nematode Caenorhabditis elegans encodes at least 160 predicted peptide precursor genes that can generate over 300 bioactive peptides, the functions of most of which remain unknown. Phenotypes resulting from deletion or transgenic expression of peptide genes are readily assayed, but genetic dissection of individual peptide activities is often confounded when a single gene encodes multiple peptides or when distinct peptides act redundantly. Here, we describe a protocol for direct microinjection of chemically synthesized peptides into individual worms. This approach permits investigation of the effects of an individual peptide while providing precise temporal control over peptide delivery.
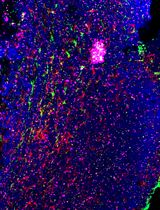
Simultaneous Immunofluorescence-Based In Situ mRNA Expression and Protein Detection in Bone Marrow Biopsy Samples
Fluorescence in situ hybridization (FISH) can be employed to study the expression and subcellular localization of nucleic acids by using labeled antisense strands that hybridize with the target RNA or DNA molecules. Likewise, immunofluorescence antibody staining (IF) takes advantage of the specific interaction between a fluorophore-labeled antibody and its corresponding antigen. This protocol reports the combination of RNA-FISH and IF antibody staining for simultaneous detection of both RNA transcripts and proteins of interest in routine formalin-fixed paraffin-embedded (FFPE) bone marrow biopsy samples. Herein, we provide a detailed description of the methodology that we have developed and optimized to study the spatial expression of two transcripts—TGFB1 and PDGFA1—in human hematopoietic (CD45+) and non-hematopoietic (CD271+) cells in the bone marrow of patients with acute lymphoblastic leukemia (ALL).

Electrophoretic Mobility Shift Assay (EMSA) for Assessing RNA–Protein Binding and Complex Formation Using Recombinant RNA-Binding Proteins and In Vitro–Transcribed RNA

Evaluating RNA–protein interactions is key to understanding post-transcriptional gene regulation. Electrophoretic mobility shift assays (EMSAs) remain a widely used technique to study these interactions, revealing information about binding affinities and binding modalities, including cooperativity and complex formation. Here, we detail, in a step-by-step protocol, how to perform EMSAs. We describe how to generate, purify, and quantitate 32P-radiolabeled RNA by in vitro transcription, as well as the expression and purification of recombinant RNA-binding proteins in E. coli using ELAV as an example. We then describe how to set up binding reactions using serial dilutions in a microtiter plate format of recombinant ELAV and in vitro–transcribed RNA and how to perform EMSAs using native low-crosslinked acrylamide gels, with detailed graphically supported instructions and troubleshooting guides.
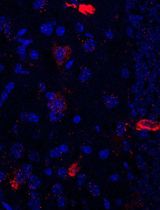
Using combined fluorescent in situ hybridization with Immunohistochemistry to co-localize mRNA in diverse neuronal cell types
Understanding gene expression within defined neuronal populations is essential for dissecting the cellular and molecular diversity of the brain. mRNA assays provide a direct readout of gene expression, capturing transcriptional changes that may precede or occur independently of protein abundance, whereas protein assays reflect the cumulative effects of translation, modification, and degradation. Moreover, in histological analysis, immunohistochemical protein detection results in visually diffuse labeling, which makes it difficult to quantitatively assess levels and locations of expression at high resolution. Here, we present a protocol that allows for mRNA detection in single neuronal cell types with a high degree of sensitivity and anatomical resolution. This protocol combines fluorescent in situ hybridization (FISH) with immunohistochemistry (IHC) on the same tissue section. Briefly, FISH is carried out by ACDBio RNAscope® fluorescent in situ hybridization technology, which involves processing the tissue sections, followed by signal amplification. This involves target retrieval, probe hybridization, and signal enhancement. Then, the tissue section is processed for IHC, which involves blocking nonspecific sites and incubation with primary antibodies, followed by development of a fluorescent signal with secondary antibodies. Typically, visual mRNA detection with FISH can be seen as individual puncta, whereas targeting the protein with an antibody results in filled cells or processes. The variation in staining pattern allows for the quantification of distinct mRNA transcripts within different neuronal populations, which renders co-localization analyses easy and efficient.
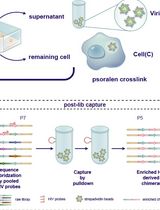
High-resolution mapping of RNA-RNA interactions across the HIV-1 genome with HicapR
The genomes of RNA viruses can fold into dynamic structures that regulate their own infection and immune evasion processes. Proximity ligation methods (e.g., SPLASH) enable genome-wide interaction mapping but lack specificity when dealing with low-abundance targets in complex samples. Here, we describe HiCapR, a protocol integrating in vivo psoralen crosslinking, RNA fragmentation, proximity ligation, and hybridization capture to specifically enrich viral RNA–RNA interactions. Captured libraries are sequenced, and chimeric reads are analyzed via a customized computational pipeline to generate constrained secondary structures. HiCapR generates high-resolution RNA interaction maps for viral genomes. We applied it to resolve the in vivo structure of the complete HIV-1 RNA genome, identifying functional domains, homodimers, and long-range interactions. The protocol's robustness has been previously validated on the SARS-CoV-2 genome. HiCapR combines proximity ligation with targeted enrichment, providing an efficient and specific tool for studying RNA architecture in viruses, with broad applications in virology and antiviral development.

Enhanced RNA-Seq Expression Profiling and Functional Enrichment in Non-model Organisms Using Custom Annotations
Functional enrichment analysis is essential for understanding the biological significance of differentially expressed genes. Commonly used tools such as g:Profiler, DAVID, and GOrilla are effective when applied to well-annotated model organisms. However, for non-model organisms, particularly for bacteria and other microorganisms, curated functional annotations are often scarce. In such cases, researchers often rely on homology-based approaches, using tools like BLAST to transfer annotations from closely related species. Although this strategy can yield some insights, it often introduces annotation errors and overlooks unique species-specific functions. To address this limitation, we present a user-friendly and adaptable method for creating custom annotation R packages using genomic data retrieved from NCBI. These packages can be directly imported as libraries into the R environment and are compatible with the clusterProfiler package, enabling effective gene ontology and pathway enrichment analysis. We demonstrate this approach by constructing an R annotation package for Mycobacterium tuberculosis H37Rv, as an example. The annotation package is then utilized to analyze differentially expressed genes from a subset of RNA-seq dataset (GSE292409), which investigates the transcriptional response of M. tuberculosis H37Rv to rifampicin treatment. The chosen dataset includes six samples, with three serving as untreated controls and three exposed to rifampicin for 1 h. Further, enrichment analysis was performed on genes to demonstrate changes in response to the treatment. This workflow provides a reliable and scalable solution for functional enrichment analysis in organisms with limited annotation resources. It also enhances the accuracy and biological relevance of gene expression interpretation in microbial genomics research.
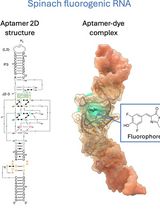
Visualizing diverse RNA functions in living cells with Spinach™ family of fluorogenic aptamers
RNA is now recognized as a highly diverse and dynamic class of molecules whose localization, processing, and turnover are central to cell function and disease. Live-cell RNA imaging is therefore essential for linking RNA behavior to mechanism. Existing approaches include quenched hybridization probes that directly target endogenous transcripts but face delivery and sequestration issues, protein-recruitment tags such as MS2/PP7 that add large payloads and can perturb localization or decay, and CRISPR–dCas13 imaging that requires substantial protein cargo and careful control of background and off-target effects. Here, we present a protocol for live-cell RNA imaging using the SpinachTM family of fluorogenic RNA aptamers. The method details the design and cloning of SpinachTM-tagged RNA constructs, selection and handling of cognate small-molecule fluorophores, expression in mammalian cell lines, dye loading, and image acquisition on standard fluorescence microscopes, followed by quantitative analysis of localization and dynamics. We include controls to verify aptamer expression and signal specificity, guidance for multiplexing with related variants (e.g., Broccoli, Corn, Squash, Beetroot), and troubleshooting for dye permeability and signal optimization. Application examples illustrate use in tracking cellular delivery of mRNA therapeutics, monitoring transcription and decay in response to perturbations, and the forming of toxic RNA aggregates. Compared with prior methods, SpinachTM tags are compact, genetically encodable, and fluorogenic, providing high-contrast imaging in both the nucleus and cytoplasm with single-vector simplicity and multiplexing capability. The protocol standardizes key steps to improve robustness and reproducibility across cell types and laboratories.
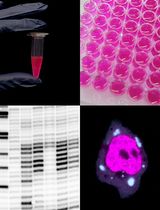
Enhancement of RNA Imaging Platforms by the Use of Peptide Nucleic Acid-Based Linkers
RNA imaging techniques enable researchers to monitor RNA localization, dynamics, and regulation in live or fixed cells. While the MS2-MCP system—comprising the MS2 RNA hairpin and its binding partner, the MS2 coat protein (MCP)—remains the most widely used approach, it relies on a tag containing multiple fluorescent proteins and has several limitations, including the potential to perturb RNA function due to the tag’s large mass. Alternative methods using small-molecule binding aptamers have been developed to address these challenges. This protocol describes the synthesis and characterization of RNA-targeting probes incorporating a peptide nucleic acid (PNA)-based linker within the cobalamin (Cbl)-based probe of the Riboglow platform. Characterization in vitro involves a fluorescence turn-on assay to determine binding affinity (KD) and selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE) footprinting analysis to assess RNA-probe interactions at a single nucleotide resolution. To show the advancement of PNA probes in live cells, we present a detailed approach to perform both stress granule (SG) and U-body assays. By combining sequence-specific hybridization with structure-based recognition, our approach enhances probe affinity and specificity while minimizing disruption to native RNA behavior, offering a robust alternative to protein-based RNA imaging systems.
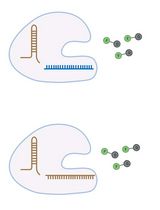
Amplification-Free Detection of Highly Structured RNA Molecules Using SCas12aV2
The CRISPR/Cas12a system has revolutionized molecular diagnostics; however, conventional Cas12a-based methods for RNA detection typically require transcription and pre-amplification steps. Our group has recently developed a diagnostic technique known as the SCas12a assay, which combines Cas12a with a split crRNA, achieving amplification-free detection of miRNA. However, this method still encounters challenges in accurately quantifying long RNA molecules with complex secondary structures. Here, we report an enhanced version termed SCas12aV2 (split-crRNA Cas12a version 2 system), which enables direct detection of RNA molecules without sequence limitation while demonstrating high specificity in single-nucleotide polymorphism (SNP) applications. We describe the general procedure for preparing the SCas12a system and its application in detecting RNA targets from clinical samples.

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics





