- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Creating a RAW264.7 CRISPR-Cas9 Genome Wide Library
Published: Vol 7, Iss 10, May 20, 2017 DOI: 10.21769/BioProtoc.2320 Views: 13772
Reviewed by: Ivan ZanoniAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Oct 2016
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
The bacterial clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9 genome editing tools are used in mammalian cells to knock-out specific genes of interest to elucidate gene function. The CRISPR-Cas9 system requires that the mammalian cell expresses Cas9 endonuclease, guide RNA (gRNA) to lead the endonuclease to the gene of interest, and the PAM sequence that links the Cas9 to the gRNA. CRISPR-Cas9 genome wide libraries are used to screen the effect of each gene in the genome on the cellular phenotype of interest, in an unbiased high-throughput manner. In this protocol, we describe our method of creating a CRISPR-Cas9 genome wide library in a transformed murine macrophage cell-line (RAW264.7). We have employed this library to identify novel mediators in the caspase-11 cell death pathway (Napier et al., 2016); however, this library can then be used to screen the importance of specific genes in multiple murine macrophage cellular pathways.
Keywords: CRISPRBackground
Historically, understanding the contribution of specific genes to phenotypes of interest in eukaryotic cells was possible using RNA interference (RNAi) or cells derived from knockout mice. However, within the last few years the new genome editing technique CRISPR-Cas9 has allowed for easy and efficient generation of knockout cell lines and genome-wide screens within eukaryotic cells. CRISPR-Cas9 genome-wide screens have expanded the toolbox for mammalian genetics and for the identification of novel proteins and their contributions to specific phenotype. Using this method, researchers have been able to identify novel genes involved in tumor growth (Chen et al., 2015; Kiessling et al., 2016; Steinhart et al., 2017), microbial entry and replication (Popov et al., 2015; Marceau et al., 2016), cell death pathways (Shi et al., 2015; Napier et al., 2106), and much more. Here we harness the CRISPR-Cas9 system, to create a genome-wide knockout library in a murine macrophage cell line. Macrophages are the crux of many innate immune responses to invading pathogens or danger signals. By creating a genome-wide knockout library in macrophages we can now begin to identify novel mediators of these innate immune responses to identify novel diagnostic and therapeutic targets for acute and chronic inflammation.
Materials and Reagents
- Pipette tips
- 10 cm bacteriological tissue culture (TC) treated Petri dish (Corning, Falcon®, catalog number: 353003 )
- 6-well plates TC treated (Corning, Costar®, catalog number: 353502 )
- 15 ml Falcon tube (E&K Scientific Products, catalog number: EK-4020 )
- 50 ml Falcon tube (E&K Scientific Products, catalog number: EK-4023 )
- Cell scraper (SARSTEDT, catalog number: 83.1830 )
- 0.45 μm syringe filters (Corning, catalog number: 431225 )
- T-75 flask (Corning, Falcon®, catalog number: 353136 )
- T-125 flask (Corning, Falcon®, catalog number: 353112 )
- Cryovials (Corning, catalog number: 430659 )
- RAW264.7 cells (ATCC, catalog number: TIB-71 )
- 293T cells (ATCC, catalog number: CRL-3216 )
- hCas9 plasmid (Addgene, catalog number: 52962 )
- FuGene (Promega, catalog number: E2311 )
- VSV-G (Addgene, catalog number: 8454 )
- pAdVAntage (Promega, catalog number: E1711 )
- △VPR (Addgene; catalog number: 8455 )
- Genome-wide Mouse lentiviral CRISPR gRNA library v1 (Addgene, catalog number: 50947 )
- Filtered DMEM (Thermo Fisher Scientific, GibcoTM, catalog number: 11995073 )
- Filtered (0.2 μm filter) heat-inactivated (30 min at 37 °C) FBS (Thermo Fisher Scientific, GibcoTM; stock specific)
- Protamine sulphate (Sigma-Aldrich, catalog number: P3369-10G ), keep stock at 8 mg/ml at 4 °C
- Blasticidin hydrochloride (MP Biomedicals, catalog number: 02150477-25 mg ), keep aliquoted stock at 10 mg/ml at -20 °C
- DMSO (Fisher Scientific, catalog number: BP231-100 )
- Puromycin dihydrochloride from Streptomyces alboniger (Sigma-Aldrich, catalog number: P8833 )
- RAW264.7 and 293T tissue culture media (see Recipes)
- Blasticidin selection media (see Recipes)
- Puromycin selection media (see Recipes)
- Tissue culture freezing media (see Recipes)
Equipment
- Pipettes
- Fluorescence microscope that can visualize BFP
- Incubator at 37 °C with 5% CO2
- Centrifuge
Procedure
- Making hCas9-expressing RAW264.7 cells
- D0: Plate 5 x 106 293T cells in a tissue culture (TC) treated 10 cm plate in 10 ml DMEM + 10% FBS, and incubate overnight at 37 °C with 5% CO2. We aim for 30-40% confluency.
- D1: Create hCas9-expressing retrovirus in 293T cells.
- Premix plasmids in 150 μl of serum-free DMEM
For a 10 cm dish (retrovirus PMX-hCas9-blasti):
1.3 μg △VPR
0.87 μg VSV-G
0.55 μg p-Advant
0.73 μg PMX-hCas9-blasti (For the plasmid map see http://www.addgene.org/52962/) - Add 15 μl of Fugene and let sit at 25 °C for 20 min.
- During incubation replace 293T media (see Recipes) with 10 ml of fresh 37 °C DMEM + 10% FBS.
- Add mixture drop by drop on 293T cells and mix by gently rocking plate back and forth.
- Incubate at 37 °C + 5% CO2 overnight.
- D2:
- Gently aspirate media off transfected 293T cells and replace with 6-8 ml of 37 °C DMEM + 10% FBS. The monolayer will be delicate (see Note 1).
- Plate RAW264.7 cells for transduction with Cas9-expressing virus on D3, at 3 x 105 cells/well in a 6-well plate, 1 well per sample. Additionally, plate 3x RAW264.7 cells at 3 x 105 cells/well in a 6-well plate for assessing viral titer.
- D3: Harvest virus and transduce RAW cells
- Harvest virus in the morning and the evening, from the same well (see Note 2).
- At each time point, gently remove supernatant from 293T cells that contains virus. For the morning time point, after removing supernatant add 6 ml of 37 °C DMEM + 10% FBS media to the 293T virus-producing cells. Again, be very careful with this monolayer.
- Filter virus-containing media with 0.45 μm syringe filter to ensure removal of cellular debris.
- Add protamine sulphate at 8 μg/ml to the filtered virus-containing media.
- Aspirate media off seeded RAW264.7 cells and add 2-5 ml of supernatant containing viruses (see Note 3). The control will just be DMEM only (see Note 4).
- For viral titer, prepare 3 10-fold dilutions of virus in serum-free DMEM and continue with assessing viral titer by plaque assay as previously published (Watanabe et al., 2016).
- Incubate at 37 °C + 5% CO2 overnight.
- D4: Aspirate media off RAW264.7 cells and add new 37 °C DMEM + 10% FBS media to transduced RAW cells.
- D5: Transfer transduced RAW264.7 cells from 6-well plate to TC treated 10 cm dish. This split should yield an ideal 30% confluence for antibiotic selection the next day. At this time, plate non-transduced RAW264.7 cells in 10 cm at a similar confluency as a negative control for your antibiotic selection (~1 x 106 cells per 10 cm dish).
- Remove media from transduced RAW264.7 cells, replace with 3 ml of fresh 37 °C DMEM + 10% FBS media.
- Using cell scrapers, scrape adherent cells off of bottom of wells and resuspend in media by pipetting up and down.
- Replate cells in a TC treated 10 cm dish in 10 ml of fresh 37 °C DMEM + 10% FBS media, aiming for 30% confluency. No cell count is necessary; this split should yield an ideal 30% confluency.
- Incubate at 37 °C + 5% CO2 overnight.
- D6: Antibiotic selection
- Removed media from transduced RAW264.7 cells.
- Add 4 ml of blasticidin selection media (see Recipes) (37 °C DMEM + 10% FBS + 10 μg/ml blasticidin) to transfected RAW264.7 cells and non-transfected RAW264.7 cells for a negative control.
- Incubate at 37 °C + 5% CO2 for 24-48 h.
- D7-8:
- Aspirate media off selected RAW264.7-hCas9 expressing cells and replace with 4 ml of fresh 37 °C DMEM + 10% FBS + 10 μg/ml blasticidin.
- Incubate at 37 °C + 5% CO2 for 24-48 h to expand cell line. Incubation period will vary, however you are aiming for > 90% cell death in your non-transfected RAW264.7 cell negative control plate.
- D8-9:
- Aspirate media off selected RAW264.7-hCas9 expressing cells and add 10 ml of 37 °C DMEM + 10% FBS. Incubate these cells for 2-4 days to allow RAW264.7 cell recovery from the selection process before replating for experiments or freezing back.
- When freezing back: Use the cell scrapers, scrape off selected RAW264.7-hCas9 expressing cells and either freeze back cells in tissue culture freezing media (see Recipes) (50% DMEM + 40% FBS + 10% DMSO) or re-plate for future experiments.
- D0: Plate 5 x 106 293T cells in a tissue culture (TC) treated 10 cm plate in 10 ml DMEM + 10% FBS, and incubate overnight at 37 °C with 5% CO2. We aim for 30-40% confluency.
- Amplifying CRISPR gRNA library
- Follow protocol previously published (Koike-Yusa et al., 2014; https://media.addgene.org/data/67/85/9641c46c-8523-11e3-b6c5-000c298a5150.pdf). Use the ‘on-plate culture’ method of amplifying the library, as the ‘liquid culture’ method can lead to exclusion of some of the plasmids in the library (Figure 1).
- Freeze back amplified CRISPR gRNA plasmid library.
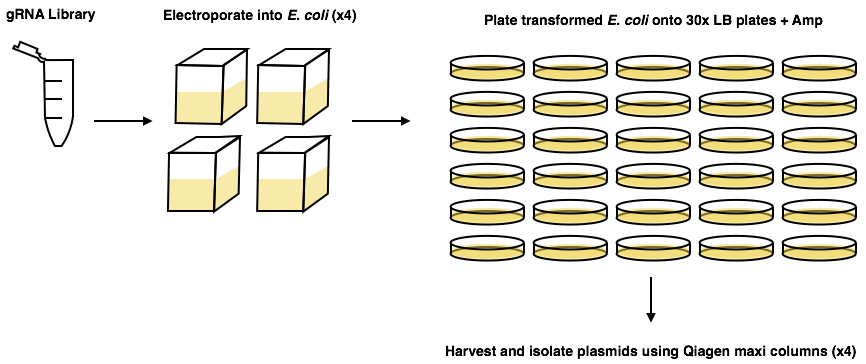
Figure 1. Graphical abstract of Koike-Yusa et al. (2014) amplification protocol of the gRNA library
- Follow protocol previously published (Koike-Yusa et al., 2014; https://media.addgene.org/data/67/85/9641c46c-8523-11e3-b6c5-000c298a5150.pdf). Use the ‘on-plate culture’ method of amplifying the library, as the ‘liquid culture’ method can lead to exclusion of some of the plasmids in the library (Figure 1).
- Creating CRISPR gRNA lentiviral library
- Before starting, see Note 5.
- D0: Plate 1 x 106 293T cells per T-175 in 25 ml of DMEM + 10% FBS.
- D1: Creating CRISPR gRNA lentiviral library.
- Premix plasmids in 150 μl of serum-free DMEM.
For a T-175 flask:
4 μg △VPR
2.6 μg VSV-G
1.66 μg p-Advant
6.6 μg CRISPR library - Add 45 μl of Fugene and let sit at 25 °C for 20 min.
- During incubation replace 293T media with 25 ml of fresh, 37 °C DMEM + 10% FBS.
- Add mixture drop by drop on 293T cells and mix with soft horizontal movements.
Incubate at 37 °C + 5% CO2 overnight. - D2:
- Gently replace media on transfected 293T cells with 20 ml 37 °C DMEM + 10% FBS (see Note 3).
- Plate RAW264.7-hCas9 expressing cells for transfection on D3. To calculate needed RAW264.7 cells, see Procedure C. Additionally, plate 3x RAW264.7 cells at 3 x 105 cells/well in a 6-well plate for assessing viral titer.
- D3: Harvest virus and transduce RAW264.7 cells.
- Harvest virus in the AM and PM.
- Removed supernatant from 293T cells that contains virus.
- Filter with 0.45 μm syringe filter.
- Add protamine sulphate at 8 μg/ml.
- Aspirate media off seeded cells and add X ml of supernatant containing viruses, so that you have 30% transfection efficiency, and add 37 °C DMEM + 10% FBS up to 20 ml per T-175 flask (see Note 5).
- At this point, you can freeze back your virus to check for viral titer if you haven’t checked the transfection efficiency previously.
- Incubate at 37 °C + 5% CO2.
- D4: Plate 5 x 106 RAW264.7-hCas9 expressing cells in a T-175 flask in 25 ml of 37 °C DMEM + 10% FBS for antibiotic selection negative control.
- D6: Check transfection efficiency using fluorescence microscope and antibiotic selection.
- BFP is constitutively expressed on the lentiviral CRISPR library plasmid (Koike-Yusa et al., 2014), and you can visualize transfection efficiency by plating an aliquot of transfected cells and calculating the % fluorescence. This will not be a quantitative measure of transfection, but will allow you to be confident that your transfection worked before moving on to the antibiotic selection. Use only the cells that have been selected using antibiotics for future work.
- For puromycin selection for RAW264.7 transfected cells:
- Aspirate media off transfected RAW264.7 transfected cells and negative control, and replace with 20 ml of 37 °C DMEM + 10% FBS + 5 μg/ml puromycin.
- Incubate for 72 h at 37 °C + 5% CO2.
- D9: Split and expand RAW264.7 cells transfected with the CRISPR gRNA lentiviral library.
- Aspirate media off puromycin-selected RAW264.7 cells transfected with the CRISPR lentiviral library.
- Add 10 ml of 37 °C DMEM + 10% FBS + 5 μg/ml puromycin.
- Scrape adherent RAW264.7 cells off of the T-175 flask into the 10 ml of media.
- Pool all of selected RAW264.7 cells transfected with the CRISPR lentiviral library and pellet at 1,000 x g for 5 min.
- Resuspend the pellet of selected RAW264.7 cells transfected with the CRISPR lentiviral library in 37 °C DMEM + 10% FBS + 5 μg/ml puromycin and plate 1 x 106 cells per T-175 flask. There should be many T-175 flasks.
- Incubate for 72 h at 37 °C + 5% CO2.
- D13: Pool and freeze RAW264.7 cells transfected with the CRISPR gRNA lentiviral library.
- Each T-175 flask should contain many millions of selected RAW264.7 cells transfected with the CRISPR lentiviral library (on average 3-5 x 107).
- Aspirate media off selected RAW264.7 cells transfected with the CRISPR lentiviral library.
- Add 10 ml of 37 °C DMEM + 10% FBS.
- Scrape adherent RAW264.7 cells off of the T-175 flask into the 10 ml of media.
- Pool all of selected RAW264.7 cells transfected with the CRISPR lentiviral library and pellet at 1,000 x g for 5 min.
- Resuspend pellet in 50 ml of 37 °C DMEM + 10% FBS and count cells.
- At this point, freeze back the library at 1.5 x 107 cells per cryovial in 50% DMEM + 40% FBS + 10% DMSO media or use these cells for screening.
- Before starting, see Note 5.
Data analysis
For sequencing your library and analyzing the coverage of your library or your library results, please refer to our original publication (Napier et al., 2016).
Notes
- Late in the day, you may start to see syncytia. Syncytia are a sign that the viruses are being produced.
- Retroviruses more easily infect mitotic cells, thus by harvesting twice from the same well in the AM and PM, you will double your chances of catching the RAW264.7 cells dividing.
- You can store the rest in 4 °C for re-infection later for up to 7 days. However, freezing decreases titers significantly.
- In our hands, transduction of RAW264.7 cells do not change the immunological profile of the cell line, however, if another cell line is being used in this protocol, it is imperative to use a retroviral control with a non-coding sequence to ensure that the phenotype of interest is not altered after transduction.
- This protocol has been standardized for cell transfection efficiency of 30% by lentivirus produced by 293T cells. This lower transfection efficiency is used to decrease the frequency of double or triple transfection events happening the same cell. You will be transfecting 1,000-fold the gRNA library in cells. For this protocol, there are 87,897 gRNAs (Koike-Yusa et al., 2014); therefore, we transfect (88 million RAW246.7 cells) x (30%) = 264 million RAW246.7 cells to maintain 1,000-fold coverage with 30% transfection efficiency.
Recipes
- RAW264.7 and 293T tissue culture media
37 °C DMEM
10% FBS - Blasticidin selection media
37 °C DMEM
10% FBS
10 μg/ml blasticidin - Puromycin selection media
37 °C DMEM
10% FBS
5 μg/ml puromycin - Tissue culture freezing media
50% DMEM
40% FBS
10% DMSO
Acknowledgments
This research was supported by the National Institute of Allergy and Infectious Diseases grants 1F32AI115959-01 (to B.A. Napier) and AI095396-05 (to D.M. Monack), and Defense Advanced Research Projects Agency (DARPA) grant DARPA-15-21-ThoR-FP-006 (to D.M. Monack).
References
- Chen, S., Sanjana, N. E., Zheng, K., Shalem, O., Lee, K., Shi, X., Scott, D. A., Song, J., Pan, J. Q., Weissleder, R., Lee, H., Zhang, F. and Sharp, P. A. (2015). Genome-wide CRISPR screen in a mouse model of tumor growth and metastasis. Cell 160(6): 1246-1260.
- Kiessling, M. K., Schuierer, Sven., Stertz, Silke., Beibel, Martin., Bergling, Sebastian., Knehr, Judith., Carbone, Walter., de Vallière, Cheryl., Tchinda, Joelle., Bouwmeester, Tewis., Seuwen, Klaus., Rogler, Gerhard. and Roma, Guglielmo. (2016). Identification of oncogenic driver mutations by genome-wide CRISPR-Cas9 dropout screening. BMC genomics 17, 723.
- Koike-Yusa, H., Li, Y., Tan, E. P., Velasco-Herrera Mdel, C. and Yusa, K. (2014). Genome-wide recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library. Nat Biotechnol 32(3): 267-273.
- Marceau, C. D., Puschnik, A. S., Majzoub, K., Ooi, Y. S., Brewer, S. M., Fuchs, G., Swaminathan, K., Mata, M. A., Elias, J. E., Sarnow, P. and Carette, J. E. (2016). Genetic dissection of Flaviviridae host factors through genome-scale CRISPR screens. Nature 535(7610): 159-163.
- Napier, B. A., Brubaker, S. W., Sweeney, T. E., Monette, P., Rothmeier, G. H., Gertsvolf, N. A., Puschnik, A., Carette, J. E., Khatri, P. and Monack, D. M. (2016). Complement pathway amplifies caspase-11-dependent cell death and endotoxin-induced sepsis severity. J Exp Med 213(11): 2365-2382.
- Popov, L. M., Marceau, C. D., Starkl, P. M., Lumb, J. H., Shah, J., Guerrera, D., Cooper, R. L., Merakou, C., Bouley, D. M., Meng, W., Kiyonari, H., Takeichi, M., Galli, S. J., Bagnoli, F., Citi, S., Carette, J. E. and Amieva, M. R. (2015). The adherens junctions control susceptibility to Staphylococcus aureus α-toxin. Proc Natl Acad Sci U S A 112(46): 14337-14342.
- Shi, J., Zhao, Y., Wang, K., Shi, X., Wang, Y., Huang, H., Zhuang, Y., Cai, T., Wang, F. and Shao, F. (2015). Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526(7575): 660-665.
- Steinhart, Z., Pavlovic, Z., Chandrashekhar, M., Hart, T., Wang, X., Zhang, X., Robitaille, M., Brown, K. R., Jaksani, S., Overmeer, R., Boj, S. F., Adams, J., Pan, J., Clevers, H., Sidhu, S., Moffat, J. and Angers, S. (2017). Genome-wide CRISPR screens reveal a Wnt-FZD5 signaling circuit as a druggable vulnerability of RNF43-mutant pancreatic tumors. Nat Med 23(1): 60-68.
- Watanabe, S., Chan, K. K. and Vasudevan, S. G. (2016). Mouse Model of Dengue Virus Infection with Serotypes 1 and 2 Clinical Isolates. Bio-protocol 6(23): e2040.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Napier, B. A. and Monack, D. M. (2017). Creating a RAW264.7 CRISPR-Cas9 Genome Wide Library. Bio-protocol 7(10): e2320. DOI: 10.21769/BioProtoc.2320.
Category
Immunology > Immune cell function > Macrophage
Molecular Biology > RNA > Transfection
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.