- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Subcellular Localization Experiments and FRET-FLIM Measurements in Plants
Published: Vol 4, Iss 1, Jan 5, 2014 DOI: 10.21769/BioProtoc.1018 Views: 18600
Reviewed by: Anonymous reviewer(s)
Original research article
The authors used this protocol in:

Apr 2013
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Determining the localization of proteins within living cells may be very essential for understanding their biological function. Usually for analysis of subcellular localization, a construct encoding the translational fusion of a cDNA of interest with a fluorescent protein (FP) is engineered, transiently expressed in plant cells and examined with confocal microscopy.
In co-localization and interaction studies, two plasmids, each encoding one of the potential interacting/binding partners tagged with an appropriate pair of fluorescence proteins (for instance CFP/YFP) are co-expressed in plant cells. If proteins co-localize in certain cellular compartments it does not necessarily mean that they bind/interact to each other, therefore an additional technique should be applied for in vivo verification of putative interaction, e.g. Fluorescence Lifetime Imaging (FLIM) to detect Fluorescence Resonance Energy Transfer (FRET).
The protocol describes in detail the method that has been used to verify interaction between the bacterial effector HopQ1 and a 14-3-3a host protein and additionally to check the necessity of the central serine in the canonical 14-3-3 binding site within HopQ1 (Giska et al., 2013) for this association.
Materials and Reagents
- 3 – 4 weeks old Nicotiana benthamiana plants grown in a greenhouse
- Agrobacterium tumefaciens strain GV3101
- For co-localization and FRET-FLIM measurement selected pGWB (Gateway) binary vectors were used (Nakagawa et al., 2007):
- pGWB454 encoding Protein1 (here HopQ1-mRFP) -mRFP
- pGWB441 encoding Protein2 (here 14-3-3a-YFP) -YFP
- pGWB444 encoding unfused, free CFP
- pGWB441 encoding unfused, free YFP
- pGWB444 encoding CFP fused to YFP
- pGWB441 encoding Protein1- (here HopQ1-YFP) -YFP
- pGWB441 encoding Protein1a-YFP (e.g. mutated variant of Protein1)
- pGWB444 encoding Protein2 (here 14-3-3a-CFP) -CFP
- pGWB454 encoding Protein1 (here HopQ1-mRFP) -mRFP
Equipment
- For Microscopic evaluation
Nikon Stereomicroscope SMZ 1500 with epi-fluorescence equipment (optional)
Nikon Eclipse TE2000-E inverted C1 confocal laser scanning microscope, equipped with PlanApo 63x immersion oil objective, solid-state Coherent Sapphire 488-nm laser, Helium-Neon (HeNe) 543 nm laser, detector unit with three photomultiplier
Useful link, http://www.microscopyu.com/ - For FRET-FLIM measurement
FRET-FLIM experimental procedure is optimized for a Zeiss LSM 510 META confocal laser scanning microscope equipped with a FLIM module SPC730 (Becker and Hickl) for time correlated single photon counting (TCSPC). Single photons were detected with a photomultiplier MCP-PMT, R3809U-52, Hamamatsu Photonics. For excitation of photons, an ultrafast oscillating multiphoton excitation laser (titanium-sapphire, Chameleon, Coherent) was used (Chameleon XS, Coherent) - Microscope slides (Gerhard Menzel GmbH, Menzel-GlaserTM, catalog number: AA00000112E )
- Microscope Cover Slips no. 1 (Gerhard Menzel GmbH, Menzel-GlaserTM, catalog number: BB024060A1 )
Software
- EZ C1 for Nikon C1 confocal – image acquisition
- EZ C1 Viewer for Nikon C1 confocal – image analysis
- LSM 510 for Zeiss Meta 510 Confocal – image acquisition
- Zeiss LSM Image Browser – image analysis
- SPC operation software for FLIM measurement (Becker & Hickl GmbH)
- SPC-Image 2.9.1 software (Becker & Hickl GmbH, http://www.becker-hickl.de/pdf/flim-zeiss-man37.pdf) – image analysis
- ImageJ freeware (http://rsbweb.nih.gov/ij/) – image analysis
- MS Excel
Procedure
- Experimental design to study subcellular localization of fusion proteins co-expressed in living plant cells
- Choosing a fluorescence protein
There is a broad range of fluorescent protein variants used for the construction of fluorescence chimeric proteins that can be expressed in living cells (http://www.microscopyu.com/articles/livecellimaging/fpintro.html). Not all have been tested in plants. Recently a set of fluorescent protein Gateway entry vectors was generated by Mylle et al., 2013 that offers a valuable resource for plant cell biologists (http://gateway.psb.ugent.be). The Green Fluorescence Protein (GFP) and its color shifted genetic derivatives are most commonly used in fusion with proteins of interest to monitor their cellular localization. For more complex imaging experiments like co-localization studies of multiple fusion proteins, the selection of FPs must be based on the technical specifications of the confocal microscope that will be used. Instruments with simple optical setup can clearly distinguish between fluorescence proteins having none or minimal spectral overlap (for example CFP/GFP/RFP), combination of GFP and YFP should be avoided. Confocal microscopes enabling spectral imaging coupled to mathematically linear unmixing of the measured spectral profiles provide the ability to distinguish between large numbers of different fluorophores with partially overlapping spectra.
In our lab for analysis of subcellular localization we usually make constructs encoding the protein of interest fused to GFP or YFP while red (e.g. mRFP, mCherry) and green fluorescence proteins are commonly used in combination for co-localization studies. The position of the fluorescence marker may affect the expression level of the gene construct. Therefore it is recommended to test the potential consequence of the N-terminal or C-terminal fusion of the FP to protein of interest on its expression level and localization pattern.
Note: If the subcellular localization pattern of protein of interest with FP at N and C-terminus is similar we can assume that fusion with FP does not influence protein sorting and protein fusions reach their destinations in the cell. However, we observed that some protein fusions with FP at N-terminus may be completely retained in the endoplasmic reticulum while FP at C-terminus does not influence their subcellular targeting. The opposite situation is also possible. - Choosing the transient gene expression system
We perform different transient expression assays; each has its own advantages and disadvantages depending on research goals:- Routinely infiltration of Nicotiana benthamiana leaves with Agrobacterium tumefaciens strain GV3101 carrying an appropriate construct is used [procedure described already by Xiyan Li (Li, 2011)]
- Advantages: Provides usually high transformation rates and allows the simultaneous expression of multiple proteins in single cells with levels sufficient for detection of fluorescence signals with confocal microscopy.
- Disadvantage: Limited applicability for other plants; Agrobacterium may also affect distribution of some proteins within plant cells, e.g. related to plant immunity.
- Advantages: Provides usually high transformation rates and allows the simultaneous expression of multiple proteins in single cells with levels sufficient for detection of fluorescence signals with confocal microscopy.
- Biolistic bombardment of plant leaves with tungsten particles coated with plasmid DNA (Lichocka, 2014)
- Advantage: Can be applied for a wide range of plants for which agroinfiltration does not work efficiently; this method is fast, fluorescence of free GFP that is expressed under control of 35S promoter can be detected already after 6 h.
- Disadvantage: Requires expensive equipment, limited transformation efficiency.
- Advantage: Can be applied for a wide range of plants for which agroinfiltration does not work efficiently; this method is fast, fluorescence of free GFP that is expressed under control of 35S promoter can be detected already after 6 h.
- Transfection of protoplasts isolated from Arabidopsis thaliana leaves or seedlings
- Advantage: Enables better single cell imaging with high resolution.
- Disadvantage: Requires substantial expertise, limited applicability e.g. cell wall proteins cannot be analyzed, subcellular morphology might be altered compared to intact cells.
Note: Only infiltration with Agrobacterium require cloning of expression cassette into binary plasmid, for biolistic bombardment and transfection of protoplast plasmids encoding the gene of interest under control of a plant promoter are sufficient.
- Advantage: Enables better single cell imaging with high resolution.
- Routinely infiltration of Nicotiana benthamiana leaves with Agrobacterium tumefaciens strain GV3101 carrying an appropriate construct is used [procedure described already by Xiyan Li (Li, 2011)]
- Choosing a fluorescence protein
- Microscopic evaluation
- Prior to confocal microscopy, plant samples can be screened under a fluorescence stereomicroscope to check if transient expression of the introduced gene construct was successful. Under low magnification, transfection rate and expression level can be estimated and an appropriate leaf peace with cells exhibiting strong fluorescence can also be cut out. Precise localization of the fusion protein within single plant cells must be done with confocal microscopy.
- Plant leaf fragments are placed on microscopic slides in a drop of water and covered with cover slips.
- For recording of images a PlanApo 63x immersion oil objective is usually used. GFP and YFP are excited with a Coherent Sapphire 488-nm laser; images of mRFP are obtained using 543 nm HeNe laser excitation. The fluorescence of GFP/YFP and mRFP is detected using the 515/30 and 605/75 band pass emission filter, respectively. The image must be optimized by adjusting the pinhole, detector gain, and laser power. If the detected fluorescence signal is low, pinhole diameter may be increased, however, this will lower optical resolution. If the pinhole is opened, more and more noise fluorescence is detected resulting in loss of confocal imaging. The laser power should be adjusted with care to avoid photodamage and photobleaching of the fluorophore. The general idea is to use the minimum amount of laser power to get sufficient signal at gain levels that does not result in too much background noise. To monitor the level of image saturation a special LUT (indicator box) should be checked which will display saturated pixels in a contrasting color. Only few pixels should be highlighted by showing the brightest signal ensuring that the full dynamic range from 0 to 255 is utilized in 8-bit color depth.
- To optimally separate the fluorescence of co-expressed fluorophores the imaging is performed in sequential mode. Images are collected in z- series with a step size of 0.5 μm. In our experiments the proteins of interest localize to cytoplasm and nucleus, therefore z-series included the epidermal cell volume including nucleus. For quantitative evaluation of fluorescence intensity within the cytoplasm and the nucleus, only certain type of cells are chosen for imaging. To eliminate the influence of the imaging depth on the fluorescence intensity only cells with nucleus located near the outer epidermal cell wall facing the coverslip are recorded. Usually plant cells with the nucleus located deeper or close to the transverse cell walls are omitted. The quality of images collected from deeper tissue layers is worse due to the scattering of laser light and definitely not appropriate for quantification and comparison of fluorescence intensity.
- It is recommended to compare single optical sections including nuclei with clearly visible nucleoli. Collected images can be processed using image analysis freeware ImageJ (http://rsbweb.nih.gov/ij/). ImageJ is a Java image processing program that can be supplemented by the user with a large number of plugins (available on ImageJ web site) that extend the capabilities of ImageJ and are useful in extracting quantitative data from confocal images.
- Prior to confocal microscopy, plant samples can be screened under a fluorescence stereomicroscope to check if transient expression of the introduced gene construct was successful. Under low magnification, transfection rate and expression level can be estimated and an appropriate leaf peace with cells exhibiting strong fluorescence can also be cut out. Precise localization of the fusion protein within single plant cells must be done with confocal microscopy.
- ImageJ for average fluorescence intensity measurement
- We applied ImageJ to measure average fluorescence intensities in cellular compartments for proteins that change their distribution in the presence of interacting partners. Sometimes it is difficult to distinguish by eye whether fluorescence intensity has changed in certain cellular compartment; therefore it is recommended to include quantitative evaluation of fluorescence images.
Note: For quantitative evaluation and comparison of fluorescence intensities it is absolutely necessary to take images with the same hardware parameters, i.e. objective, laser power, pinhole opening, gain and offset values, zoom factor, otherwise comparison of relative image brightness is not really accurate. To minimize the influence of experimental condition on the fluorescence intensity in the plant samples it is recommended to prepare and analyze all experimental variants at the same time under the same conditions. - Presented example illustrates (Figure 1) shift in the localization of 14-3-3a-YFP out of the nucleus and into the cytoplasm in the presence of the interacting partner, the bacterial effector HopQ1-mRFP. The observed phenomenon can be presented as the ratio of average fluorescence intensity in the cell nucleus versus average intensity in the cell cytoplasm (N/C ratio). Therefore average fluorescence intensity in specific cellular compartments must be quantified as follows:
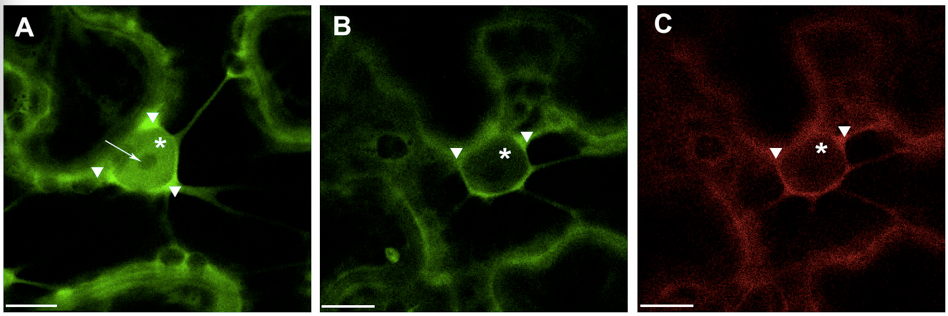
Figure 1. Transient expression of construct encoding 14-3-3a-YFP (A) and co-expression of 14 3-3a-YFP and HopQ1-mRFP (B&C) in N. benthamiana leaves. White arrowheads indicate cytoplasm, asterisks – nucleus, arrow – nucleoli. Scale bars represent 10 μm.- For fluorescence quantification confocal images with zoomed nuclear region were chosen.
- Export images to ImageJ; open Plugins list and select ROI/ Multi Measure.
Using drawing/selection tool draw small rectangular or circular region of interest (ROI) within the selected image. Add each ROI selection to the Multi Measure list; we usually select three different ROIs in each cellular compartment (i.e. nucleus and cytoplasm) and three to four in the background. To analyze ROIs of the same size one can move the ROI selection by dragging with the cursor to the next place on the image and add it to the Multi Measure list (Figure 2). - From main menu select Analyze/Set Measurements and in the check box list select Area, Integrated Density and Mean Gray Value.
Area: area of selection in square pixels
Integrated Density: the product of Area and Mean Gray Value
Mean Grey Value: the sum of the gray values of all the pixels in the selection divided by the number of pixels - In Multi Measure window press Select All and Measure, the window Results appears with quantified data that can be exported to MS Excel (Figure 2).
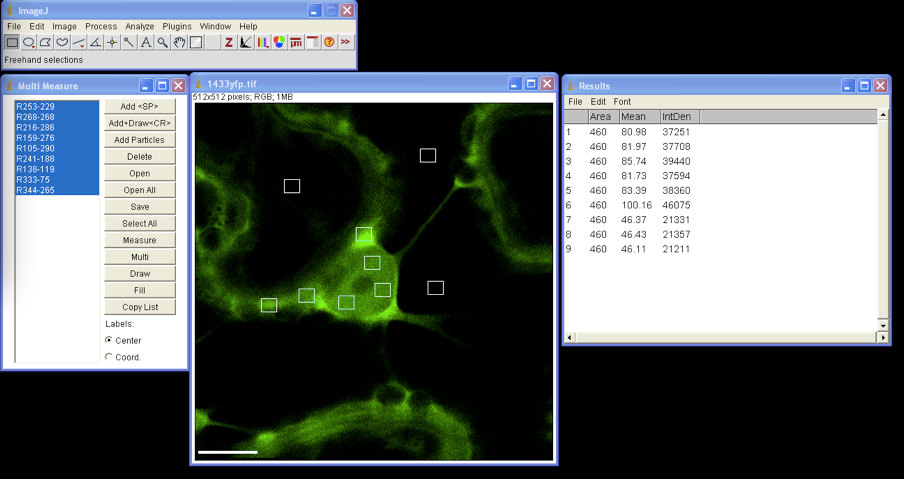
Figure 2. Multi ROIs fluorescence intensity measurement by ImageJ - All selected ROIs can be drawn on image by clicking the Draw button in the Multi Measure window.
- Background correction of fluorescence intensity must be performed before calculation of fluorescence distribution ratio (Figure 3) for each ROI as follows:
- ROI1 = Integrated Density ROI1 – (Area ROI1 * average background Mean Grey Value)
- Background corrected N/C ratios are calculated from mean fluorescence intensity in nucleus and cytoplasm. For N/C ratios calculation, ca. 25 cells from each experimental combination should be analyzed. The absolute N/C values may vary somewhat between experiments depending on the imaging conditions and thus comparisons should be made for paired experiments.
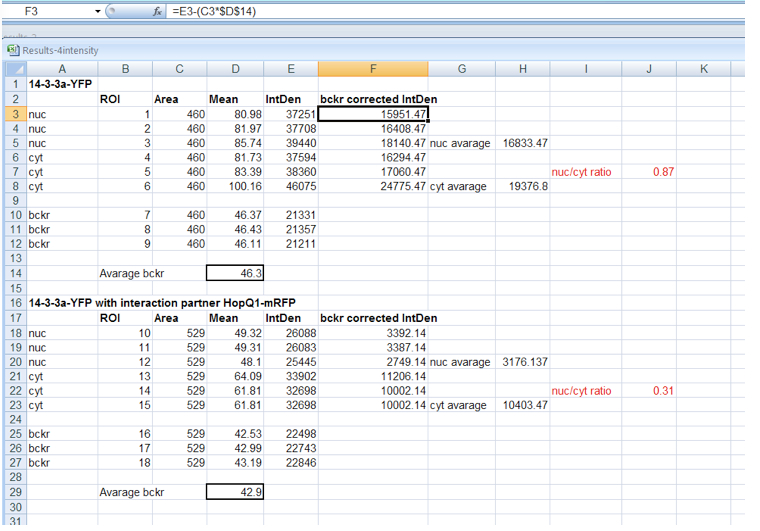
Figure 3. Background correction of fluorescence intensity within nucleus and cytoplasm and calculation of N/C ratio for 14-3-3 protein expressed alone or in HopQ1 presence
- For fluorescence quantification confocal images with zoomed nuclear region were chosen.
- We applied ImageJ to measure average fluorescence intensities in cellular compartments for proteins that change their distribution in the presence of interacting partners. Sometimes it is difficult to distinguish by eye whether fluorescence intensity has changed in certain cellular compartment; therefore it is recommended to include quantitative evaluation of fluorescence images.
- FRET-FLIM for analysis of interaction between proteins fused to YFP and CFP, co-expressed in leaf epidermal cells of Nicotiana benthamiana
- Design of FRET-FLIM experiments
- For FRET-FLIM experiments each of the potentially interacting proteins must be fused to appropriate fluorophores displaying spectral properties that together constitute a "FRET pair". Here we describe a protocol for one of the commonly used FRET fluorophore pair: cyan fluorescence protein (CFP, donor) and yellow fluorescence protein (YFP, acceptor). There are other combinations of FP-tagged proteins that can be used for FRET-FLIM measurements, such as eGFP (Enhanced-Green Fluorescent Protein) as a donor and the mCherry (monomeric Cherry red fluorescent variant) as an acceptor (more http://www.microscopyu.com/articles/fluorescence/fret/fretintro.html). The available microscope setup usually determines the fluorophore pair that will be used.
- FRET is a process of radiantionless energy transfer from donor to acceptor fluorophore if they are in close vicinity (distance of < 10 nm). In FRET-FLIM imaging only the donor fluorophore lifetime is measured, here CFP. This parameter refers to the duration of excited state of the fluorophore and can be influenced by external factors such as pH or in our case presence of the acceptor. When FRET occurs, shortening of fluorescence lifetime of the donor can be measured.
- Plant cells transiently expressing the following combinations of fusion proteins are required:
- Unfused, free CFP alone (used for initial imaging calibration)
- Co-expressed unfused, free CFP and unfused, free YFP (negative control no.1)
- CFP fused to YFP (positive control, separated by 11 amino acids)
- Protein2-CFP alone
- Protein2-CFP and unfused, free YFP (negative control no.2)
- Protein2-CFP and Protein1-YFP and optionally
- Protein2-CFP and mutated variant of Protein1a-YFP
- Unfused, free CFP alone (used for initial imaging calibration)
- For FRET-FLIM experiments each of the potentially interacting proteins must be fused to appropriate fluorophores displaying spectral properties that together constitute a "FRET pair". Here we describe a protocol for one of the commonly used FRET fluorophore pair: cyan fluorescence protein (CFP, donor) and yellow fluorescence protein (YFP, acceptor). There are other combinations of FP-tagged proteins that can be used for FRET-FLIM measurements, such as eGFP (Enhanced-Green Fluorescent Protein) as a donor and the mCherry (monomeric Cherry red fluorescent variant) as an acceptor (more http://www.microscopyu.com/articles/fluorescence/fret/fretintro.html). The available microscope setup usually determines the fluorophore pair that will be used.
- Transient co-expression of gene constructs in plant cells
- Infiltrations of N. benthamiana with A. tumefaciens (GV3101) were performed as already described by Xiyan Li (Li, 2011).
Notes:- For infiltration chose older leaves (but avoid cotyledons) with flat blade, younger leaves are more folded that make them not convenient for microscopy because stretching on microscope slide is not possible.
- Avoid damaging of leaf tissue with syringe during infiltration; strong autofluorescence of dead cells may be confusing; you can make small 4-5 injuries of the lower (abaxial) epidermis with pipette tip (size up to 200 ul) around the blade edge and by pressing syringe at injury sites infiltrate the whole leaf, it markedly improves infiltration of leaf tissue.
- For infiltration chose older leaves (but avoid cotyledons) with flat blade, younger leaves are more folded that make them not convenient for microscopy because stretching on microscope slide is not possible.
- Prior to infiltration Agrobacterium cultures carrying plasmid encoding proteins fused to CFP and YFP are mixed in combinations in equal amounts to a final OD600 0.6-1.0.
- After three days leaves are examined under fluorescence stereomicroscope to check for plant cells expressing both CFP and YFP fusion proteins at acceptable levels. If expression level is very low, the number of detected photons during FLIM measurements might be not sufficient for lifetime calculations.
- Infiltrations of N. benthamiana with A. tumefaciens (GV3101) were performed as already described by Xiyan Li (Li, 2011).
- FLIM measurement
- N. benthamiana leaf fragments are placed on slides in a drop of water and covered with coverslips.
- Measurements are performed on plant samples prepared as described in section "design of FRET-FLIM experiment" and confocal microscope settings are as described earlier by Kwaaitaal et al. (2010).
- For confocal imaging of plant cells co-expressed CFP and YFP fusion proteins, a 40x plan-apochromat water immersion objective is used. CFP and YFP are excited with the 458 nm and 514 nm laser line from the Agron-laser, respectively. Excitation of CFP for FLIM requires the ultrafast, multiphoton excitation Titanium:Sapphire laser tuned to a wavelength of 840 nm for 2-photon excitation of CFP, adjusted to 15-20% of transmission. Higher laser transmission can produce local heating which cause cell damage leading to autofluorescences and artificial photon emission.
- A selected area of interest is scanned continuously for 120 sec with a resolution of 128 x 128 pixels and 256 time channels. Usually this is sufficient to obtain a histogram of fluorescence decay, representing the number of photons recorded at each detection time in each pixel of the scanned ROI.
- The histograms are analyzed with the SPC-Image 2.9.1 software. The raw data are displayed as an intensity image (Figure 4, b + w image, left), from this a lifetime image is calculated (color coded image Figure 4 right), showing the distribution of lifetimes of the scanned ROI. In the images (Figure 4) sub-ROIs can be selected to display respective lifetime distributions. To calculate fluorescence lifetimes the decay curves in the individual pixels are fitted to an appropriate model. For estimations the brightest area on the intensity image must be selected. Typical models are either single exponential or multiexpotential decay curves. We assumed that the donor alone displays a monoexponential decay whereas a bi-expotential model adequately describes the FRET-situation. The
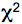 value is a measure for the fitting of data to the model, values of < 1.2 are regarded as good fit.
value is a measure for the fitting of data to the model, values of < 1.2 are regarded as good fit.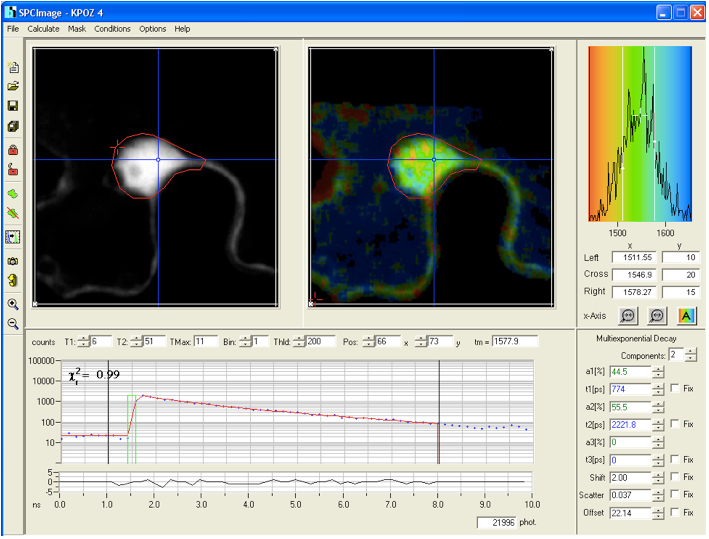
Figure 4. The main panel of the SPCImage analysis software with displayed data from FLIM measurement collected for the positive control CFP-YFP protein fusion - As Instrument Response Function (IRF), which is defined as the pulse shape of the FLIM system records for an infinitely short fluorescence lifetime, the value calculated by the SPCImage data analysis software was taken which in our experiments was sufficient for obtaining relevant lifetime data.
- The fit of fluorescence decay function can be improved by using introducing a binning factor and threshold level to exclude pixels with low photon number (more http://www.becker-hickl.de/pdf/flim-zeiss-man37.pdf). For calculation of mean lifetime of the individual cell the data are exported to Ms-Excel program.
- N. benthamiana leaf fragments are placed on slides in a drop of water and covered with coverslips.
- FRET efficiency calculation
- FLIM data should be recorded for a sufficient number of cells (>10) from each experimental combination to obtain statistically significant results. By comparing the average lifetime of the donor in the presence of an acceptor tDA τ with the average lifetime measured in absence of the acceptor tD, FRET efficiency can be calculated as follows:
EFRET = 1- (tDA FRET/tD) * 100%
Note: FLIM theory is hard-core physics, which is hard (or even not) to understand for biologists, we should be cautious with theoretical explanations and better refer to literature Becker & Hickl Manual (Becker, 2010) that is very useful in this regard.
- FLIM data should be recorded for a sufficient number of cells (>10) from each experimental combination to obtain statistically significant results. By comparing the average lifetime of the donor in the presence of an acceptor tDA τ with the average lifetime measured in absence of the acceptor tD, FRET efficiency can be calculated as follows:
- Design of FRET-FLIM experiments
Acknowledgments
This protocol is based on the procedure described by Giska et al. (2013).
References
- Becker W. (2010). The bh TCSPC Handbook. Fourth edition. Becker and Hickl GmbH.
- Giska, F., Lichocka, M., Piechocki, M., Dadlez, M., Schmelzer, E., Hennig, J. and Krzymowska, M. (2013). Phosphorylation of HopQ1, a type III effector from Pseudomonas syringae, creates a binding site for host 14-3-3 proteins. Plant Physiol 161(4): 2049-2061.
- Kwaaitaal, M., Keinath, N. F., Pajonk, S., Biskup, C. and Panstruga, R. (2010). Combined bimolecular fluorescence complementation and Forster resonance energy transfer reveals ternary SNARE complex formation in living plant cells. Plant Physiol 152(3): 1135-1147.
- Lichocka, M. (2014). Biolistic bombardment for co-expression of proteins fused to YFP and mRFP in leaf epidermal cells of Phaseolus vulgaris 'red mexican'. Bio-Protocol 4(1): e1019.
- Li X. (2011). Infiltration of Benthamiana protocol for transient expression via Agrobacterium. Bio-Protocol 1(14): e95.
- Mylle, E., Codreanu, M. C., Boruc, J. and Russinova, E. (2013). Emission spectra profiling of fluorescent proteins in living plant cells. Plant Methods 9(1): 10.
- Nakagawa, T., Suzuki, T., Murata, S., Nakamura, S., Hino, T., Maeo, K., Tabata, R., Kawai, T., Tanaka, K., Niwa, Y., Watanabe, Y., Nakamura, K., Kimura, T. and Ishiguro, S. (2007). Improved Gateway binary vectors: high-performance vectors for creation of fusion constructs in transgenic analysis of plants. Biosci Biotechnol Biochem 71(8): 2095-2100.
Article Information
Copyright
© 2014 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Lichocka, M. and Schmelzer, E. (2014). Subcellular Localization Experiments and FRET-FLIM Measurements in Plants. Bio-protocol 4(1): e1018. DOI: 10.21769/BioProtoc.1018.
- Giska, F., Lichocka, M., Piechocki, M., Dadlez, M., Schmelzer, E., Hennig, J. and Krzymowska, M. (2013). Phosphorylation of HopQ1, a type III effector from Pseudomonas syringae, creates a binding site for host 14-3-3 proteins. Plant Physiol 161(4): 2049-2061.
Category
Plant Science > Plant cell biology > Cell imaging
Cell Biology > Cell imaging > Fluorescence
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.