
- Home
- Protocols

-

Co-immunoprecipitation (co-IP) Following SUMOylation/Ubiquitination Perturbation


Last updated date: Jan 26, 2026 Views: 31 Forks: 0
Co-immunoprecipitation (co-IP) Following SUMOylation/Ubiquitination Perturbation
(TAK-981/TAK-243 treatment with NEM-preserved, non-denaturing lysis)
Overview
This protocol describes the workflow for treating cells with TAK-243 (ubiquitin E1/UBA1 inhibitor) and/or TAK-981 (SUMO E1 inhibitor), harvesting cells under cold conditions, preparing NEM-preserved non-denaturing lysates, and performing co-immunoprecipitation (co-IP) for downstream immunoblot analysis. Because SUMO conjugates can be lost during lysis, N-ethylmaleimide (NEM) is included by default in the lysis buffer. The optimal inhibitor concentration and treatment duration can vary by cell line and readout; therefore, dose/time optimization is recommended in each new cell line.
1. Reagents
2. Solutions
3. Optimization
4. Procedure
1. Reagents
• Cells and appropriate medium
• TAK-243 (ubiquitin E1/UBA1 inhibitor; Selleckchem, S8341)
• TAK-981 (SUMO E1 inhibitor; Selleckchem, S8829)
• PBS (ice-cold, home-made)
• NP-40 (Sigma-Aldrich, I8896)
• Protease inhibitor cocktail (Sigma-Aldrich, P8340)
• PMSF (Sigma-Aldrich, P7626)
• DTT (Sigma-Aldrich, D9779)
• N-ethylmaleimide (NEM; Sigma-Aldrich, 04259)
• Protein A/G agarose (ThermoFisher, 20421, or Sigma-Aldrich, A2220)
• Primary antibody for IP; control IgG
• Glycine elution buffer (0.2 M)
• Neutralization buffer (1 M Tris-HCl, pH 8.0)
• (Optional) MG132, E64, CHX, doxycycline
2. Solutions
2.1 NP-40 lysis buffer (non-denaturing)
• 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1% NP-40, 1 mM PMSF (add fresh), 1 mM DTT (add fresh), Protease inhibitor cocktail (add fresh)
2.2 SUMO-preserving lysis buffer (default)
• NP-40 lysis buffer, NEM (25 mM; add freshly immediately before use)
2.3 Elution and neutralization buffers
• Glycine-HCl elution buffer: 0.2 M glycine (pH2.5)
• Neutralization buffer: 1 M Tris-HCl (pH 8.0)
3. Optimization
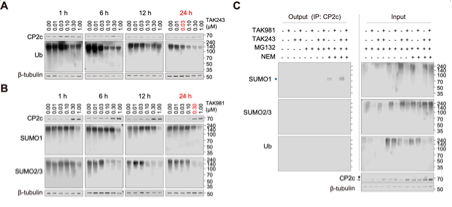
[Cell line–specific optimization and working conditions (example: MDA-MB-231 LM1)]
Working concentrations and treatment times for TAK-243 and TAK-981 should be determined empirically, as optimal conditions can differ across cell lines and experimental aims. The following example is based on optimization experiments performed in MDA-MB-231 LM1 cells (see Fig. S4 in the manuscript and the corresponding figure provided at the end of this document).
To determine appropriate working conditions in MDA-MB-231 LM1 cells, dose and time titration experiments were performed using 0, 0.01, 0.03, 0.1, 0.3, and 1 μM of TAK-243 or TAK-981, and treatment durations of 1, 6, 12, and 24 h, followed by representative Western blot analyses. Based on these data, conditions were selected to inhibit ubiquitination and SUMOylation of endogenous substrates while maintaining interpretable target readouts. Accordingly, for all subsequent experiments in MDA-MB-231 LM1 cells, the following conditions were used:
TAK-243: 0.03 μM for 24 h; TAK-981: 0.30 μM for 24 h
These optimization experiments further supported that CP2c degradation was TAK-981–sensitive but TAK-243–insensitive, consistent with a SUMOylation-dependent, rather than ubiquitin-dependent, mechanism under these conditions (Fig. S4).
Note: For other cell lines, inhibitor dose and treatment time should be optimized empirically (e.g., using a 0–1 μM titration across 6–24 h) and guided by pathway inhibition markers (global SUMO1/2/3 conjugate smears for TAK-981 and global ubiquitin conjugates for TAK-243), target stability, and cell viability.
4. Procedures
1. Maintain cells in an appropriate medium under standard culture conditions.
2. Treat cells with TAK-243 and/or TAK-981 under optimized conditions.
Example: TAK-243: 0.03 μM, 24 hours; TAK-981: 0.30 μM, 24 hours
Optional: If stabilization of short-lived intermediates is required, treat cells with MG132 using an assay-appropriate schedule (e.g., 50 μM for 6 h prior to harvest).
3. Place culture dishes on ice and aspirate the medium.
4. Wash cells with ice-cold PBS.
5. Add ice-cold PBS and detach cells using a cell scraper.
6. Transfer the suspension to a pre-chilled tube.
7. Pellet cells at 500 × g, 3 min.
8. Remove the supernatant completely.
9. Resuspend the cell pellet in SUMO-preserving lysis buffer (NP-40 lysis buffer supplemented with 25 mM NEM, freshly added).
Typical volume: 100 μL per 1x106 cells
10. Incubate lysates on ice for 10 min.
11. Clarify lysates by centrifugation at 15,000 × g, 4°C, 15 min.
12. Transfer the supernatant to a new pre-chilled tube.
13. Save ~10% of the clarified lysate as an input control for immunoblotting.
Optional: If lysates are highly viscous (e.g., transcription factors), apply DNaseI or mild shearing (short low-power sonication pulses) under conditions that preserve the interaction of interest.
14. Add protein A/G beads (e.g., 20–30 μL bead slurry) to clarified lysates.
15. Rotate at 4°C for 30 min.
16. Pellet beads briefly and transfer the pre-cleared supernatant to a fresh tube.
17. Add primary antibody (e.g., ~2 μg per sample) to pre-cleared lysates and rotate overnight at 4°C.
18. Add protein A/G beads and rotate for an additional 1 h at 4°C.
19. Wash beads three times with NP-40 lysis buffer.
Note: If background is high, increase wash stringency by adjusting NaCl (e.g., 150 → 300 mM) and/or detergent concentration (e.g., 0.5–1% NP-40/Triton), provided the interaction is retained.
20. Elute proteins using one of the following methods:
20A. Low-pH elution
- Add 0.2 M glycine-HCl (pH 2.5) to the beads (typically 50–100 μL per sample, depending on bead volume).
- Gently mix by brief low-speed vertexing and incubate for 5 min at room temperature.
- Pellet beads (e.g., 1,000–2,000 × g for 1 min for agarose beads; or place tubes on a magnetic rack for magnetic beads).
- Transfer the eluate to a new tube containing neutralization buffer to immediately restore pH.
- Add 1/10 volume of 1 M Tris-HCl (pH 8.0) (e.g., add 10 μL neutralization buffer to 100 μL glycine eluate).
- Add 2X SDS sample buffer and heat at 95°C for 5 min prior to SDS-PAGE and immunoblotting.
Notes:
Low-pH elution is broadly compatible with endogenous IPs and avoids contamination by eluted antibody heavy/light chains when the downstream detection strategy is sensitive.
Immediate neutralization is critical to prevent acid-induced protein degradation or loss of labile modifications.
20B. Tag-specific peptide elution (example: FLAG)
This option applies only when immunoprecipitation is performed using tag affinity resin (e.g., anti-FLAG M2 agarose).
- Prepare FLAG peptide elution buffer by diluting FLAG peptide to 100 μg/mL in NP-40 lysis buffer kept cold.
- Add 50–100 μL of FLAG peptide elution buffer to the beads.
- Rotate or gently mix at 4°C for 20–30 min.
- Pellet beads and transfer the eluate to a new tube.
- Add SDS sample buffer and heat at 95°C for 5 min.
Notes:
Peptide elution is preferred when preserving native complexes is important, as it avoids extreme pH. Use the peptide corresponding to the tag (e.g., FLAG, HA) and follow the resin manufacturer’s recommendations when available.
Figure : Optimization of TAK inhibitor working conditions (Figure S4 in manuscript)
- Son, S and Kim, C(2026). Co-immunoprecipitation (co-IP) Following SUMOylation/Ubiquitination Perturbation. Bio-protocol Preprint. bio-protocol.org/prep2893.
- Son, S. H., Kim, M. Y., Lim, Y. S., Jin, H. C., Shin, J. H., Yi, J. K., Choi, S., Park, M. A., Chae, J. H., Kang, H. C., Lee, Y. J., Uversky, V. N. and Kim, C. G.(2023). SUMOylation-mediated PSME3-20S proteasomal degradation of transcription factor CP2c is crucial for cell cycle progression. Science Advances 9(4). DOI: 10.1126/sciadv.add4969
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.