
- Home
- Protocols

-

Modeling and computational docking
Last updated date: Nov 12, 2025 Views: 602 Forks: 0
1. Docking method
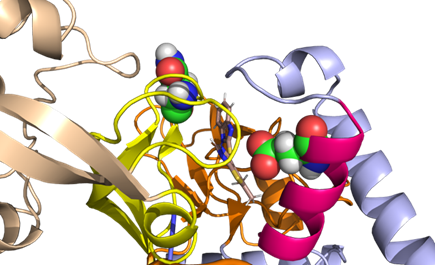
- A homology model of PfcIRS (Q8IDZ9; PF3D7_1332900) was created using SccIRS (7d5c.1) as a template using Swiss-Model (79). The SccIRS structure is bound by RM-A and an ILE-AMP intermediate located within the catalytic site of the Rossman fold and importantly, no tRNA is bound for this published structure. To help position tRNA within the catalytic domain in our homology model, we used the structure of EcLRS (4AQ7). Features such as the N-terminal catalytic Rossman fold domain, the editing or proofreading connective peptide 1 (CP1) domain inserted within the aminoacylating Rossman fold domain, a C-terminal a-helical anticodon-binding domain, and a tRNA stem loop acceptor (stem-contact fold) domain (31, 36) are all readily identifiable on our model (Fig. 4A). The the sequence for PfcIRS may be downloaded from Plasmodb.org or NCBI: https://plasmodb.org/plasmo/app/record/gene/PF3D7_1332900 Create a Homology model using Swiss-Model (https://swissmodel.expasy.org/) using the PfcIRS sequence PF3D7_1332900.
- We used the Open Chemical Computing Group's Molecular Operating Environment (MOE). This requires a paid subscription to the software. More information regarding MOE and its built-in algorithms can be obtained from https://www.chemcomp.com/en/index.htm. (Molecular Operating Environment (MOE), 2020.09; Chemical Computing Group ULC, 1010 Sherbrooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2020.)
- Within MOE, the PfcIRS homology model was energy minimized and protonated.
- Within MOE, SiteFinder was used to identify potential binding sites from the whole protein. Briefly, a global search for possible binding sites based on a geometric method utilizing a version of convex hulls that does not require calculation of interaction energies or protonation states was used to locate putative inhibitor binding sites (80).
- Within MOE, based on a ranked list of Propensity for Ligand Binding (PLB) scores > 0.5 and locations of putative binding domains that were not situated is disordered regions, calculated binding energies were calculated for the 4 thienopyrimidines. First, random poses are generated for each ligand and evaluated using a London dG scoring function. Next, the poses are refined using the GBVI/WSA dG Scoring function. The Amber14 : EHT (Extended Hueckel Theory) force field was used to approximate the potential energy of the molecular system (92). A video briefly describing this tool can be found through MOE’s commercial website: https://video.chemcomp.com/watch/2VtMGBYvvMkumZqo8A3yJN?second=268&autoplay=1. Small molecule ligand Docking was performed in MOE using the Dock (docking) interface which can be seen on https://video.chemcomp.com/watch/6dGEChdi82ZfRr1XrV1JrY?second=537&autoplay=1.
Regarding whether the method was blind or targeted:
Please see above for further details. The first pass method was blind and used MOE’s SiteFinder tool. The subsequent methods were targeted based on the rankings of found putative binding sites listed by SiteFinder.
The first pass method was blind and used MOE’s SiteFinder tool. The subsequent methods were targeted based on the rankings of found putative binding sites listed by SiteFinder.
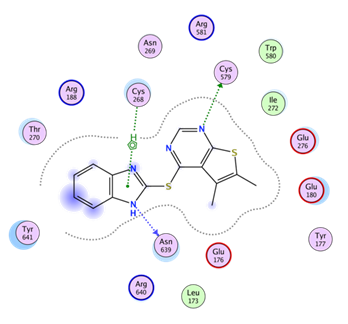
Residues in proximity to the allosteric site (occupied by MMV019266) are: Thre270, Arg188, Asn269, Cys268, Arg 581, Cys569, Trp580, Ile272, Glu276, Glu180, Tyr177, Glu176, Asn639, Arg640, Leu173, Tyr641.
All three compounds are likely adopting similar poses in the allosteric site. No specific model with MMV1081413 was prepared.
The allosteric site is near the Znf/ hinge region that bridges the Rossman fold to the CP1 domain (Fig. 4A). The hinge region is highly conserved in isoleucyl, leucyl, and valyl tRNA synthetases which are all members of IA subclass of synthases (39-41) and is crucial for structural orientation and inter-domain interactions (42, 43). The allosteric site is situated near the Znf/hinge region where the Editing and Rossman fold domains interact and where homologous/orthologous Znf regions coordinate Zn. The residues are numbered (per the P. falciparum homology model numbering):
i. # Within Editing Domain:...
1. # Core1/2 / Znf / Hinge: resi 254-280 and resi 578-597
ii. # Within Rossman fold:...
1. # alpha helix: 4 resi 173-183
2. # loop near alpha helix 4 region: resi 184-192
 |  |
- Istvan, E S, Goldberg, D E and Winzeler, E A(2025). Modeling and computational docking. Bio-protocol Preprint. bio-protocol.org/prep2874.
- Istvan, E. S., Guerra, F., Abraham, M., Huang, K., Rocamora, F., Zhao, H., Xu, L., Pasaje, C., Kumpornsin, K., Luth, M. R., Cui, H., Yang, T., Palomo Diaz, S., Gomez-Lorenzo, M. G., Qahash, T., Mittal, N., Ottilie, S., Niles, J., Lee, M. C., Llinas, M., Kato, N., Okombo, J., Fidock, D. A., Schimmel, P., Gamo, F. J., Goldberg, D. E. and Winzeler, E. A.(2023). Cytoplasmic isoleucyl tRNA synthetase as an attractive multistage antimalarial drug target. Science Translational Medicine 15(686). DOI: 10.1126/scitranslmed.adc9249
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.