- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Articles In Press
Articles In Press are peer reviewed and have been accepted for publication. Please note that these versions may be subject to further edits before their final online publication. Nevertheless, Articles In Press are citable using the DOI. Upon the formal online publication, the article will no longer be listed here, but existing links will automatically redirect to the final version in the corresponding issue.
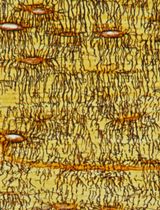
Visualizing the Osteocyte Lacuno-Canalicular System via a Rapid 10-Minute Silver Nitrate Staining Method
The conventional Ploton silver method employs a high-concentration 50% (w/v; 2.943 mol/L) silver nitrate solution for histological staining and characterization of the osteocyte lacuno-canalicular system (LCS). However, it is limited by prolonged staining times (55 min) and by risks of LCS ultrastructural damage and/or incomplete impregnation. To address these limitations, we developed the Wu–Wang silver nitrate staining method, which uses a 1 mol/L silver nitrate solution under elevated temperature (50–70 °C) to achieve rapid, effective, and high-contrast visualization of the osteocyte LCS within 10 min. We further demonstrate that this novel method enables robust LCS visualization across multiple vertebrate species. Compared with the Ploton method, the Wu–Wang method substantially reduces staining time and overcomes staining limitations inherent to prolonged exposure to concentrated silver nitrate solutions. This rapid and efficient staining method supports more accurate quantitative analysis of LCS morphology and facilitates systematic investigation of osteocyte and LCS morphogenesis, as well as the pathological mechanisms underlying bone and joint disease.
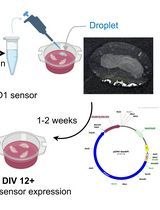
Liposome-based Expression of the PIEZO1 Sensor GenEPi in Hippocampal Neurons in Organotypic Slices

Expressing large DNA constructs in the native three-dimensional brain microenvironment remains technically challenging. Although viral vectors provide high transduction efficiency and cell-type selectivity, their genetic payload capacity is limited. Various non-viral approaches have been used in brain tissue, but they may compromise tissue viability or require specialised equipment, such as biolistic delivery or electroporation. We present an adapted protocol for delivering the large DNA vector encoding the optical PIEZO1 sensor GenEPi into brain tissue to enable sensor expression in pyramidal neurons. By applying DNA–Lipofectamine liposomes directly to the slice surface, we achieved efficient, minimally invasive transfection of pyramidal neurons in the CA1 and CA3 regions of organotypic hippocampal slices. PIEZO1 sensor expression was detectable as early as 7 days after transfection, increased with longer tissue maintenance, and was sustained for 3–4 weeks in vitro. This protocol describes a cost-effective, non-invasive approach that preserves cell viability and enables investigation of PIEZO1-mediated mechanotransduction in a native brain microenvironment.
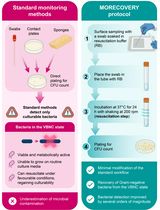
MORECOVERY: A Swab-Based Surface Sampling Protocol Incorporating a Nutrient-Free Resuscitation Step for the Detection of Clinically Relevant Gram-Negative Pathogens in the Viable but Non-Culturable State
Difficult-to-treat Gram-negative bacteria are a major cause of healthcare-associated infections due to multidrug resistance and limited therapeutic options. The hospital environment plays a central role in the persistence and transmission of infection, making environmental monitoring an essential component of infection prevention and control plans. Standard surface sampling techniques, including swabs, contact plates, and sponges, are widely used for environmental surveillance and are all based on culture-dependent methods. However, these techniques may underestimate the actual level of bacterial contamination since they fail to detect bacteria in the viable but non-culturable (VBNC) state, a reversible physiological condition in which bacterial cells remain viable but do not grow on conventional culture media. An innovative environmental sampling protocol, herein named MORECOVERY, has been developed to improve the detection of VBNC bacteria. The protocol integrates an essential resuscitation step, which improves the recovery of VBNC Gram-negative bacteria by a few orders of magnitude, into the standard swab-based sampling workflow. Following sample collection, swabs are incubated for 24 h at 37 °C in a carbon-free resuscitation buffer before plating, enabling the recovery of VBNC bacterial pathogens that would otherwise remain undetectable. By improving the recovery of VBNC cells, the MORECOVERY protocol allows a more accurate assessment of bacterial contamination of critical surfaces in healthcare settings. Its simplicity and minimal variation from standard workflows facilitate easy implementation in routine environmental monitoring.
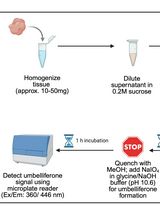
Fluorogenic Tissue-Based Assessment of Acid Ceramidase Activity
Acid ceramidase (aCDase) is a lysosomal amidase that catalyzes the hydrolysis of sphingolipids (SphL), including ceramides and glucosylceramides. Altered expressions of aCDase are associated with several pathological conditions, such as cancer, inflammation, pain, and pulmonary disorders. aCDase activity is reduced in Farber disease, spinal muscular atrophy with progressive myoclonic epilepsy, diabetes, and cardiovascular disease. Recent reports suggest that aCDase inhibition may be an emerging strategy for treating several SphL-related neurodegenerative conditions, such as Krabbe, Gaucher, and Parkinson’s disease, due to its role in the accumulation of glycosphingolipids. Therefore, the development of a tissue-based aCDase activity assay has potential applications in clinical diagnostics and drug discovery, enabling the evaluation of the onset and progression of disease from biological samples of patients, drug-target engagement analysis, and identification of biomarkers. Here, we report a detailed protocol for detecting aCDase activity in tissue lysates, using Rbm14-12 as a specific fluorogenic substrate for aCDase. Assay protocol optimization, including a procedure for the preparation and storage of tissue lysates and the identification of optimal protein tissue lysate amounts and substrate concentrations based on kinetic enzymatic parameter analyses, is described.
Sample Preparation for Imaging-Based Spatial Transcriptomics in Rigid Plant Tissues (Roots, Shoots)
Plant roots dynamically respond to environmental changes and serve as an ideal system for studying cell development and gene regulation. Recent advances in imaging-based spatial transcriptomics have enabled high-resolution mapping of gene expression while preserving spatial context. However, existing sample preparation techniques remain inadequate for handling rigid plant tissues such as crop roots. Here, we present a detailed and practical protocol for preparing rigid plant tissue samples for imaging-based spatial transcriptomics. The workflow ensures effective tissue handling while maintaining RNA integrity and spatial organization. Within approximately eight days, samples can be processed and mounted onto commercial slides, making them ready for subsequent probe hybridization and imaging. This protocol also includes an integrated sample attachment test performed to assess slide quality. It has been optimized to produce consistent and reliable results across experiments. Overall, our method provides a robust solution for spatial transcriptomic analysis in rigid plant tissues, facilitating broader application of these technologies in plant research.
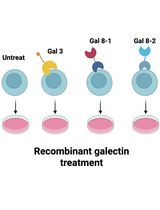
Protocol for In Vitro Activation of Jurkat E6-1 Cells Using Recombinant Human Galectin
Surface receptor engagement governs T-cell activation. Since these surface receptors undergo extensive glycosylation, lectin-mediated crosslinking of these glycosylated surface receptors has the potential to modulate signaling. Here, we systematically evaluate the abilities of recombinant human galectins in triggering immune responses. We describe how to apply the human galectins to modulate Jurkat E6-1 cell activation by measuring the expression level of cellular surface CD69 and the mRNA of IL-2. To validate the protocol, we confirmed that galectin-3 and galectin-8 variants 1 and 2 reproducibly induce CD69 and IL-2 expression on Jurkat E6-1 cells. Our approach offers a galectin-based toolset to study how glycosylation modulates human adaptive immunity.
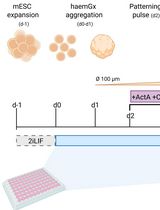
Generation of 3D Hemogenic Gastruloids From Mouse Embryonic Stem Cells
Embryonic blood formation encompasses the independent generation of different cell types in distinct cellular and anatomical environments, reflecting highly coordinated specific hierarchies of interacting tissues. Despite widespread use of embryonic stem cells (ESC) and induced pluripotent stem cell (iPSC)-based models to attempt to capture blood development in vitro and generate hematopoietic stem cells (HSC), a system that fully captures the spatial and temporal complexity of embryonic hematopoiesis is still lacking. In recent years, gastruloid models have emerged as powerful representations of early development, demonstrating self-organizing behaviors such as symmetry breaking, elongation, multi-axis formation, somitogenesis, and early organogenesis, with striking parallels to embryonic processes. Here, we present a protocol to generate hemogenic gastruloids (haemGx) from mouse ESC (mESC) that closely recapitulates the multi-stage, multi-niche process of blood formation and generates developmentally accurate hematopoietic progenitors. The haemGx model has been proven valuable in understanding embryonic hematopoiesis, as well as an in vitro model of forms of infant leukemia with an embryonic, in utero origin.
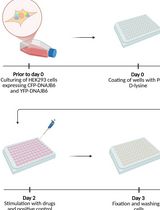
A Novel Plate Reader–Based Protocol for Measurement of DNAJB6 Dimerization Activity
Progressive neurodegeneration linked to the accumulation of misfolded proteins is a hallmark of several neurodegenerative disorders, including Parkinson’s disease, Huntington’s disease, and Alzheimer’s disease. Dysfunction in the protein homeostasis machinery correlates with pathology. The chaperone protein DNAJB6 is expressed in neurons and oligodendrocytes and has been shown to play a key role in preventing amyloid aggregation by binding to amyloidogenic proteins and facilitating their refolding or degradation, in cooperation with other chaperones. Here, we describe a simple and feasible assay that enables high-throughput screening for DNAJB6 activity in a plate reader format. We use genetically engineered HEK293 cells that stably express DNAJB6 fused to either CFP or YFP. These cells can be plated into multi-well plates, and the fluorescence resonance energy transfer (FRET) signal can be measured for analysis of DNAJB6 dimerization, which is linked to DNAJB6 activity. The protocol can be used for drug screening and to identify compounds that increase DNAJB6 dimerization, and can serve as a starting point for finding new medicines that act through modulating DNAJB6 activity.
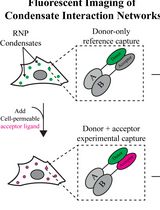
Automated FLIM-FRET Segmentation Within RNP Condensates
Ribonucleoprotein (RNP) condensates are membraneless organelles that exist alongside many RNA-driven processes, such as transcription and splicing. Despite their ubiquity, the biological necessity of forming a condensed phase remains unclear, particularly because the same RNP components exist both within these organelles and in the surrounding dilute phase. Most current methods for studying biochemical interaction dynamics within condensates rely on in vitro reconstitution of minimal factors or low-throughput single-molecule studies. However, RNP condensates are complex organelles containing tens to hundreds of proteins and hundreds to thousands of different RNAs. Here, we describe a scalable, high-throughput fluorescence microscopy–based approach to analyze protein–protein interaction networks, allowing for the rigorous assessment of dynamic, process-critical interactions within RNP condensates from live cells. This method takes advantage of fluorescence lifetime imaging (FLIM) and phasor plot analysis to automate segmentation of condensate-localized fluorescence signals. Using suitable FLIM–Förster resonant energy transfer (FLIM-FRET) fluorescent pairs fused to proteins of interest, protein–protein interactions can be actively monitored throughout various conditions via changes in fluorescence lifetime. Results from this assay yield valuable insight into the organization and assembly of essential factors for different condensate-associated processes to infer the functional consequences of RNP granule partitioning. Although this protocol is tailored for studying protein interactions within condensates, the design and execution framework can be adapted to investigate protein–protein interactions across a wide variety of compartments within different biological systems.
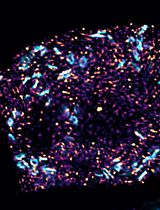
Dual Color tau-STED Super Resolution Microscopy in Arabidopsis Root Tip

Super-resolution microscopy has transformed our ability to visualize subcellular structures, but its application in plant biology remains challenging due to the optical complexity of plant tissues. Here, we present a detailed protocol for tau-STED microscopy (Leica Microsystems), which combines stimulated emission depletion (STED) with fluorescence lifetime imaging (FLIM) to achieve nanoscale resolution while minimizing phototoxicity. This method leverages time-correlated single-photon counting (TCSPC) to separate fluorescence signals based on their lifetimes, enhancing signal specificity and enabling the visualization of elusive subcellular compartments in Arabidopsis thaliana root tips. The protocol covers sample preparation, fluorophore selection, microscope configuration, image acquisition, and data analysis, providing a step-by-step guide to optimize tau-STED imaging for plant cell biology. By addressing the unique challenges of plant tissue imaging, such as autofluorescence, refractive index mismatches, and light scattering, this approach facilitates super-resolution imaging of intracellular structures, including the plant endoplasmic reticulum–Golgi intermediate compartment (ERGIC). This protocol is designed to be accessible to researchers with basic microscopy experience and offers a robust framework for exploring subcellular dynamics in plants with unprecedented detail.

Measurement of Net NH4+ Fluxes Using the Non-invasive Micro-Test Technology (NMT) System in Rice

Ammonium (NH4+) is the primary inorganic nitrogen source for rice (Oryza sativa L.). Substantial progress has been made in characterizing the functions of ammonium transporters (AMTs) in roots; however, the regulatory dynamics governing subcellular ammonium compartmentation after its entry into cells, particularly its vacuolar sequestration and efflux back to the external environment, remain poorly understood. This knowledge gap stems mainly from two factors: the difficulty of applying conventional detection methods at the organellar scale and interference caused by nonspecific ion adsorption to the cell wall of intact roots. To address these challenges, we present a detailed and reproducible protocol for real-time measurement of net NH4+ fluxes in rice roots, root protoplasts, and isolated vacuoles using non-invasive micro-test technology (NMT). The protocol covers the preparation of protoplasts and vacuoles from rice roots, the configuration and calibration of the NMT system, and the step-by-step measurement of net NH4+ fluxes at three distinct biological levels (intact roots, protoplasts, and vacuoles). By employing a unified sample preparation and measurement strategy, this protocol enables quantification of net uptake fluxes across the plasma membrane, characterization of net efflux dynamics under specific conditions, and indirect estimation of vacuolar sequestration capacity using the isolated vacuole system. Overall, this protocol provides a flexible and robust framework for studying NH4+ homeostasis in plants and is readily adaptable to different crop species, treatment conditions, and experimental objectives. Owing to its modular design and compatibility with standard NMT equipment, it can be readily adopted by laboratories seeking to investigate nitrogen transport mechanisms in plants.
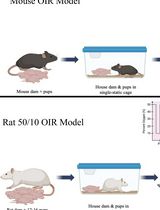
Mouse and Rat Oxygen-Induced Retinopathy Models to Study Vascular Features Seen in Retinopathy of Prematurity
Retinopathy of prematurity (ROP), a retinovascular disease, is a leading cause of childhood blindness worldwide. Given the constraints of studying molecular mechanisms in preterm infants, reproducible animal models are important to understand ROP pathophysiology. Mouse and rat oxygen-induced retinopathy (OIR) models are the most commonly used and recapitulate key vascular features seen in ROP. However, these models are susceptible to inherent variability that limits reproducibility, including inter-litter variability, consistency of oxygen delivery across experiments, retinal dissection technique, and immunohistochemistry. Here, we describe a comprehensive protocol for performing the most common mouse and rat OIR models, and procedures such as eye enucleation, retinal dissection and flat mounting, isolectin GS-IB4 staining, whole retina stitched fluorescence imaging from Z-stacks, and quantification of vascular features. This protocol provides important materials and procedural details to increase the reproducibility of the mouse and rat OIR models.

In Vivo and In Vitro SUMOylation Assays in Arabidopsis
Small ubiquitin-like modification (SUMOylation) is a crucial post-translational modification that modulates protein stability, localization, and interaction dynamics. Despite the identification of thousands of putative small ubiquitin-like modifier (SUMO) substrates, functional validation remains challenging due to the low abundance and highly dynamic nature of SUMOylated proteins. Here, we present a protocol for detecting protein SUMOylation, integrating bioinformatic site prediction, and rapid substrate screening via in vivo tobacco transient expression and in vitro E. coli assay, followed by precise validation using transgenic Arabidopsis lines. However, detection of low-abundance SUMOylated proteins may require coupling with mass spectrometry, and the in vitro system does not fully recapitulate the complex regulatory network in vivo. This workflow provides a useful tool for studying SUMOylation in plants.
Coupled Enzyme Assay for Measuring Ornithine Decarboxylase Activity in Cell Lysates Using a Liquid-Stable CO2 Detection Reagent
Ornithine decarboxylase (ODC) is a rate-limiting enzyme in polyamine biosynthesis that plays a critical role in cell proliferation and tumorigenesis. Reliable quantification of ODC activity is essential for mechanistic and therapeutic studies. Traditional assays often rely on radiolabeled substrates or discontinuous endpoint measurements. Here, we describe a non-radioactive, continuous spectrophotometric assay for measuring ODC activity in cell lysates using a commercially available liquid-stable CO2 detection reagent. In this assay, CO2 generated by ODC is captured as bicarbonate and utilized in a coupled enzymatic system containing phosphoenolpyruvate carboxylase (PEPC) and malate dehydrogenase (MDH), leading to oxidation of thio-NADH. The decrease in absorbance at 405 nm due to thio-NADH oxidation is monitored in real time and is proportional to ODC activity. The protocol is performed in a 96-well plate format, requires minimal reagent preparation, and is suitable for medium- to high-throughput applications.
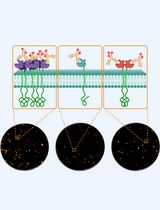
direct Stochastic Optical Reconstruction Microscopy to Determine the Oligomeric State of Proteins on the Plasma Membrane and Their Accessibility for Immunotherapeutic Antibodies
Super-resolution fluorescence microscopy enables the visualization of protein structures at nanometer resolution, providing insights into receptor organization on the plasma membrane that are essential for the development and optimization of immunotherapies. In this context, monoclonal antibodies are employed, which typically bind only a subset of available membrane receptors, due to steric hindrance or otherwise limited epitope accessibility, to quantify the accessible targets. These accessible targets, rather than the total receptor density, are critical for determining therapeutic efficacy. Here, we present a simplified, robust protocol to quantify antibody-accessible endogenous receptors using monoclonal antibodies directly labeled with fluorescent dyes in combination with total internal reflection fluorescence (TIRF) direct stochastic optical reconstruction microscopy (dSTORM). The method employs optimized labeling and fixation conditions to preserve the native receptor distribution, enabling precise quantification of accessible receptors and their stoichiometry at single-molecule resolution. Omitting secondary antibodies and minimizing fixation-induced artifacts prevents artificial clustering and maintains the physiological binding pattern of therapeutic antibodies. The standardized workflow delivers therapy-relevant information about receptor accessibility and organization underlying therapeutic antibody binding, thereby advancing the mechanistic understanding of immunotherapy resistance and personalized treatment strategies across diverse membrane protein targets.
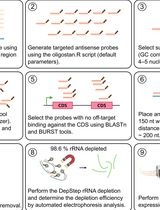
DepStep: An Efficient One-Step rRNA Depletion Workflow for RNA Sequencing in Non-model Organisms
RNA sequencing (RNA-seq) has revolutionized transcriptomics, ribosome footprinting, and polysome profiling, providing a wealth of data. Many RNA-based omics typically remove ribosomal RNA (rRNA) or select for messenger RNA (mRNA) prior to sequencing, thereby enriching reads that map to the translationally active part of the transcriptome. Prokaryotic mRNA lacks the 3′ polyadenylated tail, which excludes the use of poly(A)-based selection methods. While commercial rRNA depletion products exist for prokaryotes, their proprietary nature and potential inefficiency with non-model organisms are factors that may limit broad-scale application. To mitigate this issue, we designed DepStep, a consolidated workflow for one-step rRNA depletion using species-specific biotinylated antisense probes for selective hybridization and removal of the target rRNA molecules. As a proof-of-concept, RNA-seq libraries of the psychrophilic gram-negative bacterium Shewanella glacialimarina TZS-4T were prepared using both DepStep and a commercial rRNA depletion kit for gram-negative bacteria, to which DepStep was benchmarked. DepStep compares favorably to the commercial depletion kit; it removes >98.6% of the rRNA content in the sample, resulting in sequencing libraries where the coding DNA sequence (CDS) reads account for >80% of the total read count. Importantly, DepStep’s cost-per-sample is three times lower than the commercial kit, establishing DepStep as a simple yet cost-effective alternative to commercial solutions.

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics





