- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

An Ex Vivo Protocol to Assess IRE1α-Dependent RNA Cleavage Using Total RNA Isolated from Mouse Tissues
(*Contributed equally to this work, §aTechnical contact: mamun.phiiuc@gmail.com, §bTechnical contact: syrah76@jbnu.ac.kr) Published: Vol 15, Iss 16, Aug 20, 2025 DOI: 10.21769/BioProtoc.5414 Views: 2051
Reviewed by: Al Borhan BayazidNavnita DuttaAnonymous reviewer(s)
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Regulated IRE1-dependent decay (RIDD) is a critical cellular mechanism mediated by the endoplasmic reticulum (ER) stress sensor IRE1α, which cleaves a variety of RNA targets to regulate ER homeostasis. Current in vitro assays to study IRE1α activity largely rely on synthetic or in vitro transcribed RNA substrates, which may not fully replicate the physiological complexities of native RNA molecules. Here, we present a comprehensive protocol to assess IRE1α-dependent RNA cleavage activity using total RNA isolated directly from mouse tissues. This protocol provides a step-by-step guide for tissue collection, RNA isolation, an ex vivo RIDD assay, cDNA synthesis, and subsequent RT-PCR analysis of target mRNA cleavage products. Key reagents include active IRE1α protein, the RIDD-specific inhibitor 4μ8C, and target-specific primers for RIDD-regulated genes such asBloc1s1 and Col6a1. Quantitative assessment is achieved using agarose gel electrophoresis and imaging software. This methodology enables the study of IRE1α's RNA cleavage activity under conditions that closely mimic in vivo environments, providing a more physiologically relevant approach to understanding the role of RIDD in cellular and tissue-specific contexts.
Key features
• Uses total RNA from mouse tissues instead of synthetic RNA to better reflect in vivo conditions.
• Includes RIDD-specific controls such as IRE1α inhibitor (4μ8C) and RNase A to confirm targeted RNA cleavage.
• Combines agarose gel electrophoresis and ImageJ quantification for both qualitative and statistical validation.
• Allows comparative studies of IRE1α activity across multiple mouse tissues in different biological contexts.
Keywords: IRE1αGraphical overview
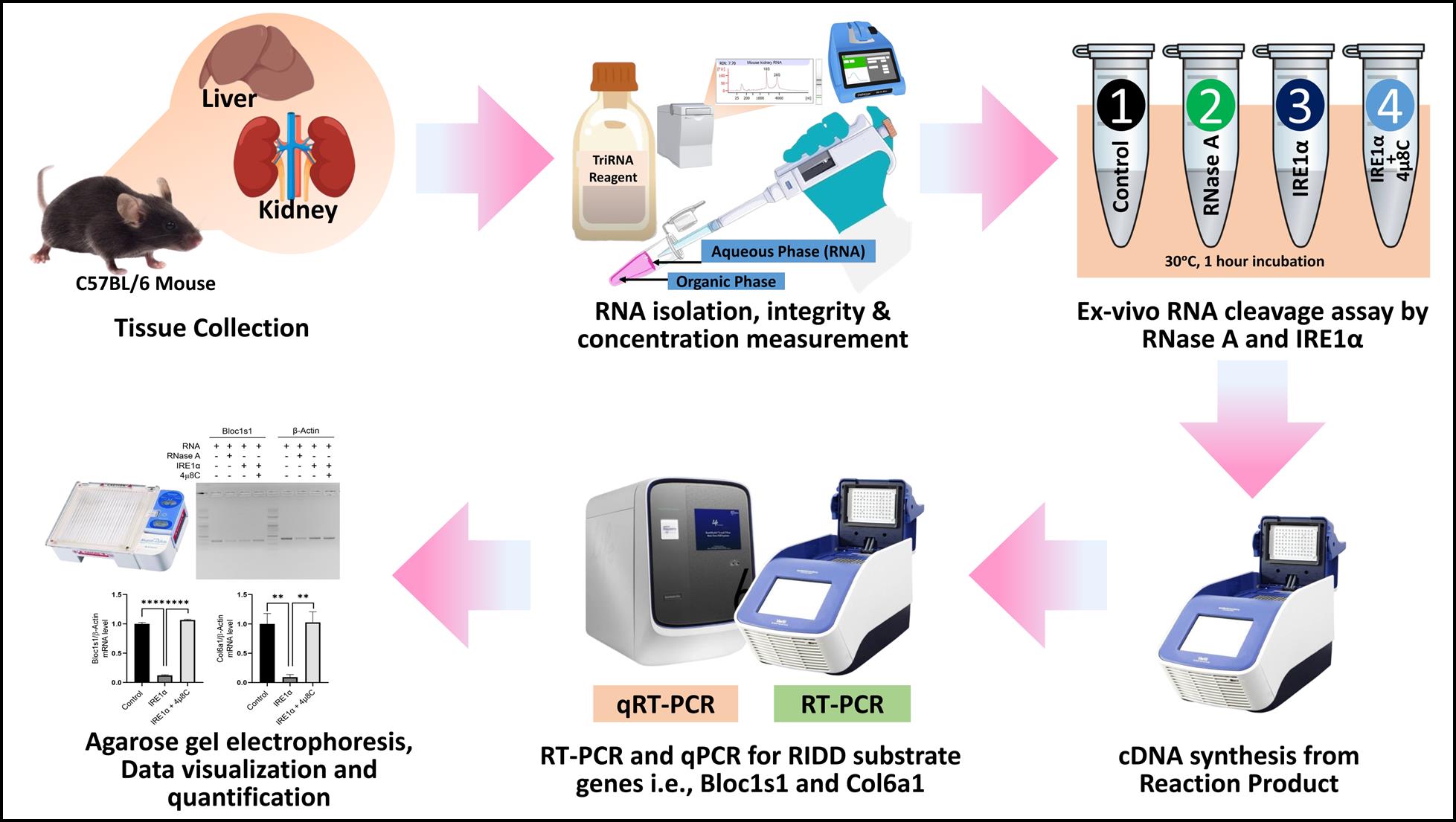
Ex vivo regulated IRE1-dependent decay (RIDD) assay workflow
Background
Regulated IRE1-dependent decay (RIDD) is a cellular mechanism that was first described inDrosophila melanogaster cells, where the endoplasmic reticulum (ER) stress sensor IRE1 was observed to induce the degradation of mRNAs encoding proteins that transit through the ER [1]. Subsequent studies in mammalian cells reported that IRE1α suppressed the expression not only of secretory pathway cargo proteins but also of ER-resident proteins that handle the folding and trafficking of cargo proteins [1].
Under high ER stress, IRE1α acquires endonucleolytic activity against a large array of RNA targets, first identified inD. melanogaster and termed RIDD, including ER-localized mRNAs and non-coding RNAs in mammals [2]. The RIDD pathway was later confirmed in mammalian cells, but it has yet to be fully determined in fungi [3].
The physiological significance of RIDD has been explored in various contexts. RIDD has been shown to regulate IgM and IgG2b expression in B cells, while IgG1 response relies on XBP-1 [4]. RIDD has also been implicated in curtailing immunoglobulin secretion from plasma cells and regulating autophagy in plants by degrading the mRNAs of several negative regulators of autophagy [5,6]. Additionally, RIDD has been suggested to play cytoprotective roles, such as contributing to feedback control on proinsulin expression in pancreatic beta-cells or protecting liver cells from acetaminophen toxicity [7].
Previous studies have established robust in vitro protocols to assess the RNA cleavage activity of IRE1α, predominantly utilizing synthetic RNA substrates [8,9]. These methods often rely on short, specifically designed RNA sequences or in vitro–transcribed RNAs mimicking XBP1 mRNA or validated RIDD targets. Such approaches, while valuable, allow precise kinetic measurements of IRE1α activity and have significantly contributed to understanding its RNase function. However, these methods may oversimplify the complex structural and contextual variations found in native RNA molecules within a cellular or tissue environment. This limitation underscores the need for methodologies that better represent the physiological conditions in which IRE1α operates.
While existing protocols effectively demonstrate IRE1α's RNA cleavage activity using synthetic or specific RNA substrates in vitro, these approaches may not fully capture the physiological context of RNA folding and complexity inherent in native tissues. To address this limitation, we propose a methodology utilizing total RNA isolated directly from various mouse tissues. This approach aims to provide a comprehensive understanding of IRE1α's substrate specificity and activity under conditions that closely mimic in vivo environments. By introducing this innovative perspective, our study seeks to expand the applicability of in vitro RNA cleavage assays, offering a versatile tool to unravel the intricate regulatory roles of IRE1α in diverse biological contexts.
Materials and reagents
Biological materials
1. Collected tissues from C57BL/6 mouse, e.g., kidney and liver
2. PCR primers (Table 1)
Table 1. List of primers
| Gene | Primer | Accession no. | Usage |
| Bloc1s1 | F: 5'-CAAGGAGCCTGCAGGAGAAGA-3' (Tm= 58.4 °C) R: 5'-CCAGGAGGGTGAAGTAAGAGG-3' (Tm = 58.2 °C) | NM_015740.3 | RT-PCR, qRT-PCR |
| Col6a1 | F: 5'-TAGCCGCGATGCAGAAGAG-3' (Tm= 59 °C) R: 5'-TTCCTCGCTCCCCCTCATA-3' (Tm = 58.4 °C) | NM_009933.5 | RT-PCR, qRT-PCR |
| Beta Actin | F: 5'-AGGCCAACCGTGAAAAGATGACC-3' (Tm= 62.7 °C) R: 5'-ACCGCTCGTTGCCAATAGTGATGA-3' (Tm= 62.7 °C) | NM_007393.5 | RT-PCR |
| Beta Actin | F: 5'-GGCTGTATTCCCCTCCATCG-3' (Tm= 59 °C R: 5'-CCAGTTGGTAACAATGCCATGT-3' (Tm= 58.5 °C) | NM_007393.5 | qRT-PCR |
| Stock concentration: 100 µM. Source: Bioneer Corporation, Daejeon, Republic of Korea. | |||
Reagents
A. RNA isolation from different mouse tissues
1. Tri-RNA reagent (Favorgen Biotech Corp., catalog number: FATRR 001) (store at 4 °C; shelf life: 12 months)
2. Chloroform (Sigma-Aldrich, catalog number: C2432) (store at room temperature)
3. Isopropanol (Sigma-Aldrich, catalog number: I9616) (store at room temperature)
4. Ethanol (Sigma-Aldrich, catalog number: E7023) (store at room temperature)
5. UltrapureTM DNase/RNase-free distilled water (Invitrogen, catalog number: 10977015) (store at room temperature)
6. Agilent RNA 6000 Nano kit (Agilent Technologies, catalog number: 5067-1511) (store according to the manufacturer's instructions)
7. 1× PBS (Enzynomics, catalog number: EBP008-1000)
B. RNA cleavage assay of isolated RNA with IRE1α peptide
1. IRE1α protein, active (SignalChem, catalog number: E31-11G) (store at -80 °C)
2. IRE1α inhibitor 4μ8C (MedChemExpress, catalog number: HY-19707) (store at -20 °C)
3. RNase A 100 mg/mL (QIAGEN, catalog number: 1007885) (store at -20 °C; shelf life: 12 months)
4. Sodium chloride (NaCl) (Sigma-Aldrich, catalog number: S3014) (store at room temperature)
5. Magnesium chloride (MgCl2) (Sigma-Aldrich, catalog number: M8266) (Store at room temperature)
6. Dithiothreitol (DTT) (Sigma-Aldrich, catalog number: D0632) (Store at -20 °C)
7. Glycerol (Sigma-Aldrich, catalog number: G5516) (Store at room temperature)
8. Adenosine 5’-triphosphate (ATP) (Sigma-Aldrich, catalog number: A2383) (Store at -20 °C)
C. cDNA synthesis after RNA cleavage
1. PrimeScriptTM RT Reagent kit (Takara, catalog number: RR037A) (store at -20 °C)
D. Checking mRNA expression of RIDD target mRNA
1. SolgTM 2× Taq PCR Smart mix 1 (SolGent Co. Ltd., catalog number: STD01-M50h) (store at -20 °C)
2. 100 bp DNA ladder (Bioneer Corporation, catalog number: D-1035) (store at -20 °C)
3. UltrapureTM agarose (InvitrogenTM, catalog number: 16500-100) (store at room temperature)
E. qRT-PCR
1. Power SYBRTM Green PCR Master Mix (Applied BiosystemsTM, catalog number: 4367659) (store at -20 °C)
2. 1 M HEPES buffer pH 7.4 (Bio-solution Co., Ltd., catalog number: BH048) (store at room temperature)
3. 50× TAE buffer (Bio-Rad Laboratories, Inc., catalog number: 1610743) (store at room temperature)
4. UltraPureTM ethidium bromide, 10 mg/mL (InvitrogenTM, catalog number: 15585011) (store at room temperature)
Solutions
1. 1 M NaCl (see Recipes)
2. 100 mM ATP (see Recipes)
3. 1 M MgCl2 (see Recipes)
4. 1 M DTT (see Recipes)
5. 5× Reaction buffer (see Recipes)
6. 0.5× TAE buffer (see Recipes)
Recipes
1. 1 M NaCl
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| NaCl | 1 M | 58.44 mg/1 mL |
| UltrapureTM DNase/RNase-free distilled water | - | 1 mL |
| Storage condition: Room temperature; shelf life: 1 month |
2. 100 mM ATP
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| ATP | 100 mM | 55.114 mg/1 mL |
| UltrapureTM DNase/RNase-free distilled water | - | 1 mL |
| Storage condition: -20 °C, avoid repeated freeze-thaw cycles; shelf life: 1 month |
3. 1 M MgCl2
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| MgCl2 | 1 M | 95.21 mg/1 mL |
| UltrapureTM DNase/RNase-free distilled water | - | 1 mL |
| Storage condition: Room temperature; shelf life: 1 month |
4. 1 M DTT
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| DTT | 1 M | 15.425 mg/0.1 mL |
| UltrapureTM DNase/RNase-free distilled water | - | 0.1 mL |
| Storage condition: -20 °C, avoid repeated freeze-thaw cycles; shelf life: 1 month |
5. 5× Reaction buffer
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| 1 M HEPES pH 7.4 | 100 mM | 10 μL |
| 1 M NaCl | 350 mM | 35 μL |
| 100 mM ATP | 10 mM | 10 μL |
| 1 M MgCl2 | 10 mM | 1 μL |
| 1 M DTT | 25 mM | 2.5 μL |
| Glycerol | 25 % v/v | 25 μL |
| UltrapureTM DNase/RNase-free distilled water | - | 16.5 μL |
| Total volume | 100 μL | |
| *5× reaction buffer is prepared freshly for every experiment. |
6. 0.5× TAE buffer
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| 50× TAE buffer | 0.5× | 10 mL |
| Distilled water | - | 990 mL |
| Total volume | 1 L | |
| Storage condition: Room temperature; shelf life: 1 month. |
Laboratory supplies
1. RNase-free 1.5 mL microcentrifuge tubes (Corning Life Sciences Co., Ltd, catalog number: MCT-150-C)
2. Pipette tips 1 mL (Corning Life Sciences Co., Ltd, catalog number: T-1000-B)
3. Pipette tips 200 μL (Corning Life Sciences Co., Ltd, catalog number: T-200-C)
4. Pipette tips 10 μL (Corning Life Sciences Co., Ltd, catalog number: T-300)
5. PCR tubes (Hundaimicro, catalog number: 203017)
6. MicroAmpTM optical 384-well reaction plate with barcode (Applied BiosystemTM, catalog number: 4309849)
7. MicroAmpTM optical adhesive film (Applied BiosystemTM, catalog number: 4311971)
Equipment
1. Veriti 96-well thermal cycler (Applied Biosystem, model: 9902)
2. -80 °C freezer (Nihon Freezer Co., LTD., model: CLN-71UWM)
3. FrescoTM 17 microcentrifuge (Thermo ScientificTM, catalog number: 75002402)
4. Mupid-2plus electrophoresis system (Takara, model: AD110)
5. DeNovix spectrophotometer (DeNovix Inc., model: DS-11+)
6. UltraSlim UV transilluminator (Maestrogen Inc., model: UUV-01)
7. Agilent 2100 bioanalyzer (Agilent Technologies, model: G2939B)
8. QuantStudioTM 6 Flex real-time PCR systems, 384-well, laptop (Applied BiosystemTM, model: 4485691)
Software and datasets
1. ImageJ image processing software (NIH, USA, 1.53e)
2. GraphPad Prism 10 (GraphPad Software, LLC, Boston, USA, 10.2.3)
Procedure
A. Tissue collection
1. Euthanize one healthy C57BL/6 mouse by using an approved method following the institutional animal care and use guidelines.
2. After confirmation of euthanasia, open the abdominal cavity under sterile conditions.
3. Excise the kidney and liver tissues using sterile surgical scissors and forceps.
4. Wash the tissues immediately with ice-cold PBS to remove blood residues and place them in cold PBS.
5. Chop the tissue with small scissors to wash the remaining blood.
6. Centrifuge at 500× g for 5 min at 4 °C.
7. Discard the supernatant and keep the tissue for RNA extraction.
B. RNA extraction
1. Add 1 mL of Tri-RNA reagent to the tissue and grind it using a pestle to prepare a homogenous mixture.
2. Incubate the homogenate for 5 min at room temperature.
3. Add 200 μL of chloroform, vortex vigorously for 15 s, and incubate for 2 min at room temperature.
4. Centrifuge at 12,000× g for 15 min at 4 °C.
5. Transfer the aqueous phase to a new tube, add 500 μL of isopropanol, and gently mix the solution by inverting 3–5 times.
6. Incubate the sample at -20 °C for 30 min.
7. Centrifuge at 12,000× g for 10 min at 4 °C. A white color pellet of the respective RNA is obtained.
8. Discard the supernatant and wash the pellet with 75% ethanol.
9. Centrifuge at 7,500× g for 5 min at 4 °C.
10. Air-dry the RNA pellet and resuspend it in RNase-free water.
Critical: RNA yield and integrity may vary depending on the tissue type due to differences in cellular composition, endogenous RNase activity, and fibrosis. It is recommended to optimize homogenization and extraction conditions for each tissue type to ensure high-quality RNA suitable for downstream IRE1α cleavage assays.
In addition, this protocol is potentially adaptable for use with human tissue samples or RNA extracted from cultured cells. However, differences in RNA composition, processing history, and sample origin may necessitate minor optimization of reaction conditions, such as incubation time, buffer composition, or IRE1α concentration. Ensuring high RNA integrity and purity remains essential for reproducible cleavage results across different species and sample types.
C. RNA integrity measurement and quantification
1. Measure the integrity of kidney and liver RNA using the Agilent 2100 Bioanalyzer following the manufacturer’s protocol (Figure 1). Keep the RNA integrity number (RIN) value close to 8 for further analysis.
2. Quantify the RNA concentration using the DeNovix spectrophotometer. RNA with an A260/A280 ratio greater than 1.8 and an A260/A230 ratio greater than 2.0 is used for ex vivo RIDD assay.
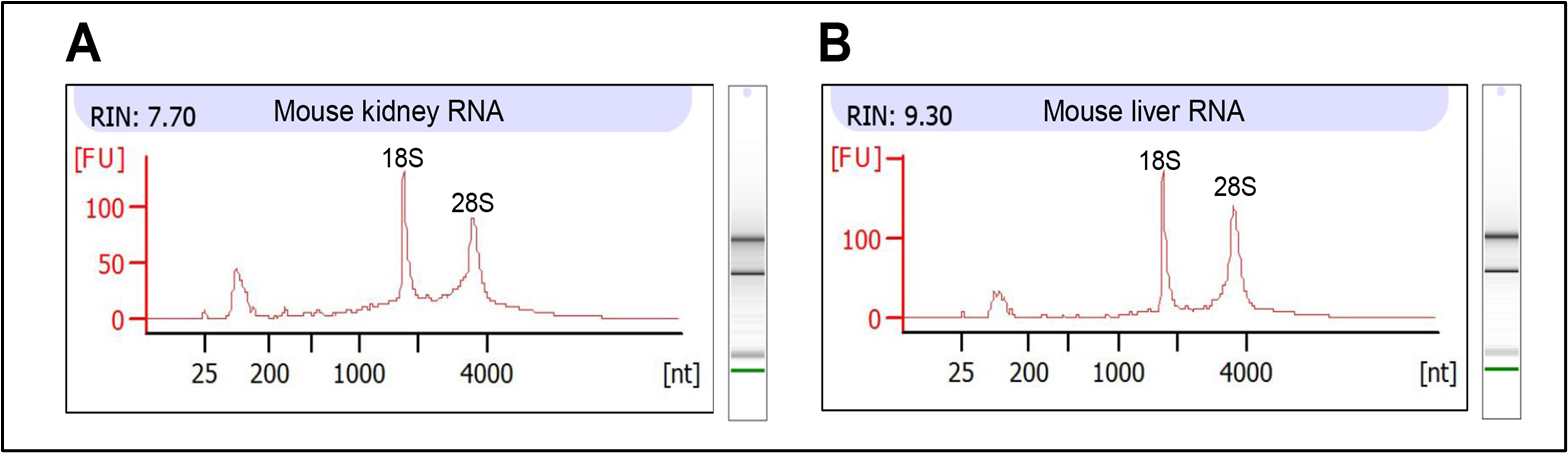
Figure 1. Integrity of mouse kidney and liver RNA. (A, B) Electropherogram showing the RNA integrity of mouse (A) kidney and (B) liver. RNA integrity number (RIN) is greater than 7 in both RNAs, which is acceptable for further analysis.
D. Preparation of reaction mix for ex vivo RIDD assay
1. Prepare the reaction mix followingTable 2.
Table 2. Preparation of reaction mixture for ex vivo RIDD reaction
| Reagent | Tube 1 | Tube 2 | Tube 3 | Tube 4 |
| RNA (Volume for 1.0 µg of RNA and RNase-free water up to 2 μL) | 2 μL | 2 μL | 2 μL | 2 μL |
| 5× reaction buffer | 4 μL | 4 μL | 4 μL | 4 μL |
| 0.5 mM 4μ8C | 0 μL | 0 μL | 0 μL | 1.6 μL |
| 1 mg/mL RNase A | 0 μL | 3.2 μL | 0 μL | 0 μL |
| UltrapureTM DNase/RNase-free distilled water | 10 μL | 6.8 μL | 6 μL | 4.4 μL |
| IRE1α purified peptide (10 µg/200 μL) | 0 μL | 0 μL | 4 μL | 4 μL |
| Total volume | 16 μL | 16 μL | 16 μL | 16 μL |
Critical: Ensure the RNA concentration is measured prior to setting up the reaction. If the RNA stock concentration is higher than 1 µg/µL (as in our case), adjust the RNA volume accordingly to add exactly 1.0 µg of total RNA to the reaction. The remaining volume should be adjusted with nuclease-free water up to 2 μL. Accurate RNA quantification is essential for consistent cleavage efficiency.
2. Mix well and keep these four tubes in a 30 °C incubator for 60 min. To inhibit IRE1α, preincubate IRE1α with 4µ8C for 5 min at room temperature.
3. Keep these four tubes in an ice bath for 10 min to stop the enzymatic reaction.
E. cDNA synthesis
1. Add 7 μL of Takara cDNA synthesis (PrimeScriptTM RT Reagent kit) master mix (Table 3) to 13 μL of the reaction mix of Table 2 in a PCR tube. The final volume is 20 μL.
2. Put the PCR tubes in the Veriti 96-well thermal cycler for synthesizing cDNA by following the company protocol.
3. Store the cDNA in a -80 °C freezer for further RT-PCR.
Table 3. Preparation of cDNA synthesis master mix
| Reagent | Required volume |
|---|---|
| 5× PrimeScript buffer | 4 μL |
| PrimeScript RT enzyme mix I | 1 μL |
| Oligo dT primer | 1 μL |
| Random 6 mers | 1 μL |
| Total volume | 7 μL |
F. RT-PCR for RIDD target genes, Bloc1s1 and Col6a1
1. Primer design for RIDD target gene
Design the primer of a RIDD target gene by considering the IRE1α-dependent RNA cleavage site of the respective gene. For instance, Figure 2 shows the primer design for mouse-Bloc1s1 and Col6a1 genes.
Critical: During primer design, the RIDD target site of the gene must be carefully considered. Primers should be designed flanking the RIDD cleavage site by placing the forward and reverse primers on either side of the target sequence. This strategy ensures accurate detection of RNA degradation following in vitro RIDD reactions, enabling precise identification and validation of RIDD-mediated RNA cleavage.
2. Prepare RT-PCR mix with synthesized cDNA as follows:
2× Taq Master mix: 10 μL
Forward primer (10 μM): 1 μL (final concentration 500 nM)
Reverse primer (10 μM): 1 μL (final concentration 500 nM)
RNase-free water: 3 μL
Template cDNA: 5 μL
Total volume: 20 μL
3. Run the PCR in a thermocycler by following the PCR conditions shown inTable 4.
Table 4. PCR thermocycling conditions used for the amplification of Blos1s1, Col6a1, and β-actin
| Temperature | Time | Cycle |
| 95 °C | 5 min | 1 |
| 95 °C | 30 s | 30 |
| 56 °C* | 30 s | |
| 72 °C | 30 s | |
| 72 °C | 2 min | |
| 4 °C | ∞ |
*Critical: Annealing temperatures were determined based on the melting temperatures (Tm) of the respective primers. For the Bloc1s1 and Col6a1 genes, the primer Tm was calculated to be 58–59 °C; therefore, the annealing temperature was set to 56 °C. For the negative control gene β-actin (non-RIDD target gene), the annealing temperature was set at 60 °C, considering its higher primer Tm. This approach ensures optimal primer binding and amplification efficiency by adjusting the annealing temperature relative to each primer pair’s melting temperature.
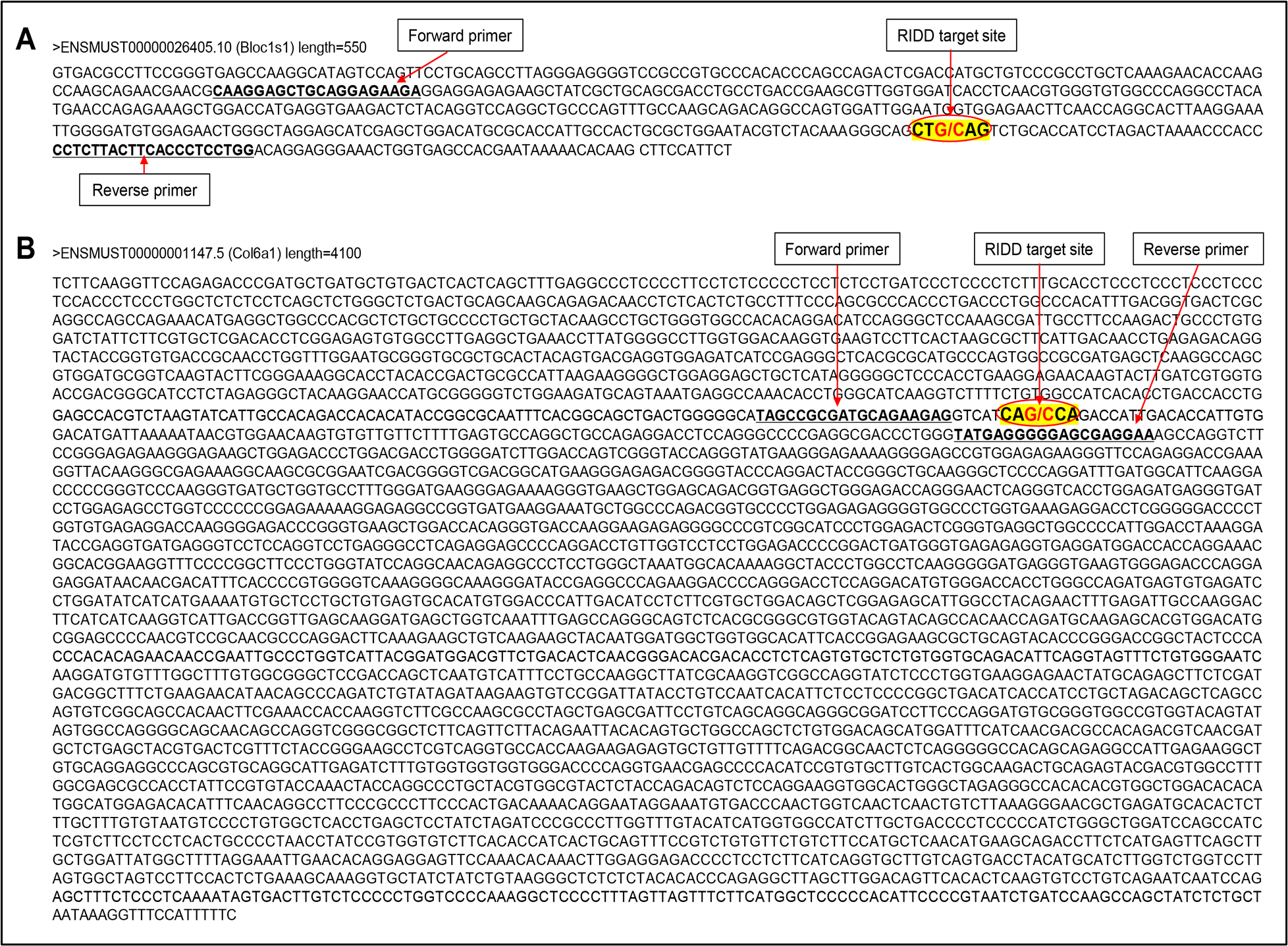
Figure 2. Primer design approach for Bloc1s1 (A) and Col6a1 (B) genes by keeping the RIDD target site highlighted in yellow. Cleavage sites (G/C sites) by IRE1α are marked with a red slash. The sizes of PCR products are 365 bp for Bloc1s1 and 158 bp for Col6a1.
G. Agarose gel electrophoresis
1. Prepare a 1.5% agarose gel by dissolving 1.5 g of UltrapureTM agarose powder in 100 mL of 0.5× TAE buffer. Heat the solution in a microwave oven until completely dissolved. Cool this solution to ~60 °C. Add 10 μL of ethidium bromide solution (final concentration: 1 μg/mL). Pour the gel into a casting tray with a comb and allow it to solidify at room temperature.
2. Set the gel into the Mupid-2plus electrophoresis system.
3. Submerge the gel in 0.5× TAE buffer.
4. Load the PCR product into wells as follows:
Well 1: Ladder
Well 2: Tube 1 (Control)
Well 3: Tube 2 (RNase A-treated)
Well 4: Tube 3 (IREα-treated)
Well 5: Tube 4 (IRE1α and 4µ8C-treated)
5. Run the electrophoresis in full voltage for 25 min or until adequate separation of bands is achieved.
6. Visualize the bands using the UltraSlim UV transilluminator and capture the image using a camera (Figure 3).
7. Quantify data using ImageJ.
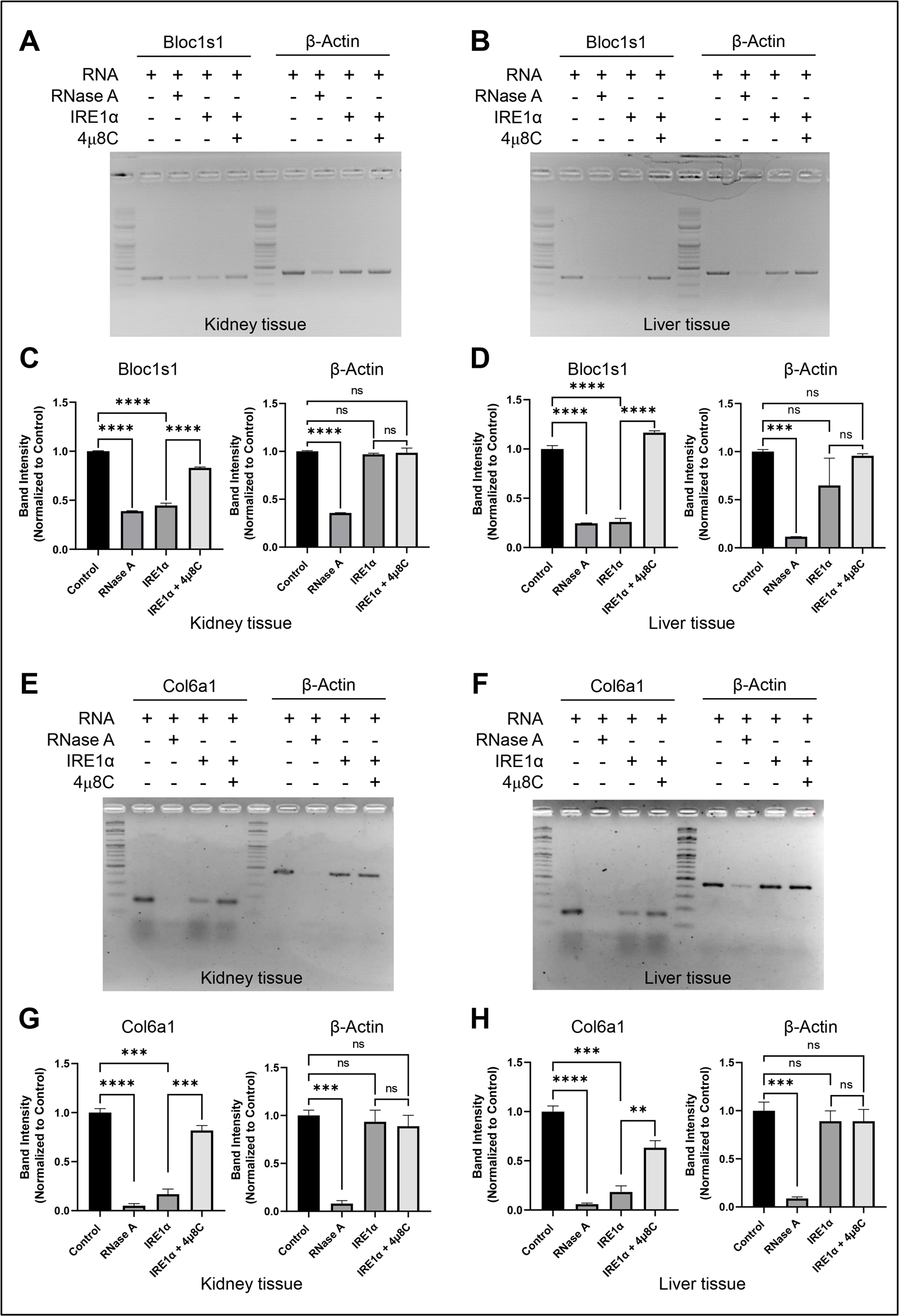
Figure 3. IRE1α-dependent RNA decay visualization by agarose gel electrophoresis. (A, B) Agarose gel showing the IRE1α-dependent cleavage of selected IRE1α target, Bloc1s1, and negative control, β-Actin, in mouse RNA that was isolated from (A) kidney and (B) liver. 0.2 μg of IRE1α was used to cleave 1.0 µg of total RNA for 60 min. The IRE1α inhibitor 4µ8C was utilized as a control for specificity. (C, D). Quantification of A and B using ImageJ. (E, F) Agarose gel showing the IRE1α-dependent cleavage of selected IRE1α target, Col6a1, and negative control, β-Actin, in mouse RNA that was isolated from (E) kidney and (F) liver. 0.2 μg of IRE1α was used to cleave 1.0 μg of total RNA for 60 min. The IRE1α inhibitor 4µ8C was utilized as a control for specificity. (G, H) Quantification of E and F using ImageJ and GraphPad Prism. The values are shown as mean ± SEM. Statistical significance was quantified by one-way ANOVA with Dunnett’s multiple comparison test. ****p < 0.0001, ***p < 0.001, **p < 0.01, and not significant (ns). n = 3.
H. qRT-PCR for RIDD target genes, Bloc1s1 and Col6a1
1. Prepare qRT-PCR mix with synthesized cDNA (Section E) as follows:
2× SYBR Green qPCR master mix: 5 μL
Forward primer (2 µM): 1 μL (final concentration 200 nM)
Reverse primer (2 µM): 1 μL (final concentration 200 nM)
RNase-free water: 3 μL
Synthesized cDNA: 2 μL
Total volume: 10 μL
Critical: 1 µg of RNA was reverse-transcribed in a 20 μL volume through a cDNA synthesis reaction (Section E). Dilute the cDNA product (Section E) 1/5 using RNase-free water and use 2 μL for the qRT-PCR reaction. Therefore, 20 ng of reverse-transcribed total RNA was used for the qRT-PCR reaction.
2. Add the qRT-PCR reaction mixtures in MicroAmpTM optical 384-well reaction plate and cover the plate with MicroAmpTM optical adhesive film.
3. Put the plate in the QuantStudioTM 6 Flex real-time PCR machine and run the PCR conditions shown in Table 5.
4. Collect the Ct (cycle threshold) values using the QuantStudioTM 6 Flex software.
5. Calculate the relative mRNA expression levels of Bloc1s1 and Col6a1 using the 2^(-ΔΔCt) method, normalizing to housekeeping gene, i.e., β-actin [10].
6. Export relative expression values to GraphPad Prism and generate bar charts representing the Bloc1s1 and Col6a1 mRNA levels (Figure 4).
Table 5. qRT-PCR cycling conditions for analysis of Bloc1s1 and Col6a1 mRNA levels after ex vivo RIDD assay
| Temperature | Time | Cycle |
| 50 °C | 2 min | 1 |
| 95 °C | 10 min | |
| 95 °C | 15 s | 40 |
| 60 °C | 60 s | |
| 95 °C | 15 s | Melt curve (1) |
| 60 °C | 60 s | |
| 95 °C | 15 s |
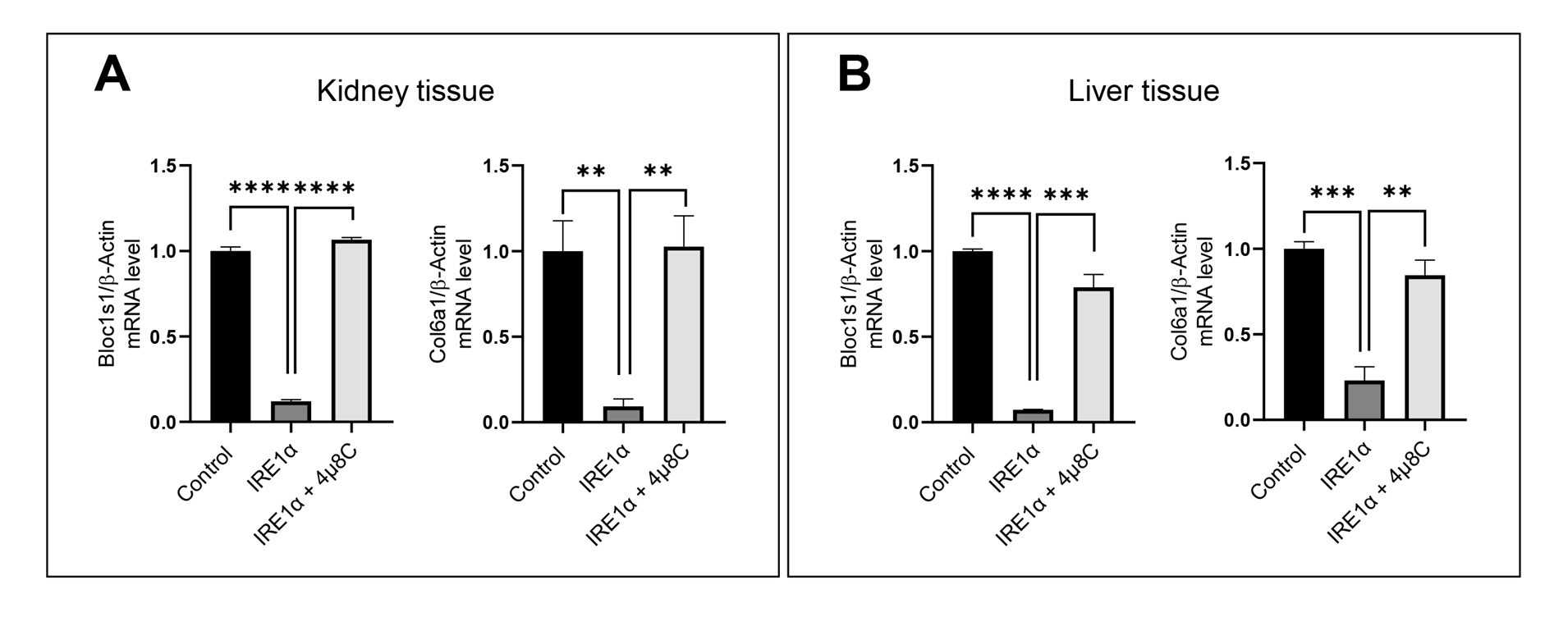
Figure 4. IRE1α-dependent RNA decay quantification by qRT-PCR. (A) qRT-PCR of Bloc1s1 and Col6a1, IRE1α target genes, after the RIDD reaction with mouse kidney RNA. (B) qRT-PCR of Bloc1s1 and Col6a1, IRE1α target genes, after the RIDD reaction with mouse liver RNA. The values are shown as mean ± SEM. Statistical significance was quantified by one-way ANOVA with Dunnett’s multiple comparison test. ****p < 0.0001, ***p < 0.001, **p < 0.01. n = 3.
Validation of protocol
To validate the ex vivo protocol for IRE1α-dependent RNA cleavage, we examined its specificity, reproducibility, and robustness across various biological replicates and tissue types. We employed total RNA from mouse kidney and liver tissues, with subsequent confirmation of the cleavage of a validated RIDD target, Bloc1s1 and Col6a1, by RT-PCR and qRT-PCR after incubation with recombinant IRE1α. Specificity was examined by employing the RNase inhibitor 4µ8C and the non-target gene β-actin as a negative control. The IRE1α-dependent cleavage of Bloc1s1 and Col6a1 RNA was highly diminished following treatment with 4µ8C, implicating that the degradation seen was a result of IRE1α activity.
Reproducibility of the assay was determined by carrying out three independent biological replicates, each of which yielded similar cleavage patterns. Quantitative analysis done using ImageJ revealed statistically significant degradation of Bloc1s1 and Col6a1 in IRE1α-treated samples as opposed to control samples. Further, the protocol could identify RIDD-mediated cleavage in two distinct tissues (kidney and liver), thereby indicating its applicability in diverse physiological milieus.
To ensure the fidelity of the results, the integrity of RNA was checked by an Agilent 2100 bioanalyzer, ensuring that the RIN is greater than 7. Moreover, the quality of RNA was inspected by spectrophotometric techniques to overcome DNA contamination prior to cDNA synthesis. Overall, the described protocol yields a reliable and physiologically relevant strategy for analyzing IRE1α-mediated RNA decay using native RNA substrates, effectively bridging the gap between in vitro synthetic assays and in vivo conditions.
General notes and troubleshooting
General notes
1. Ensure all reagents and consumables used for RNA work are RNase-free. Even trace amounts of RNase contamination can lead to degradation of RNA and compromise the assay result.
2. Freshly isolate RNA and use it immediately or store it at -80 °C in RNase-free water to preserve the integrity. Avoid repeated freeze-thaw cycles.
3. Always design primers to flank the predicted cleavage site of the target RNA to accurately detect cleavage efficiency.
Troubleshooting
| Problem | Possible cause | Recommended solutions |
|---|---|---|
| Weak or no PCR amplification | Low RNA quality and quantity | Verify RNA integrity using gel electrophoresis or spectrophotometer (A260/A280 > 1.8). Use at least 1 µg of RNA per reaction. |
| Nonspecific bands in RT-PCR | Suboptimal primer design or incorrect annealing temperature | Redesign primers to flank the RIDD site and re-optimize PCR conditions, particularly the annealing temperature. |
| No cleavage observed in IRE1α-treated samples | Inactive IRE1α enzyme or incorrect reaction buffer | Confirm enzyme activity with a known substrate, ensure buffer components are fresh, and check that incubation was at 30 °C. |
| RNA degradation in control samples | RNase contamination | Use RNase-free tubes, tips, and reagents. Wear gloves and work in a clean RNase-free area. |
| Incomplete inhibition by 4µ8C | Insufficient inhibitor concentration or incomplete mixing | Ensure proper preincubation of IRE1α with 4µ8C for at least 5 min at room temperature before adding RNA. Confirm inhibitor quality and use freshly prepared solutions. |
| Inconsistent RNA cleavage result | Lot-to-lot variation in recombinant IRE1α | Use a pre-validated positive control RNA (e.g., Bloc1s1) to confirm enzyme activity with each new lot. Store single-use aliquots to avoid freeze-thaw degradation. |
Acknowledgments
This research was supported by the National Research Council of Science & Technology (NST) grant by the Korean government (MSIT) (CAP 23052-000) and also by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Korean government (MSIT) (RS-2024-00335573). Conceptualization, M.M.U.R & S.Y.R; Investigation, M.M.U.R, S.Y.R & S.W.K; Writing —Original Draft, M.M.U.R & S.Y.R; Writing —Review & Editing, M.M.U.R, S.Y.R & H.J.C; Funding acquisition, H.J.C; Supervision, H.J.C.
Competing interests
The authors declare no conflicts of interest.
Ethical considerations
All animal procedures and experiments were performed according to NIH guidelines for the care and use of laboratory animals. Protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of Jeonbuk National University (CBU 2023-005).
References
- Hur, K. Y., So, J. S., Ruda, V., Frank-Kamenetsky, M., Fitzgerald, K., Koteliansky, V., Iwawaki, T., Glimcher, L. H. and Lee, A. H. (2012). IRE1α activation protects mice against acetaminophen-induced hepatotoxicity. J Exp Med. 209(2): 307–318. https://doi.org/10.1084/jem.20111298
- Ghosh, R., Wang, L., Wang, E. S., Perera, B. G. K., Igbaria, A., Morita, S., Prado, K., Thamsen, M., Caswell, D., Macias, H., et al. (2014). Allosteric Inhibition of the IRE1α RNase Preserves Cell Viability and Function during Endoplasmic Reticulum Stress. Cell. 158(3): 534–548. https://doi.org/10.1016/j.cell.2014.07.002
- Miyazaki, T., Nakayama, H., Nagayoshi, Y., Kakeya, H. and Kohno, S. (2013). Dissection of Ire1 Functions Reveals Stress Response Mechanisms Uniquely Evolved in Candida glabrata. PLoS Pathog. 9(1): e1003160. https://doi.org/10.1371/journal.ppat.1003160
- Choi, H. J., Tang, C. H., Tian, L., Wu, Y., Sofi, M. H., Ticer, T., Schutt, S. D., Hu, C. C. and Yu, X. Z. (2021). XBP-1s Promotes B Cell Pathogenicity in Chronic GVHD by Restraining the Activity of Regulated IRE-1α-Dependent Decay. Front Immunol. 12: e705484. https://doi.org/10.3389/fimmu.2021.705484
- Benhamron, S., Hadar, R., Iwawaky, T., So, J., Lee, A. and Tirosh, B. (2013). Regulated IRE1‐dependent decay participates in curtailing immunoglobulin secretion from plasma cells. Eur J Immunol. 44(3): 867–876. https://doi.org/10.1002/eji.201343953
- Yang, M., Bu, F., Huang, W. and Chen, L. (2019). Multiple Regulatory Levels Shape Autophagy Activity in Plants. Front Plant Sci. 10: e00532. https://doi.org/10.3389/fpls.2019.00532
- Kimmig, P., Diaz, M., Zheng, J., Williams, C. C., Lang, A., Aragón, T., Li, H. and Walter, P. (2012). The unfolded protein response in fission yeast modulates stability of select mRNAs to maintain protein homeostasis. eLife. 1: e00048. https://doi.org/10.7554/elife.00048
- Almanza, A., Mnich, K., Blomme, A., Robinson, C. M., Rodriguez-Blanco, G., Kierszniowska, S., McGrath, E. P., Le Gallo, M., Pilalis, E., Swinnen, J. V., et al. (2022). Regulated IRE1α-dependent decay (RIDD)-mediated reprograming of lipid metabolism in cancer. Nat Commun. 13(1): e1038/s41467–022–30159–0. https://doi.org/10.1038/s41467-022-30159-0
- Karagöz, G., Peschek, J., Walter, P. and Acosta-Alvear, D. (2019). In vitro RNA Cleavage Assays to Characterize IRE1-dependent RNA Decay. Bio Protoc. 9(14): e3307. https://doi.org/10.21769/bioprotoc.3307
- Rao, X., Huang, X., Zhou, Z. and Lin, X. (2013). An improvement of the 2^(-delta delta CT) method for quantitative real-time polymerase chain reaction data analysis. Biostat Bioinforma Biomath. 3(3): 71–85. https://pubmed.ncbi.nlm.nih.gov/25558171/
Article Information
Publication history
Received: Apr 21, 2025
Accepted: Jul 15, 2025
Available online: Jul 24, 2025
Published: Aug 20, 2025
Copyright
© 2025 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Rashid, M. M. U., Rah, S., Ko, S. and Chae, H. (2025). An Ex Vivo Protocol to Assess IRE1α-Dependent RNA Cleavage Using Total RNA Isolated from Mouse Tissues. Bio-protocol 15(16): e5414. DOI: 10.21769/BioProtoc.5414.
Category
Molecular Biology > RNA > qRT-PCR
Biochemistry > Protein > Interaction > Protein-RNA interaction
Molecular Biology > RNA > Reverse transcription
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.