- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

A Fluorescence-Based Flippase Assay to Monitor Lipid Transport by Drs2-Cdc50
Published: Vol 15, Iss 14, Jul 20, 2025 DOI: 10.21769/BioProtoc.5393 Views: 3677
Reviewed by: Anonymous reviewer(s)
Original research article
The authors used this protocol in:
Dec 2023
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Flippases, a functionally distinct group of transmembrane proteins that flip lipids from the extracellular or luminal side to the cytosolic side of biological membranes, are key players in many important physiological processes, such as membrane trafficking and cellular signaling. To study the function of these membrane proteins under chemically defined conditions, reconstituting them into artificial vesicles is a crucial and effective approach. There are various methods for protein reconstitution involving different detergents and detergent removal techniques to integrate membrane proteins into artificial vesicles. In this protocol, we describe the reconstitution of the yeast flippase complex Drs2-Cdc50, which translocates phosphatidylserine across membranes of the trans-Golgi network at the expense of ATP hydrolysis. The flippase complex is incorporated into liposomes using a zwitterionic detergent, followed by detergent removal via dialysis—a gentle and effective strategy that helps preserve protein function. To evaluate the activity of the reconstituted flippase complex, two complementary assays are employed: (1) a fluorescence-based quenching assay to measure lipid transport; and (2) an ATPase assay using an ATP-regenerating system to measure ATP hydrolysis. Together, these methods provide a robust platform for analyzing the functional reconstitution of Drs2-Cdc50 in a defined membrane environment.
Key features
• Provides a gentle reconstitution approach via detergent removal by dialysis.
• Measures ATPase activity using an NADH-coupled assay with an ATP-regenerating system.
• Assesses lipid flippase activity with a sodium dithionite-based assay with fluorescent lipid derivatives.
• Provides a well-defined experimental setup for direct characterization of lipid flippases.
Keywords: Large unilamellar vesiclesGraphical overview
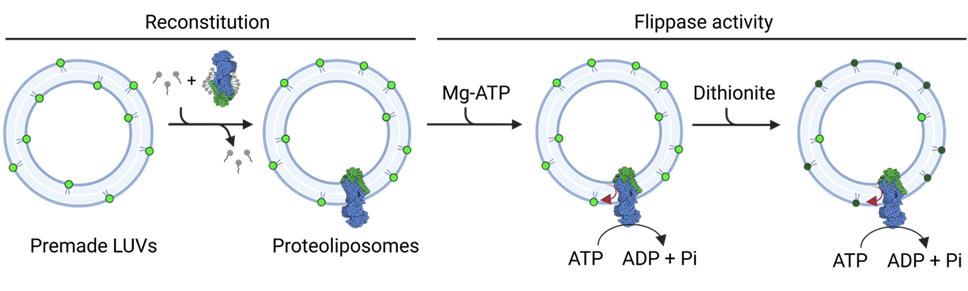
Background
The asymmetrical distribution of lipids across biological membranes is a key characteristic of cells, playing a crucial role in various important physiological processes, including membrane trafficking and cellular signaling [1–5]. Flippases and floppases are essential for the maintenance of phospholipid asymmetry by unidirectional transport of lipids across bilayer leaflets [6]. Eukaryotic flippases, many belonging to the P4-ATPase family, and floppases, which belong to the ABC transporter family, require energy in the form of ATP to move lipids against electrochemical gradients. In contrast, scramblases can transport lipids in both directions without the need of energy, thereby disrupting transbilayer lipid asymmetry [7]. Drs2-Cdc50, a well-studied member of the P4-ATPase family, is primarily localized in the trans-Golgi network of yeast cells. It generates and maintains membrane lipid asymmetry by transporting phosphatidylserine (PS) from the luminal to the cytosolic leaflet of the membrane. This asymmetry is essential for various cellular processes, including vesicle budding and intracellular vesicle trafficking [8]. The role of flippases in these important cellular functions is highlighted by the fact that mutations in human P4-ATPases can lead to pathological conditions in humans. It also makes them attractive drug targets [9].
The molecular study of lipid flippases is challenging due to the complexity of their native membrane environments. Therefore, an important step toward a detailed functional understanding of these proteins is their purification and reconstitution into well-defined, tunable systems such as liposomes. As a first step toward purification, detergent molecules are traditionally used to extract the transmembrane protein from cell membranes, replacing the lipids in lipid–protein interactions [10,11]. This step is critical because, in the absence of lipids, transmembrane proteins are prone to denaturation [12]. To restore their original structure and activity, the purified transmembrane proteins need to be reintroduced into a lipid environment. In traditional reconstitution approaches, the solubilized and purified transmembrane proteins are incorporated into premade large unilamellar vesicles (LUVs) by first destabilizing LUVs with a detergent and then removing the detergent molecules [13]. Typically, non-ionic or zwitterionic, mild detergents are used for vesicle destabilization to preserve the structure and function of the protein. The detergent can then be removed by dialysis, gel filtration, or the use of polystyrene beads that remove the detergent molecules by adsorption [14,15].
In this protocol, a mild detergent is chosen, and the detergent is removed by dialysis. This approach ensures slow removal, which may help to preserve protein function. However, not all detergents are effectively removed by dialysis. The removal rate depends on the detergent’s critical micelle concentration (CMC), being lower for detergents with low CMCs [14]. As to the study of P4-ATPases flippase activity, many approaches are based on fluorescent nitrobenzoxadiazole (NBD)-labeled lipids [16–18]. These lipids can be incorporated into vesicles either during the preparation of LUVs or during the reconstitution process. In proteoliposomes containing active flippases, the addition of ATP and Mg2+ to the medium will trigger the inward-to-outward movement of NBD lipids. This occurs in vesicles where the catalytic domains of the flippases are facing the outside. To assess flippase activity, the fluorescence of NBD lipids remaining in the outer leaflet is quenched by adding dithionite, a membrane-impermeable reducing agent. Proteoliposomes with functional lipid flippases show increased quenching after incubation with Mg-ATP compared to control vesicles [19]. Alternatively, bovine serum albumin (BSA) back extraction of accessible NBD-lipids can be used to determine lipid flippase activity [19,20]. Another approach to study protein activity is to measure its ability to hydrolyze ATP. This can be done using an ATP-regenerating system that couples ATP hydrolysis to the conversion of NADH to NAD+. This decline of NADH can be tracked by measuring the absorbance at 340 nm [21].
It is important to note that the approach described here relies on ensemble-averaged biochemical assays to assess lipid flippase activity. While these bulk measurements provide valuable quantitative estimates of protein function, they are subject to limitations due to the heterogeneous nature of liposome preparations. Variability in vesicle size, lamellarity, lipid composition, membrane integrity, protein-to-lipid ratio, and the number and orientation of reconstituted proteins can affect data interpretation. For example, only a subset of vesicles may contain functional or correctly oriented proteins, while others may be empty, leaky, or structurally compromised [22–24]. As a result, the activity measured in ensemble assays may not accurately reflect the true behavior of individual protein molecules, leading to underestimation of functional parameters such as transport rates and substrate specificity.
Materials and reagents
Biological materials
The yeast flippase complex Drs2-Cdc50 serves as the exemplary protein in this protocol, with Drs2 carrying an N-terminal biotin acceptor domain (BAD)-tag used for affinity purification. Drs2 was truncated by 104 residues at the N-terminus and 65 residues at the C-terminus (Δ104, Δ65). This truncation is necessary to relieve autoinhibition by the N- and C-termini. The resulting BAD-ΔN104ΔC65Drs2-Cdc50 (from here on referenced as Drs2-Cdc50) was overexpressed from a single co-expression pYeDP60 plasmid in the Saccharomyces cerevisiae strain W303.1b/GAL4 (MAT α, leu2-3, his3-11, trp1-1::TRP1-GAL10-GAL4, ura3-1, ade2-1, canr, cir+) and purified on streptavidin beads [25,26]. The resulting protein stock in elution buffer [50 mM MOPS-Tris, pH 7.0; 100 mM KCl; 10% (v/v) glycerol (Fisher, catalog number: 56-81-5), 0.05% (w/v) n-Dodecyl β-D-maltoside (DDM) (Glycon Biochemicals, catalog number: D97002)] was snap-frozen in liquid N2 and stored at -80 °C.
Note: The flippase complex Drs2-Cdc50 was solubilized using DDM and subsequently stored in DDM to maintain protein stability during storage. For other flippase complexes, optimal solubilization and storage conditions may vary and should be determined empirically.
Reagents
1. 1,2-Dioleoyl-sn-glycero-3-phosphocholine (DOPC) (Avanti Polar Lipids, catalog number: 850375)
2. 1-Palmitoyl-2-{12-[(7-nitro-2-1,3-benzoxadiazol-4-yl)amino]dodecanoyl}-sn-glycero-3-phosphoserine (NBD-PS) (Avanti Polar Lipids, catalog number: 810193)
3. 1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) (Avanti Polar Lipids, catalog number: 850457)
4. 1-Palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (POPG) (Avanti Polar Lipids, catalog number: 840457)
5. 3-((3-Cholamidopropyl) dimethylammonio)-1-propansulfonate (CHAPS) (Glycon Biochemicals, catalog number: D99009)
6. 3-(N-morpholino)propanesulfonic acid (MOPS) (Applychem, catalog number: A1076.0100)
7. 4-(2-Hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) (Roth, catalog number: 6763.3)
8. Adenosine 5′-triphosphate disodium salt (ATP) (Roth, catalog number: HN35.3)
9. BeSO4·4H2O (Sigma, catalog number: 14270)
10. Chloroform, ethanol-stabilized and certified for absence of phosgene and HCl (VWR chemicals, catalog number: 22711.260)
11. Deionized water
12. HCl (1 M solution) (VWR chemicals, catalog number: 35328)
13. KOH (1 M solution) (VWR chemicals, catalog number: 35113)
14. Lactate dehydrogenase (LDH, 5 mg/mL) (Sigma, catalog number: 10127230001)
15. L-α-phosphatidylinositol-4-phosphate (PI4P) (Avanti Polar Lipids, catalog number: 840045)
16. Methanol (VWR chemicals, catalog number: 20847.307)
17. MgCl2·6H2O (Sigma, catalog number: M2670)
18. NaCl (Roth, catalog number: 9265.2)
19. NaF (Sigma, catalog number: S1504)
20. NaOH (1 M solution) (VWR chemicals, catalog number: 35256)
21. Nicotinamide adenine dinucleotide disodium salt (NADH) (Sigma, catalog number: 481913)
22. Phosphoenol pyruvate (PEP) (Sigma, catalog number: P0564)
23. Pyruvate kinase (PK, 10 mg/mL) (Sigma, catalog number: P9136-25KU)
24. Sodium dithionite (Fisher, catalog number: CL000239)
25. Tris(hydroxymethyl)aminomethane (Tris) (Roth, catalog number: 4855.1)
26. TritonTM X-100, extra pure (Roth, catalog number: 3051.3)
Solutions
1. Reconstitution buffer (see Recipes)
2. 0.5 M CHAPS (see Recipes)
3. 50 mM MgCl2 (see Recipes)
4. 100 mM PEP (see Recipes)
5. 6.7 mM NADH (see Recipes)
6. 0.5 M ATP (see Recipes)
7. 45 mM beryllium fluoride (see Recipes)
8. Mg-ATP buffer (see Recipes)
9. Na-ATP buffer (see Recipes)
10. 1 M sodium dithionite (see Recipes)
11. 20% (w/v) Triton X-100 (see Recipes)
12. 1 M HEPES-NaOH, pH 4.7 (see Recipes)
13. 1 M NaCl (see Recipes)
14. 1 M MgCl2 (see Recipes)
15. 1 M MOPS-KOH, pH 7 (see Recipes)
16. 1 M Tris-HCl, pH 9 (see Recipes)
17. 0.9 M NaF (see Recipes)
18. 2 M BeSO4 (see Recipes)
Recipes
1. Reconstitution buffer
Adjust the pH to 7.4 with 1 M NaOH before bringing the solution to its final volume with ddH2O. Then, filter sterilize using a 0.22 μm filter.
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| 1 M HEPES-NaOH, pH 7.4 (see Recipe 12) | 50 mM | 150 mL |
| 1 M NaCl (see Recipe 13) | 100 mM | 300 mL |
| ddH2O | n/a | 2.55 L |
| Total | n/a | 3 L |
2. 0.5 M CHAPS
Store in aliquots at -20 °C.
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| CHAPS | 0.5 M | 4.61 g |
| ddH2O | n/a | Fill up to 15 mL |
| Total | n/a | 15 mL |
3. 50 mM MgCl2
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| 1 M MgCl2 (see Recipe 14) | 50 mM | 750 μL |
| ddH2O | n/a | 14.25 mL |
| Total | n/a | 15 mL |
4. 100 mM PEP
Before adjusting the solution to its final volume with ddH2O, check the pH and adjust it to 7 using 1 M KOH. Store in aliquots at -20 °C.
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| 1 M MOPS-KOH, pH 7 (see Recipe 15) | 20 mM | 200 μL |
| PEP | 100 mM | 0.19 g |
| ddH2O | n/a | 9.8 mL |
| Total | n/a | 10 mL |
5. 6.7 mM NADH
Before adjusting the solution to its final volume with ddH2O, check the pH and adjust it to 7 using 1 M KOH.
Store in aliquots at -20 or -80 °C and protect from light to prevent degradation.
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| 1 M MOPS-KOH, pH 7 (see Recipe 15) | 20 mM | 1 mL |
| NADH | 6.7 mM | 0.24 g |
| ddH2O | n/a | 49 mL |
| Total | n/a | 50 mL |
6. 0.5 M ATP
Before adjusting the solution to its final volume with ddH2O, check the pH and adjust it to 7 using 1 M KOH.
Store in aliquots at -20 °C.
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| ATP | 0.5 M | 2.25 g |
| ddH2O | n/a | Fill up to 10 mL |
| Total | n/a | 10 mL |
7. 45 mM beryllium fluoride
Caution: Beryllium and its compounds are highly toxic and can cause severe skin burns and eye damage. Please wear appropriate protective gear including gloves, safety goggles, and a lab coat when handling this chemical and perform necessary tasks under a fume hood if possible.
Store in aliquots at -80 °C or prepare a fresh batch before each measurement.
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| 0.9 M NaF (see Recipe 17) | 0.88 M | 9.775 mL |
| 2 M BeSO4 (see Recipe 18) | 0.045 M | 225 μL |
| Total | n/a | 10 mL |
8. Mg-ATP buffer
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| Reconstitution buffer (see Recipe 1) | n/a | 496.55 μL |
| 1 M MgCl2 (see Recipe 14) | 3.3 mM | 1.65 μL |
| 0.5 M ATP (see Recipe 6) | 1.8 mM | 1.8 μL |
| Total | n/a | 500 μL |
9. Na-ATP buffer
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| Reconstitution buffer (see Recipe 1) | n/a | 496.55 μL |
| 1 M NaCl (see Recipe 13) | 3.3 mM | 1.65 μL |
| 0.5 M ATP (see Recipe 6) | 1.8 mM | 1.8 μL |
| Total | n/a | 500 μL |
10. 1 M Sodium dithionite
Caution: Sodium hydrosulfide (sodium dithionite) powder is highly toxic if swallowed and can cause severe skin burns and eye damage. Please wear appropriate protective gear, including gloves, safety goggles, and a lab coat, when handling this chemical and prepare the solution under a chemical hood.
Note: Sodium dithionite is unstable in solution; keep the stock on ice and use it within 20–30 min.
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| 1 M Tris-HCl, pH 9 (see Recipe 16) | 1 M | 500 μL |
| Sodium dithionite | 1 M | 87 mg |
| Total | n/a | 500 μL |
11. 20 % (w/v) Triton X-100
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| TritonTM X-100 | 20% | 20 g |
| ddH2O | n/a | Fill up to 100 mL |
| Total | n/a | 100 mL |
12. 1 M HEPES-NaOH, pH 7.4
Before adjusting the solution to its final volume with ddH2O, check the pH and adjust it to 7.4 using 1 M NaOH.
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| HEPES | 1 M | 23.83 g |
| ddH2O | n/a | Fill up to 100 mL |
| Total | n/a | 100 mL |
13. 1 M NaCl
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| NaCl | 1 M | 29.22 g |
| ddH2O | n/a | Fill up to 500 mL |
| Total | n/a | 500 mL |
14. 1 M MgCl2
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| MgCl2·6H2O | 1 M | 20.33 g |
| ddH2O | n/a | Fill up to 100 mL |
| Total | n/a | 100 mL |
15. 1 M MOPS-KOH, pH 7
Before adjusting the solution to its final volume with ddH2O, check the pH and adjust it to 7 using 1 M KOH.
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| MOPS | 1 M | 20.93 g |
| ddH2O | n/a | Fill up to 100 mL |
| Total | n/a | 100 mL |
16. 1 M Tris-HCl, pH 9
Before adjusting the solution to its final volume with ddH2O, check the pH and adjust it to 9 using 1 M HCl.
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| Tris | 1 M | 12.11 g |
| ddH2O | n/a | Fill up to 100 mL |
| Total | n/a | 100 mL |
17. 0.9 M NaF
Caution: NaF is toxic and can cause skin irritation. In contact with acids, it develops highly toxic gases. Please wear appropriate protective gear including gloves, safety goggles, and a lab coat when handling this chemical and perform necessary tasks under a fume hood if possible.
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| NaF | 0.9 M | 0.567 g |
| ddH2O | n/a | Fill up to 15 mL |
| Total | n/a | 15 mL |
18. 2 M BeSO4
Caution: Beryllium and its compounds are highly toxic and can cause severe skin burns and eye damage. Please wear appropriate protective gear including gloves, safety goggles, and a lab coat when handling this chemical and perform necessary tasks under a fume hood if possible.
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| BeSO4·4H2O | 2 M | 3.543 g |
| ddH2O | n/a | Fill up to 10 mL |
| Total | n/a | 10 mL |
Laboratory supplies
1. Glass cylinder 1 L (e.g., Roth, catalog number: K261.1)
2. Aluminum foil (e.g., VWR chemicals, catalog number: 293-4183)
3. Clamps for dialysis tubes (e.g., Scienova, catalog number: 40330)
4. Dialysis tubing (SERPAVOR, SERVA, catalog number: 44145.01)
5. Filtropur BT25, bottle top filter 0.22 μm (SARSTEDT, catalog number: 83.3940.511)
6. Glass beads, 3 mm (Supelco, catalog number: 1040150500)
7. Glas screw top vials ROTILABO® ND8 (Roth, catalog number: KE30.1)
8. Macro polystyrene cuvettes (SARSTEDT, maximum 4.5 mL volume, catalog number: 67.745)
9. Micro test plate, 96 wells (SARSTEDT, catalog number: 82.1581)
10. Pipette tips 10 μL, 200 μL, and 1,000 μL (SARSTEDT, catalog numbers: 70.3010.305, 70.3030.020, and 70.3050.020)
11. Reaction tubes 1.5 mL (SARSTEDT, catalog number: 72.690.001)
12. Round bottom glass tube (16 × 150 mm, Roth, catalog number: NY90.1)
13. Screw caps ROTILABO® ND8, without borehole, polypropylene (PP), black (Roth, catalog number: KE39.1)
Equipment
1. Analytical balance (e.g., Sartorius Entris-I II, 220 g/0.1 mg; Buch Holm, catalog number: 4669128)
2. Avanti mini extruder set (Avanti Polar Lipids, catalog number: 610000)
a. 1 mL gas-tight syringes (Avanti, no: 610017)
b. 10 mm filter supports (Avanti, no: 610014)
c. 19 mm Nucleopore tracketch membrane with a pore size of 0.2 μm (Schleicher & Schuell)
3. Computer with monitor (e.g., DELL OptiPlex 5090)
4. End-over-end turning device (neoLab, catalog number: 7-0045)
5. Eppendorf Research plus pipettes P2.5, P10, P20, P200, P1000 (Eppendorf, catalog numbers: 3123000012, 3123000020, 3123000039, 3123000055, and 3123000063)
6. Flow cabinet to work with organic solvents
7. Fluorometer (PTI QuantaMaster 800 fluorometers) with integrated FelixGX software, equipped with single cuvette Peltier K-155-C temperature control and magnetic stirrer (Horiba)
8. Freezer (-20 °C) (e.g., Liebherr, model: FNc 4625)
9. Freezer (-80 °C) (e.g., Panasonic, model: VIP plus)
10. Hamilton syringes 25 μL, 50 μL, 100 μL, 500 μL, and 1,000 μL (Hamilton Company, catalog numbers: 20972, 20788, 20790-U, 24539, and 81317)
11. Ice bucket (e.g., Sigma-Aldrich, catalog number: BAM168072002, model: Magic Touch 2)
12. Magnetic stir bar (e.g., Merck, catalog number: HS120548)
13. Magnetic stirrer (e.g., IKA, model: IKAMAG)
14. pH meter (pH-Meter 761 Calimatic, Knick)
15. Refrigerator (4 °C) (e.g., Liebherr, model: FKS 5000)
16. Scissors (e.g., VWR chemicals, catalog number: 233-1280)
17. Microplate reader (ClarioStar, BMG LABTECH)
18. Thermomixer (e.g., Eppendorf, model: Thermomixer 5436)
19. Vacuum pump V-100 with interface I-100 and rotary evaporator Rotavapor® R-100, SJ29/32, V, 220–240V (Buchi, catalog numbers: 11593636, 11593655D, 11100V111, and 11061895)
20. Vortex mixer (e.g., Scientific Industries, model: Vortex Genie 2)
Software and datasets
1. FelixGX software for the control of the PTI QuantaMaster 8000 fluorometer and accessories (Version 4)
2. CLARIOstar software for the control of the CLARIOstar plate reader (Version 5.40 R2)
3. CLARIOstar MARS software for data presentation of plate reader measurements (Version 3.31)
4. Microsoft Excel for data analysis (Version 2505)
Procedure
The following procedure outlines five main steps: (A) Preparation of LUVs, (B) detergent titration assay, (C) reconstitution procedure, (D) NADH-coupled ATPase assay to test for protein activity, and (E) dithionite-based flippase assay to measure lipid transport.
A. Preparation of LUVs
1. Clean the Hamilton syringes by flushing them 5–10 times with chloroform/methanol (1:1, volume/volume) under a fume hood.
Caution: Chloroform is a hazardous solvent. Conduct all work in a fume hood while wearing appropriate personal protective equipment.
2. Lipids are received as a powder or dissolved in chloroform and packaged in sealed glass ampoules; store at -20 °C until use.
Note: For long-term storage, evaporate the solvent and store the lipids at -20 °C to avoid oxidation of unsaturated lipids.
3. To prepare lipid stocks, transfer 10 mg aliquots from the Avanti glass ampoule into glass screw-top vials. Evaporate the solvent from the glass vials in a desiccator at 250 mbar for 3 h with an additional incubation for 1 h at 30 mbar. Close the vials with PP-lined screw caps and store at -20 °C until further use (PTFE-lined glass vials can also be used). Remove desired lipid stocks from the freezer, place on ice, and dissolve in chloroform:methanol (1:1, v/v) to a final lipid concentration of 10 mg/mL.
Notes:
1. It is highly recommended to use ethanol-stabilized chloroform with a certificate for the absence of phosgene and HCl.
2. Lipids other than DOPC, POPC, and POPG may have limited or very poor solubility in chloroform:methanol and may require only chloroform, only methanol, or a mixture of chloroform, methanol, and water. For example, PI4P lipids dissolve well in a mixture of chloroform:methanol:water (20:9:1, v/v/v).
4. Using Hamilton syringes, transfer 440 μL of DOPC, 425 μL of POPC, 99 μL of POPG, 19 μL of PI4P, and 17 μL of NBD-PS from 10 mg/mL lipid stocks into a round-bottom glass tube placed on ice. This results in a 10 mg lipid film with a molar ratio of 43.5/43.5/10/1.5/1.5 DOPC/POPC/POPG/PI4P/NBD-PS.
Note: Avoid any use of plasticware when handling organic solvents.
5. Evaporate the organic solvent at room temperature (RT) in a rotary evaporator at the reduced pressure of 250 mbar overnight, followed by evaporation at ~50 mbar for 1 h.
Pause point: The dried lipid film can be sealed and stored at -20 °C up to a month.
6. Add 667 µL of reconstitution buffer (see Recipe 1) and a 3-mm glass bead to the lipid film. Vortex for 10 min, which results in a final lipid concentration of 15 mg/mL.
7. Assemble the mini-extruder, consisting of two Hamilton glass syringes, a metal casing, an internal membrane support with a rubber ring, two filter supporters on each side, and two 200 nm polycarbonate membranes in between them; see Figure 1.

Figure 1. Assembly of the mini extruder set. Two polycarbonate membranes are used with the blank sides facing each other, and two filter supports are used on each side. More detailed information on the assembly can be found on the manufacturer's website (https://avantiresearch.com/services/equipment-products/mini-extruder-assembly-instructions).
8. Check the tightness of the mini-extruder by flushing with 1 mL of reconstitution buffer (see Recipe 1), i.e., pass back and forth between the syringes three to five times. Continue only if the buffer volume stays the same for each pass.
9. Pull the lipid suspension into one of the Hamilton glass syringes and place the filled syringe into one end of the mini-extruder.
10. Carefully place the second empty syringe into the opposite end of the mini-extruder. The plunger of the empty syringe should be depressed completely into the syringe barrel.
11. Extrude lipid suspension by passing through the filters 21 times, starting in syringe 1 and finishing in syringe 2. The lipid suspension appears milky at first and becomes more transparent after the first extrusion passes.
Note: The number of passages through the extruder needs to be uneven so that the final liposome sample is collected in syringe 2, which is uncontaminated by residual multilamellar vesicles that have never passed through the extruder. Placing the stabilizer block-extruder assembly on a hot plate enables precise and rapid temperature regulation, which is particularly crucial for processing lipids with transition temperatures exceeding ambient conditions. The phase transition temperatures of the main lipids in this study are as follows: DOPC: -20 °C [27], POPC: -2 °C [28], and POPG: -2 °C [29]. These low transition temperatures allow the extrusion to be performed at room temperature.
12. Inject the final liposome solution into a 1.5 mL reaction tube, cover it with aluminum foil to protect the NBD-lipids from light, and store it at 4 °C.
Pause point: The liposome solution can be stored at 4 °C for up to two weeks.
Note: If the lipid film does not contain any negatively charged lipids, the liposome solution should only be stored for a maximum of two days since the absence of electrostatic repulsion accelerates aggregation and possible fusion, compromising the stability and uniformity of the liposomes over time. The lipid concentration may change due to loss during the extrusion. It is possible to check the lipid concentration, e.g., using a colorimetric Bartlett assay [30]. The size of the liposomes depends on both the pore size of the membrane used during extrusion and the lipid composition. Dynamic light scattering (DLS) can be used to measure their size [24]. More information on DLS and instructions can be found in section B of Parker et al. [31].
B. Detergent titration assay
1. To each of three 1.5 mL reaction tubes, add 83.3 µL of the premade LUV solution.
2. Add 0, 1, and 2 µL of a 0.5 M CHAPS solution (see Recipe 2), and 41.7, 40.7 and 39.7 µL of reconstitution buffer (see Recipe 1), respectively, and incubate with end-over-end rotation at 30 rpm and room temperature for 25 min.
Note: The three tubes now contain CHAPS concentrations of 0, 4, and 8 mM.
3. Pipette 100 µL from each reaction tube into a well of a 96-well plate. Measure the absorbance at 550 nm in the plate reader.
Note: Destabilization of the vesicles can also be followed by light scattering measurements at 550 nm on a fluorescence spectrometer.
4. Pipette the 100 µL from each well back into their original tubes and add 3 µL of a 0.5 M CHAPS solution (see Recipe 2) to each tube. Incubate with end-over-end rotation at 30 rpm for 25 min.
Note: The three tubes now contain CHAPS concentrations of 12, 16, and 20 mM.
5. Pipette 100 µL from each reaction tube into a well of a 96-well plate. Measure the absorbance at 550 nm in the microplate reader. The CHAPS concentration used for the reconstitution should correspond to a concentration between the Rhalf and Rsol, indicated by the blue area in Figure 2. For this lipid composition and concentration, 13 mM CHAPS is required.
Note: Please ensure that the selected detergent does not absorb light at 550 nm, as this may interfere with spectrophotometric measurements. For optimal reproducibility, we recommend performing a detergent titration assay whenever a new lipid stock, lipid mixture, or different detergent is used. This allows accurate determination of the required detergent concentration for each specific condition. However, if the same lipid stock and composition are used, it is not necessary to repeat the assay before each reconstitution.
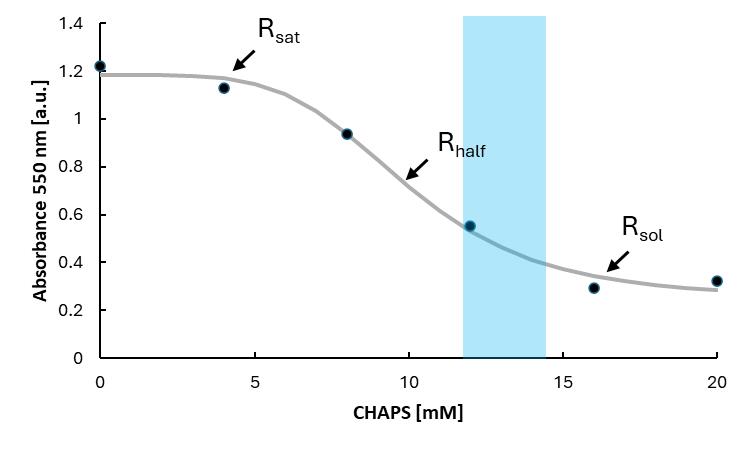
Figure 2. Solubilization curve of large unilamellar vesicles. Large unilamellar vesicles (LUVs) are solubilized with the detergent 3-[(3-cholamidopropyl)dimethylammonio]-1-propansulfonate (CHAPS). Destabilization of the vesicles is followed by absorbance measurements at 550 nm in a plate reader. Increasing detergent concentrations in the liposome solution cause a decrease in absorbance, corresponding to clarification. This occurs because the partitioning of detergent molecules in the membrane leads to the progressive formation of micelles, which are smaller than liposomes and therefore scatter less light. The points of saturation (Rsat), half-maximal solubilization (Rhalf), and complete solubilization (Rsol) are marked. In this reconstitution procedure using a detergent-mediated vesicle destabilization, a concentration between the Rhalf and Rsol was chosen, indicated by the blue area. The grey curve only serves as a visual aid; the concentration used can be estimated.
C. Reconstitution procedure
1. In a 1.5 mL reaction tube and to a final volume of 250 µL, add:
a. 166.6 µL of the LUV solution to a final concentration of 10 mg/mL.
b. 0.5 M CHAPS solution (see Recipe 2) to a final concentration determined in section B.
c. Reconstitution buffer (see Recipe 1) up to a volume of 240 µL.
d. Incubate at room temperature with end-over-end rotation at 30 rpm for 25 min.
2. Add 10 µL of a 1 µg/µL Drs2-Cdc50 stock in elution buffer containing 0.05% DDM (final concentration 0.04 µg/µL) and incubate at room temperature with end-over-end rotation at 30 rpm for 5 min.
Notes:
1. It is advised to additionally prepare a mock sample with 10 µL of protein elution buffer instead of protein stock solution.
2. Detailed information regarding the protein-to-lipid ratio can be found under General note 2.
3. Put a clamp on one side of a piece of pre-rinsed dialysis tubing, carefully pipette the sample into the tubing, and seal it with a second clamp.
Note: To rinse the tubing, wash it three times in ddHHH2O for 30 min at 60 °C, then wash it in reconstitution buffer at room temperature for 15 min.
4. Add the tubing to a 1 L glass cylinder filled with 900 mL of reconstitution buffer (see Recipe 1) and incubate while stirring at 4 °C for 3–4 h (see Figure 3).
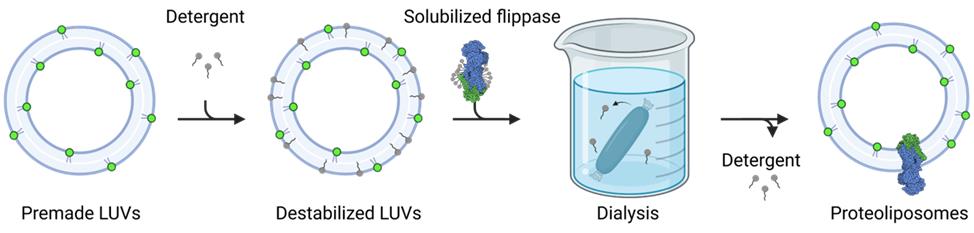
Figure 3. Schematic outline of the reconstitution procedure. Premade large unilamellar vesicles (LUVs) are destabilized by the addition of detergents. Solubilized membrane proteins are added, and the detergent is removed by dialysis to form proteoliposomes.
5. Discard the buffer and fill up the glass cylinder with 900 mL of fresh reconstitution buffer (see Recipe 1). Incubate while stirring at 4 °C overnight.
6. Discard the buffer and fill up the glass cylinder with 900 mL of fresh reconstitution buffer (see Recipe 1). Incubate while stirring at 4 °C for 4 h.
Note: Total dialysis time should be about 24 h.
7. Remove the dialysis tubing from the buffer, open one clamp, and carefully pipette the proteoliposome solution into a fresh 1.5 mL reaction tube. Cover the reaction tube with aluminum foil to protect the sample from light.
Note: The size distribution of proteoliposomes was determined by dynamic light scattering (DLS) at 25°C using a Zetasizer Nano ZS instrument (Malvern, Worcestershire, UK), yielding an average diameter of 232 ± 1.0 nm [32].
Pause point: The proteoliposomes can be stored at 4 °C for up to three days, but it is recommended to do the activity measurements as soon as possible because the protein activity decreases over time [32].
D. NADH-coupled ATPase assay
Note: The assay takes advantage of the fact that ATP hydrolysis can be coupled to NADH oxidation via an auxiliary enzyme system, such as PK and LDH. The addition of Mg2+ to ATP-containing buffer initiates the reaction because ATPases require Mg2+ and ATP for their catalytic activity.
1. Prepare the CLARIOstar microplate reader with the following steps and settings:
a. Prime pump 1 two times in ddH2O and then two times in 50 mM MgCl2 (see Recipe 3). Leave the pump inflow in the MgCl2 solution.
b. Set a discrete wavelength of 340 nm.
c. Set the number of cycles to 1,000 and the number of flashes per well and cycle to 20.
d. Set the final volume to 200 µL so the path length is corrected.
Note: If the experiment is performed with a different plate reader that does not have this function, a path length of 5.19 mm can be assumed and used to calculate the NADH concentration.
e. Adjust the layout for the sample wells and blank.
f. Set the injection timing to 40 cycles and the injection volume to 5 µL of MgCl2 per well from pump 1.
g. To mix the samples after injection, set the shaking after the injection cycle to 15 s and 300 rpm double orbital shaking.
h. Use the check timing option and set it to the lowest possible value.
Note: The cycle time depends on the number of wells and flashes per well and cycle.
2. Prepare a NADH-master mix by mixing 950 µL of reconstitution buffer (see Recipe 1), 20 µL of 100 mM PEP (see Recipe 4), 2.5 µL of 10 mg/mL PK, 2.5 µL of 5 mg/mL LDH, 20 µL of 6.7 mM NADH (see Recipe 5), and 2.5 µL of 0.5 M ATP (see Recipe 6) in a 1.5 mL reaction tube. Keep the master mix on ice.
3. In two wells of a 96-well plate, add 10 µL of proteoliposomes each. In one of the two wells, add 1.5 µL of 45 mM beryllium fluoride (see Recipe 7) and mix it thoroughly with the proteoliposomes to inhibit protein activity.
Note: Other inhibitors, such as vanadate or other metal fluorides (aluminum fluoride, magnesium fluoride), may be used in this step.
4. Add 185 µL of NADH-master mix to the well without the inhibitor and 183.5 µL to the well with the inhibitor. Add 195 µL of NADH-master mix to a third well as a blank for background subtraction. Mix by pipetting and start the measurement in the microplate reader immediately. As per the settings, 5 µL of 50 mM MgCl2 will be added to each well after 40 cycles to initiate ATPase activity.
E. Dithionite-based flippase assay
Note: To attribute changes in the fluorescence signal specifically to flippase activity, it is essential to conduct careful controls. Here, we include control measurements performed in the presence of Na-ATP, which cannot be hydrolyzed by P4-ATPases. Additional controls may involve using non-hydrolysable ATP derivatives, specific inhibitors, or reconstituted catalytically inactive transporter mutants to validate the findings further.
1. In a 1.5 mL reaction tube, mix 10 µL of proteoliposomes with 15 µL of Mg-ATP buffer (see Recipe 8) by vortexing and incubate for 30 min at 30 °C in the heat block. Cover the samples with aluminum foil to protect them from light.
2. Add 975 µL of reconstitution buffer, mix by pipetting, and transfer the solution to a cuvette.
3. Measure the fluorescence in the fluorometer at an excitation wavelength of 467 nm and an emission wavelength of 537 nm. The slit widths in the QuantaMaster should be set at 5 nm. Measure the fluorescence until the signal is stable (typically 50–100 s).
Note: If the signal is too low (below 106 cps in the QuantaMaster), the experiment can be performed with an increased volume of proteoliposomes, e.g., 15 µL of proteoliposomes, 22.5 µL of Mg-ATP buffer, and 962.5 µL of reconstitution buffer.
4. When the signal is stable, add 20 µL of a 1 M sodium dithionite solution (see Recipe 10) to obtain a final concentration of 10 mM and mix by pipetting. Continually measure for 350 s or until a stable line is obtained.
5. Add 50 µL of a 20 % (w/v) Triton X-100 solution (see Recipe 11) to a final concentration of 1% and mix by pipetting. Measure for another 50 s.
6. Repeat all the above steps with Na-ATP buffer (see Recipe 9) instead of Mg-ATP buffer.
Note: If available, repeat the experiment with a mock sample instead of proteoliposomes. Start each sample with a delay time of 12 min to ensure that you have enough time to measure the individual samples.
Data analysis
A. Solubilization curve
1. Export the measured data points into an Excel sheet.
2. Plot the concentration of CHAPS against the absorbance at 550 nm.
3. Estimate Rsat, Rhalf, and Rsol and choose an appropriate CHAPS concentration for the reconstitution.
B. NADH-coupled ATPase assay
1. Export the measured data points into an Excel sheet.
2. Calculate the NADH concentration from the measured extinction using Beer–Lambert’s law:
where c represents the concentration of NADH, A stands for the measured absorbance, ϵ is the extinction molar coefficient of NADH at 340 nm (6.22 L × mMol-1 × cm-1), and d is the path length of light in centimeters. Due to the path length correction, d equals one and can be omitted. To get a concentration in the range of µM, all values are multiplied by 1,000. The decrease in NADH over time is directly correlated with the hydrolysis of ATP by the Drs2-Cdc50 flippase complex (see Figure 4A).
Note: Automatic path length correction compensates for differences in sample fill volumes, ensuring that absorbance values and the corresponding calculated concentrations remain comparable regardless of the volume used. In a standard flat-bottom 96-well plate, a volume of 350 µL corresponds to a path length of approximately 1 cm. The path length (h) for other volumes can be estimated using the formula V = πr2h, where r is the radius of the well, which is measured at 0.34 cm.
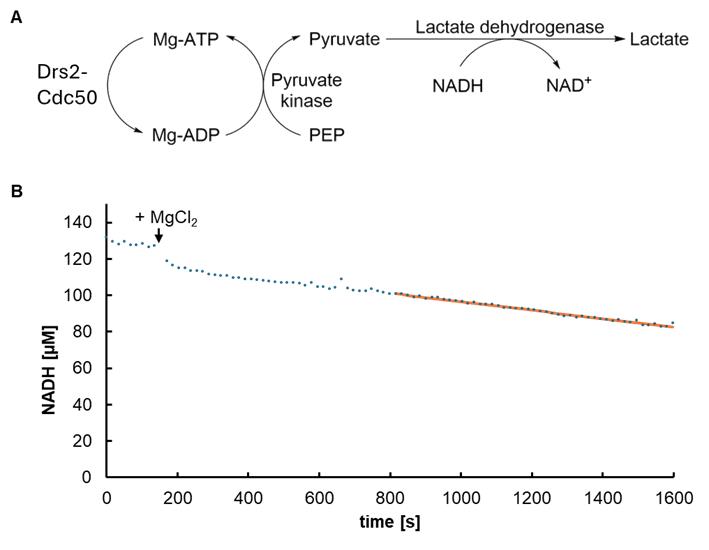
Figure 4. NADH-coupled ATPase assay. (A) Schematic outline of the assay. In the presence of Mg2+, ATP is hydrolyzed to ADP by the Drs2-Cdc50 flippase complex, and the pyruvate kinase converts ADP back to ATP, turning phospho(enol)pyruvic acid (PEP) to pyruvate in the process. Pyruvate is converted to lactate by lactate dehydrogenase, turning NADH into NAD+. (B) Exemplary trace of an ATPase assay. The NADH concentration decreases after the addition of MgCl2. The slope of the linear regression curve, which represents the rate of ATP hydrolysis, is used to calculate the specific activity.
Note: The same assay has been reported in multiple scientific articles [33,34].
3. Plot the concentration of NADH against the time and choose a linear area. Determine the slope of the regression of this area using Excel's SLOPE function (for an exemplary trace, see Figure 4B).
4. Calculate the specific protein activity in µmol Pi/min/mg as follows:
where x is the slope of the regression in µmol Pi/min, V represents the assay volume in L, and m is the amount of protein in mg.
C. Dithionite-based flippase assay
1. Save the measured data points as a text file (txt file) and import them into an Excel document.
2. Take all data points from 25 s before to the time point of dithionite addition and calculate the average. This will be the maximum fluorescence signal Fmax.
3. To normalize all data points, divide them by Fmax and multiply by 100.
F stands for the measured fluorescence, Fn stands for normalized fluorescence, and Fmax stands for the average maximum fluorescence signal.
4. The fluorescence decay curve observed in the dithionite assay typically exhibits two distinct phases: a fast and a slow decay, which correspond to the differential accessibility of fluorescently labeled lipids to dithionite. The initial rapid decrease in fluorescence (between 50 and 250 s) reflects the quenching of outer leaflet NBD-lipids, which are immediately accessible to dithionite. The subsequent slower decline (from 250 to 350 s) is typically attributed to the gradual permeation of dithionite into the vesicle interior, allowing the quenching of luminal NBD-lipids. This notion is further supported by the observation that the slow phase is present in both Mg-ATP (active transport) and Na-ATP (inactive control) samples, indicating that it is not a result of flippase-mediated lipid translocation.
5. To quantify flippase-mediated transport, take all data points between 250 and 350 s after dithionite addition and calculate the average fluorescence intensity over this time range. The extent of lipid transport is then determined by the difference in average fluorescence between the Mg-ATP (active transport) and Na-ATP (inactive control) samples (see Figure 5).
Note: If the signal decreases faster in one sample compared to the other, the difference in fluorescence at the timepoint of dithionite addition must be compared instead. To do this, fit a linear regression curve to the fluorescence data between 200 and 300 s after dithionite addition, and use the resulting equation to extrapolate the fluorescence value at the exact time of dithionite addition. This approach accounts for differences in decay rate during the slow phase and provides a more accurate comparison of initial transport activity between samples.
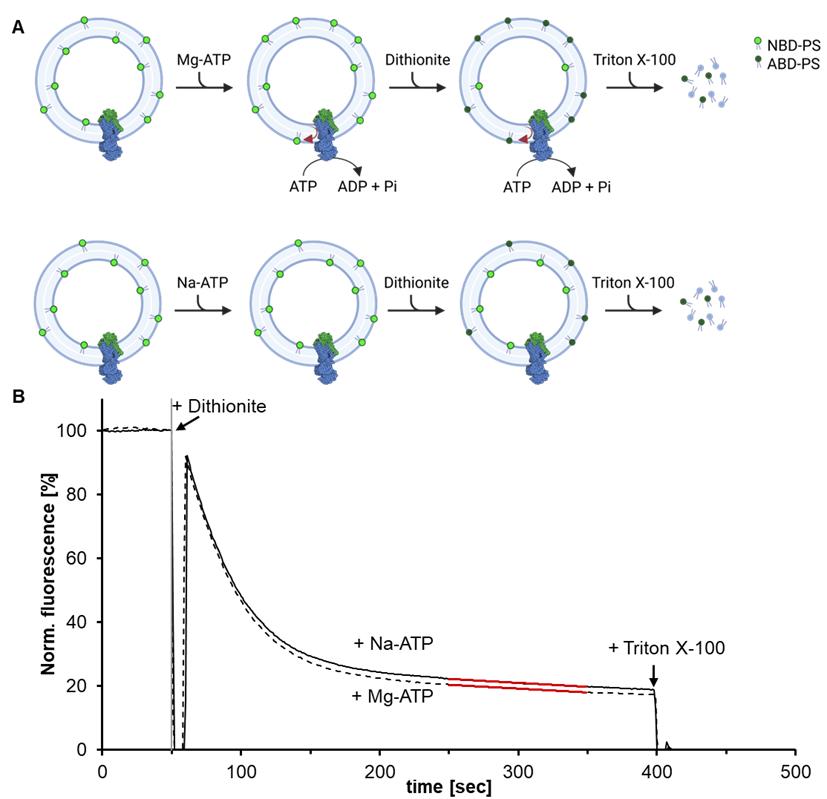
Figure 5. Dithionite-based lipid flippase assay. (A) Proteoliposomes containing fluorescent NBD-PS (7-nitro-2-1,3-benzoxadiazol-phosphatidylserine) are incubated with either Mg-ATP or Na-ATP for 30 min at 30 °C. The reconstituted Drs2-Cdc50 complex actively flips lipids from the inside to the outside of the vesicle only in the presence of Mg-ATP, whereas the Na-ATP sample remains inactive. To assess lipid translocation, outward-facing NBD-PS lipids are selectively quenched by dithionite, converting them into non-fluorescent ABD-PS (7-amino-2-1,3-benzoxadiazol-phosphatidylserine). As a result, the Na-ATP sample retains higher fluorescence compared to the Mg-ATP sample. Finally, the addition of Triton X-100 solubilizes the proteoliposomes, allowing complete quenching of all remaining NBD-labeled lipids. (B) Exemplary trace of a dithionite-based flippase assay. The extent of lipid flipping is determined by comparing the residual fluorescence between the two conditions, with the difference indicating the amount of translocated lipids.
Note: The Drs2-Cdc50 flippase complex can be incorporated in either orientation. In this figure, only the orientation with the ATP-binding domain facing outward is shown for simplicity, since it is the only one that can use Mg-ATP to translocate NBD-lipids.
Validation of protocol
This protocol or parts of it has been used and validated in the following research article:
Herrera et al. [32]. Direct evidence of lipid transport by the Drs2-Cdc50 flippase upon truncation of its terminal regions. Protein Science (Figure 3B–E).
Drs2-Cdc50 is only active in its truncated form and in the presence of PI4P [32]. Therefore, as an additional control in this case, the full-length Drs2-Cdc50 was reconstituted, and a dithionite assay was performed. Also, a catalytically inactive mutant, i.e., a dead mutant, can be reconstituted and used as a negative control. The dithionite assay can also be performed in the absence and presence of cofactors; for Drs2-Cdc50, the cofactor would be PI4P. Including a mock sample (protein-free reconstitution) is also recommended as a baseline control. To ensure reproducibility, each reconstitution experiment should be performed at least three times using large unilamellar vesicles (LUVs) prepared independently.
General notes and troubleshooting
General notes
1. This protocol is optimized for the yeast flippase Drs2-Cdc50. If this protocol is used to measure the activity of other flippases, the lipid composition of the vesicles and the choice of detergent must be considered. To choose an appropriate lipid composition, it is useful to consider the natural lipid environment of the protein. CHAPS is a zwitterionic detergent and, as such, is strong enough to solubilize many proteins. If a different detergent has to be used, it should be noted that not all detergents are dialyzable. If the detergent of choice is not dialyzable, a different detergent removal approach must be chosen. Depending on the specific properties of the detergent, a suitable approach may include gel filtration or adsorption using polystyrene beads [35].
2. In this protocol, a lipid-to-protein mass ratio of 250:1 and a molecular ratio of 51,403:1 were chosen. Based on an average vesicle diameter of 230 nm (as determined by DLS), and assuming a membrane bilayer thickness of 5 nm and a phospholipid cross-sectional area of 0.71 nm2, each vesicle is estimated to contain approximately 448,230 phospholipids with an average of 9 flippase molecules. This ratio can be adjusted individually for each protein. To achieve higher activity, the number of incorporated flippases per vesicle can be increased. This can be done by increasing the amount of protein added during reconstitution or reducing the lipid concentration, particularly when protein yields are low.
3. The NADH-coupled ATPase assay can also be used on purified, solubilized transmembrane proteins, to assess their activity before the reconstitution procedure. In this case, the NADH-master mix should contain a low concentration of DDM for protein solubility and lipids, including the proteins lipid substrate, as well as other components/co-factors necessary for protein activity. Activity strongly depends on the detergent/lipid ratio. In case of Drs2/Cdc50, use 0.98 g/L DDM, 0.145 g/L POPC, 0.075 g/L 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-L-serine, and 0.031 g/L PI4P [36].
Troubleshooting
Problem 1: No lipid transport can be measured (ATPase activity might or might not be measured).
Possible cause: Unsuccessful or low-efficiency reconstitution. The incorporation of the proteins into the vesicles can be checked by flotation on a discontinuous sucrose gradient. Instructions can be found in Section F of Bittame et al. [37]. The fractions can be analyzed by SDS-PAGE for protein content, and the presence of vesicles can be checked by measuring the fluorescence of the NBD-PS.
Solution: Try slightly increasing the concentration of detergent used for the reconstitution to bring it closer to Rsol. Alternatively, increasing the protein-to-lipid ratio may decrease the potentially empty vesicles and amplify differences in flippase activity. This adjustment aims to enhance assay sensitivity, enabling clearer detection and more accurate comparison of flippase function under different conditions.
Problem 2: No detectable ATPase activity or lipid transport, despite successful protein incorporation into liposome.
Possible cause 1: Loss of protein activity during reconstitution.
Solution: Validate the activity of the flippase complex at the solubilized stage before reconstitution into proteoliposomes by assessing lipid-stimulated ATPase activity (see General note 3). A 24-h dialysis used for detergent removal typically does not compromise protein activity once liposomes have formed. In fact, some P-type ATPases retain full activity even after extended dialysis periods of up to 72 h [38]. If dialysis adversely affects protein activity, consider lowering the detergent concentration during reconstitution or using a milder detergent. If the selected detergent is not dialyzable, switch to an alternative detergent removal method such as adsorbent beads (e.g., Bio-Beads) or size-exclusion chromatography.
Possible cause 2: ATP-binding domain oriented inward.
Solution: Verify the orientation of the flippase within the liposomes using for example a fluorescence-based method involving self-labeling enzyme tags combined with membrane-impermeable fluorescent probes, protease protection assays, or antibody accessibility in the presence and absence of membrane permeabilization [39,40].
Adjusting the lipid composition, protein-to-lipid ratio, detergent-lipid ratio, or detergent removal rate may help favor a more uniform and functional orientation.
Problem 3: A very steep slope of the fluorescence curve during the dithionite assay or a fast drop in fluorescence to zero.
Possible cause: Permeability of vesicles to dithionite resulting in the quenching of luminal NBD-lipids before Triton X-100 addition.
Solution: To assess detergent content, it is recommended to perform thin-layer chromatography to detect any residual detergent [41]. If detergent remains, add one or more additional dialysis steps to ensure complete removal. Alternatively, perform the dithionite assay at a lower temperature (e.g., 10 °C) or use a reduced dithionite concentration (e.g., 5 mM). If these adjustments do not resolve the issue, consider a different lipid composition for the vesicles (e.g. addition of sterols).
Acknowledgments
This protocol was adapted from our previous work [32]. Research in the author’s laboratory is funded by grants from the Deutsche Forschungsgemeinschaft (GU 1133/13-1; GU 1133/15-1) and from the Agence Nationale de la Recherche (ANR-21-CE11-0015-01). The figures were prepared using Biorender.com.
Competing interests
The authors declare no conflicts of interest.
References
- Clarke, R., Hossain, K. and Cao, K. (2020). Physiological roles of transverse lipid asymmetry of animal membranes. Biochimica et Biophysica Acta (BBA) – Biomembranes. 1862(10): 183382. https://doi.org/10.1016/j.bbamem.2020.183382
- Doktorova, M., Levental, I. and Heberle, F. A. (2023). Seeing the Membrane from Both Sides Now: Lipid Asymmetry and Its Strange Consequences. Cold Spring Harbor Perspect Biol. 15(12): a041393. https://doi.org/10.1101/cshperspect.a041393
- Doktorova, M., Symons, J. L. and Levental, I. (2020). Structural and functional consequences of reversible lipid asymmetry in living membranes. Nat Chem Biol. 16(12): 1321–1330. https://doi.org/10.1038/s41589-020-00688-0
- Lopez-Marques, R. L., Theorin, L., Palmgren, M. G. and Pomorski, T. G. (2014). P4-ATPases: lipid flippases in cell membranes. Pflugers Arch - Eur J Physiol. 466(7): 1227–1240. https://doi.org/10.1007/s00424-013-1363-4
- Pabst, G. and Keller, S. (2024). Exploring membrane asymmetry and its effects on membrane proteins. Trends Biochem Sci. 49(4): 333–345. https://doi.org/10.1016/j.tibs.2024.01.007
- Sprong, H., van der Sluijs, P. and van Meer, G. (2001). How proteins move lipids and lipids move proteins. Nat Rev Mol Cell Biol. 2(7): 504–513. https://doi.org/10.1038/35080071
- Sharom, F. J. (2011). Flipping and flopping-lipids on the move. IUBMB Life. 63: 736–746. https://doi.org/10.1002/iub.515
- Chen, S., Wang, J., Muthusamy, B., Liu, K., Zare, S., Andersen, R. J. and Graham, T. R. (2006). Roles for the Drs2p–Cdc50p Complex in Protein Transport and Phosphatidylserine Asymmetry of the Yeast Plasma Membrane. Traffic. 7(11): 1503–1517. https://doi.org/10.1111/j.1600-0854.2006.00485.x
- Bull, L. N., van Eijk, M. J., Pawlikowska, L., DeYoung, J. A., Juijn, J. A., Liao, M., Klomp, L. W., Lomri, N., Berger, R., Scharschmidt, B. R., et al. (1998). A gene encoding a P-type ATPase mutated in two forms of hereditary cholestasis. Nat Genet. 18(3): 219–224. https://doi.org/10.1038/ng0398-219
- Jaakola, V. P. and Scalise, M. (2019). Membrane Proteins: New Approaches to Probes, Technologies, and Drug Design, Part II. SLAS Discovery. 24(10): 941–942. https://doi.org/10.1177/2472555219881666
- Boulos, I., Jabbour, J., Khoury, S., Mikhael, N., Tishkova, V., Candoni, N., Ghadieh, H. E., Veesler, S., Bassim, Y., Azar, S., et al. (2023). Exploring the World of Membrane Proteins: Techniques and Methods for Understanding Structure, Function, and Dynamics. Molecules. 28(20): 7176. https://doi.org/10.3390/molecules28207176
- Zoonens, M., Comer, J., Masscheleyn, S., Pebay-Peyroula, E., Chipot, C., Miroux, B. and Dehez, F. (2013). Dangerous Liaisons between Detergents and Membrane Proteins. The Case of Mitochondrial Uncoupling Protein 2. J Am Chem Soc. 135(40): 15174–15182. https://doi.org/10.1021/ja407424v
- Shen, H. H., Lithgow, T. and Martin, L. (2013). Reconstitution of Membrane Proteins into Model Membranes: Seeking Better Ways to Retain Protein Activities. Int J Mol Sci. 14(1): 1589–1607. https://doi.org/10.3390/ijms14011589
- Allen, T. M., Romans, A. Y., Kercret, H. and Segrest, J. P. (1980). Detergent removal during membrane reconstitution. Biochimica et Biophysica Acta (BBA) – Biomembranes. 601: 328–342. https://doi.org/10.1016/0005-2736(80)90537-4
- Rigaud, J. L., Levy, D., Mosser, G. and Lambert, O. (1998). Detergent removal by non-polar polystyrene beads. Eur Biophys J. 27(4): 305–319. https://doi.org/10.1007/s002490050138
- Jensen, M. S., Costa, S. R., Theorin, L., Christensen, J. P., Pomorski, T. G. and López‐Marqués, R. L. (2016). Application of image cytometry to characterize heterologous lipid flippases in yeast. Cytometry Part A. 89(7): 673–680. https://doi.org/10.1002/cyto.a.22886
- Pagano, R. E. and Sleight, R. G. (1985). Defining Lipid Transport Pathways in Animal Cells. Science. 229(4718): 1051–1057. https://doi.org/10.1126/science.4035344
- RAGGERS, R. J., VOGELS, I. and van MEER, G. (2001). Multidrug-resistance P-glycoprotein (MDR1) secretes platelet-activating factor. Biochem J. 357(3): 859–865. https://doi.org/10.1042/bj3570859
- McIntyre, J. C. and Sleight, R. G. (1991). Fluorescence assay for phospholipid membrane asymmetry. Biochemistry. 30(51): 11819–11827. https://doi.org/10.1021/bi00115a012
- Connor, J. and Schroit, A. J. (1987). Determination of lipid asymmetry in human red cells by resonance energy transfer. Biochemistry. 26(16): 5099–5105. https://doi.org/10.1021/bi00390a031
- Warren, G. B., Toon, P. A., Birdsall, N. J. M., Lee, A. G. and Metcalfe, J. C. (1974). Reconstitution of a Calcium Pump Using Defined Membrane Components. Proc Natl Acad Sci USA. 71(3): 622–626. https://doi.org/10.1073/pnas.71.3.622
- Larsen, J., Hatzakis, N. S. and Stamou, D. (2011). Observation of Inhomogeneity in the Lipid Composition of Individual Nanoscale Liposomes. J Am Chem Soc. 133(28): 10685–10687. https://doi.org/10.1021/ja203984j
- Mathiasen, S., Christensen, S. M., Fung, J. J., Rasmussen, S. G. F., Fay, J. F., Jorgensen, S. K., Veshaguri, S., Farrens, D. L., Kiskowski, M., Kobilka, B., et al. (2014). Nanoscale high-content analysis using compositional heterogeneities of single proteoliposomes. Nat Methods. 11(9): 931–934. https://doi.org/10.1038/nmeth.3062
- Veit, S., Paweletz, L. C., Bohr, S. R., Menon, A. K., Hatzakis, N. S. and Pomorski, T. G. (2022). Single Vesicle Fluorescence-Bleaching Assay for Multi-Parameter Analysis of Proteoliposomes by Total Internal Reflection Fluorescence Microscopy. Acs Appl Mater Inter. 14(26): 29659–29667. https://doi.org/10.1021/acsami.2c07454
- Azouaoui, H., Montigny, C., Ash, M. R., Fijalkowski, F., Jacquot, A., Grønberg, C., López-Marqués, R. L., Palmgren, M. G., Garrigos, M., le Maire, M., et al. (2014). A High-Yield Co-Expression System for the Purification of an Intact Drs2p-Cdc50p Lipid Flippase Complex, Critically Dependent on and Stabilized by Phosphatidylinositol-4-Phosphate. PLoS One. 9(11): e112176. https://doi.org/10.1371/journal.pone.0112176
- Dieudonné, T., Jaxel, C., Lejeune, M., Lenoir, G. and Montigny, C. (2023). Expression in Saccharomyces cerevisiae and Purification of a Human Phospholipid Flippase. Methods Mol Biol. 2652: 231–246. https://doi.org/10.1007/978-1-0716-3147-8_13
- Thakur, R., Das, A. and Chakraborty, A. (2014). Interaction of human serum albumin with liposomes of saturated and unsaturated lipids with different phase transition temperatures: a spectroscopic investigation by membrane probe PRODAN. RSC Adv. 4(28): 14335–14347. https://doi.org/10.1039/c4ra01214c
- Wanderlingh, U., Branca, C., Crupi, C., Conti Nibali, V., La Rosa, G., Rifici, S., Ollivier, J. and D’Angelo, G. (2017). Molecular Dynamics of POPC Phospholipid Bilayers through the Gel to Fluid Phase Transition: An Incoherent Quasi-Elastic Neutron Scattering Study. J Chem. 2017: 1–8. https://doi.org/10.1155/2017/3654237
- Pozo Navas, B., Lohner, K., Deutsch, G., Sevcsik, E., Riske, K., Dimova, R., Garidel, P. and Pabst, G. (2005). Composition dependence of vesicle morphology and mixing properties in a bacterial model membrane system. Biochimica et Biophysica Acta (BBA) – Biomembranes. 1716(1): 40–48. https://doi.org/10.1016/j.bbamem.2005.08.003
- Mathiassen, P. and Pomorski, T. (2022). A Fluorescence-based Assay for Measuring Phospholipid Scramblase Activity in Giant Unilamellar Vesicles. Bio Protoc. 12(6): e4366. https://doi.org/10.21769/bioprotoc.4366
- Parker, E. and Lollar, P. (2021). Measurement of the Translational Diffusion Coefficient and Hydrodynamic Radius of Proteins by Dynamic Light Scattering. Bio Protoc. 11(20): e4195. https://doi.org/10.21769/bioprotoc.4195
- Herrera, S. A., Justesen, B. H., Dieudonné, T., Montigny, C., Nissen, P., Lenoir, G. and Günther Pomorski, T. (2024). Direct evidence of lipid transport by the Drs2–Cdc50 flippase upon truncation of its terminal regions. Protein Sci. 33(3): e4855. https://doi.org/10.1002/pro.4855
- Sehgal, P., Olesen, C. and Møller, J. V. (2016). ATPase Activity Measurements by an Enzyme-Coupled Spectrophotometric Assay. Methods Mol Biol. 1377: 105–109. https://doi.org/10.1007/978-1-4939-3179-8_11
- Lenoir, G., Dieudonné, T., Lamy, A., Lejeune, M., Vazquez‐Ibar, J. and Montigny, C. (2018). Screening of Detergents for Stabilization of Functional Membrane Proteins. Curr Protoc Protein Sci. 93(1): e59. https://doi.org/10.1002/cpps.59
- Lévy, D., Bluzat, A., Seigneuret, M. and Rigaud, J. L. (1990). A systematic study of liposome and proteoliposome reconstitution involving Bio-Bead-mediated Triton X-100 removal. Biochimica et Biophysica Acta (BBA) – Biomembranes. 1025(2): 179–190. https://doi.org/10.1016/0005-2736(90)90096-7
- Azouaoui, H., Montigny, C., Dieudonné, T., Champeil, P., Jacquot, A., Vázquez-Ibar, J. L., Le Maréchal, P., Ulstrup, J., Ash, M. R., Lyons, J. A., et al. (2017). High phosphatidylinositol 4-phosphate (PI4P)-dependent ATPase activity for the Drs2p-Cdc50p flippase after removal of its N- and C-terminal extensions. J Biol Chem. 292(19): 7954–7970. https://doi.org/10.1074/jbc.m116.751487
- Bittame, A., Lopez, J., Effantin, G., Blanchard, N., Cesbron-Delauw, M. F., Gagnon, J. and Mercier, C. (2016). Lipid Extraction from HeLa Cells, Quantification of Lipids, Formation of Large Unilamellar Vesicles (LUVs) by Extrusion and in vitro Protein-lipid Binding Assays, Analysis of the Incubation Product by Transmission Electron Microscopy (TEM) and by Flotation across a Discontinuous Sucrose Gradient. Bio Protoc. 6(20): e1963. https://doi.org/10.21769/bioprotoc.1963
- Apell, H. J. and Damnjanovic, B. (2016). Assaying P-Type ATPases Reconstituted in Liposomes. Methods Mol Biol. 1377: 127–156. https://doi.org/10.1007/978-1-4939-3179-8_14
- Paweletz, L., Veit, S. and Pomorski, T. (2022). A Fluorescence-based Approach Utilizing Self-labeling Enzyme Tags to Determine Protein Orientation in Large Unilamellar Vesicles. Bio Protoc. 12(21): e4542. https://doi.org/10.21769/bioprotoc.4542
- Veit, S., Paweletz, L. C. and Günther Pomorski, T. (2023). Determination of membrane protein orientation upon liposomal reconstitution down to the single vesicle level. Biol Chem. 404(7): 647–661. https://doi.org/10.1515/hsz-2022-0325
- Paweletz, L. C., Holtbrügge, S. L., Löb, M., De Vecchis, D., Schäfer, L. V., Günther Pomorski, T. and Justesen, B. H. (2023). Anionic Phospholipids Stimulate the Proton Pumping Activity of the Plant Plasma Membrane P-Type H+-ATPase. Int J Mol Sci. 24(17): 13106. https://doi.org/10.3390/ijms241713106
Article Information
Publication history
Received: Apr 1, 2025
Accepted: Jun 16, 2025
Available online: Jul 3, 2025
Published: Jul 20, 2025
Copyright
© 2025 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Van Der Linden, I. M., Herrera, S. A., Montigny, C., Lenoir, G., Pomorski, T. G. and Uzun, H. D. (2025). A Fluorescence-Based Flippase Assay to Monitor Lipid Transport by Drs2-Cdc50. Bio-protocol 15(14): e5393. DOI: 10.21769/BioProtoc.5393.
Category
Biochemistry > Protein > Activity
Biochemistry and Biophysics
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.