- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Comprehensive Mapping of EZ-Tn5 Transposon Insertion Sites in Pseudomonas argentinensis SA190 Using RATE-PCR


Published: Vol 15, Iss 14, Jul 20, 2025 DOI: 10.21769/BioProtoc.5389 Views: 2013
Reviewed by: Joyce ChiuAnonymous reviewer(s)
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Transposon mutagenesis is a powerful tool for investigating gene function in bacteria, particularly in newly discovered species. In this study, we applied the hyperactive EZ-Tn5 transposase system to Pseudomonas argentinensis SA190, an endophytic bacterium known for enhancing plant resilience under drought stress. By leveraging the random amplification of transposon ends (RATE)-PCR method, we successfully mapped the insertion sites of the transposon within the SA190 genome. This approach enabled the precise identification of disrupted genes, offering insights into their roles in bacterial function and interaction with host plants. Our comprehensive protocol, including competent cell preparation, transformation, and insertion site mapping, provides a reliable framework for future studies aiming to explore gene function through mutagenesis.
Key features
• The use of the hyperactive EZ-Tn5 transposase system ensures efficient and detectable random mutagenesis across the Pseudomonas argentinensis SA190 genome, facilitating comprehensive gene disruption studies.
• The technique is employed to identify and map the transposon insertion sites, allowing for precise determination of gene function and its impact on bacterial phenotypes.
• This method enables the exploration of a broad range of gene functions within SA190, particularly those involved in plant growth promotion and stress tolerance.
• This method can be readily adapted to generate mutant libraries in other bacterial species, emphasizing its transferability.
Keywords: Transposon mutagenesisGraphical overview
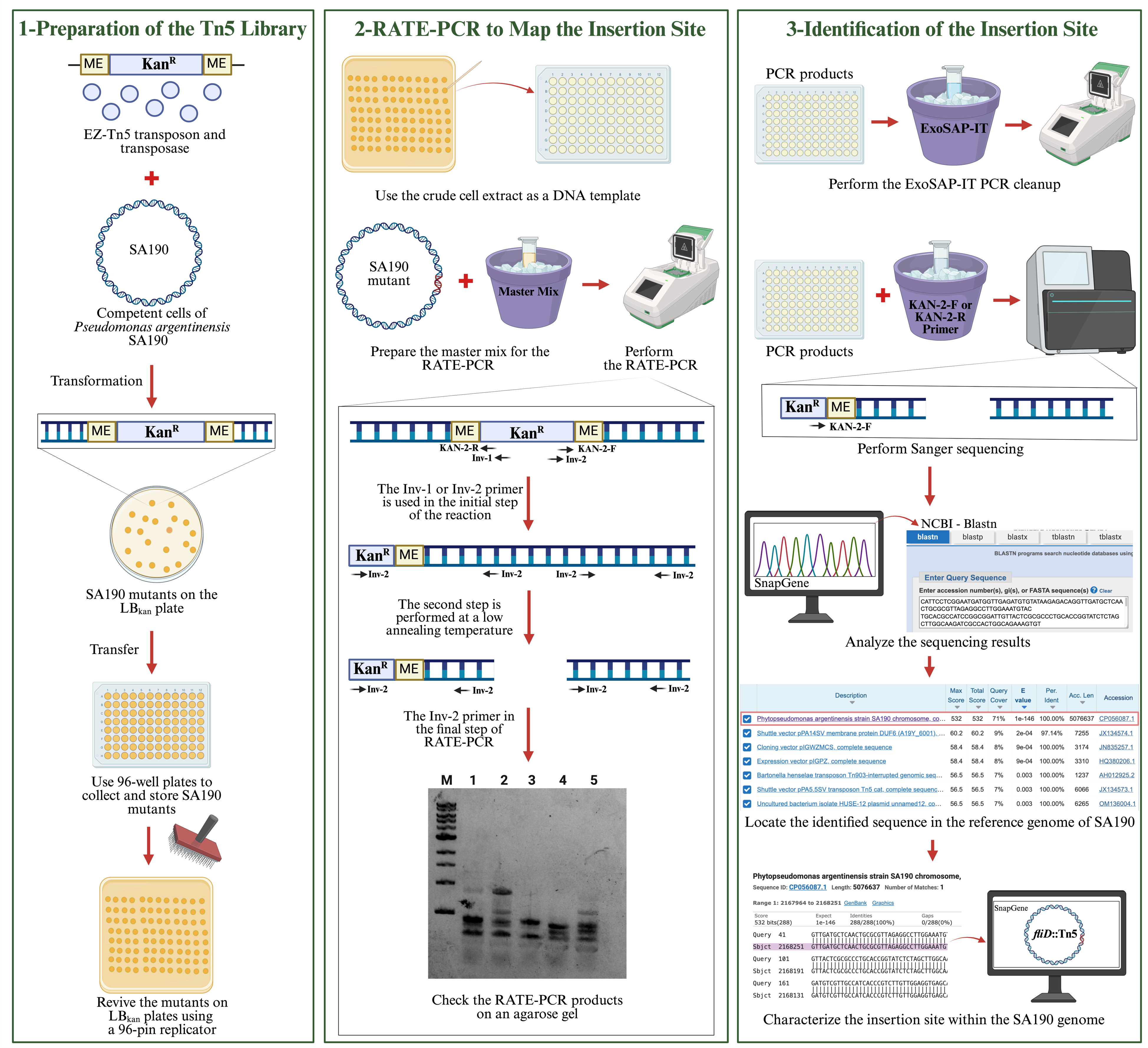
Workflow for characterizing the Tn5 insertion site in the Pseudomonas argentinensis SA190 genome using RATE-PCR
Background
Identification of essential genes by bacterial genomic studies helps to understand the functions of biological processes in bacteria [1–4]. One of the indispensable tools for studying genes in bacteria is mutagenesis, e.g., transposons insertion [1,5]. It allows us to establish a link between gene disruption and phenotypic traits. Tn5 is one of the most common transposases used to generate random mutations in the bacterial genome [6]. However, random mutagenesis methods vary in precision and efficiency, with chemical and UV mutagenesis often causing multiple mutations [7], while conventional transposons frequently exhibit lower insertion efficiency and can result in multiple insertions per genome [8]. The EZ-Tn5 transposome system addresses these limitations by incorporating a hyperactive Tn5 transposase and a kanamycin resistance cassette, allowing high-frequency, single-copy insertions that simplify mutant analysis [4,8]. Its broad host range and proven reliability across diverse bacterial species make EZ-Tn5 the preferred tool for generating effective mutant libraries in this study [8].
Mechanistically, the EZ-Tn5 transposome system, developed from the native Tn5 structure, consists of hyperactive Tn5 transposase and a kanamycin antibiotic cassette [6,9]. In the EZ-Tn5 system, the hyperactive transposase binds to the mosaic ends at the ends of the kanamycin cassette and facilitates random transposition in the genome [6,9]. The presence of the kanamycin cassette facilitates the selection of bacteria in which transposition occurs. After transposition takes place in the bacterial genome, the transposase decays [9]. In this way, transposition occurs only once in the genome.
In this work, the EZ-Tn5 transposon system was applied on the bacteria Pseudomonas argentinensis SA190, an endophytic plant growth-promoting bacteria that enhances plant performance under drought stress [10]. As SA190 is a newly discovered bacterium, it is crucial to understand its essential genes. Elucidating the role of genes in SA190 will contribute to understanding the beneficial interaction between SA190 and plants. SA190 should first be prepared as competent cells to receive the transposon DNA for transposon mutagenesis. The transposon is transferred to recipient-competent cells by transformation. Since EZ-Tn5 performs random mutagenesis in the bacterial genome, identifying the mutated gene is vital in establishing a significant relationship between the gene and its function. To achieve this, we employed the random amplification of transposon ends (RATE)-PCR method, a fast and effective approach for identifying disrupted genes in the genome [11,12]. Specific primers designed for the transposon ends were used to amplify one of the transposon-end regions. RATE-PCR involves three stages, utilizing a combination of high-stringency and low-stringency temperatures to produce both specific and nonspecific PCR products of various sizes on agarose gels. These products were then subjected to Sanger sequencing using either the forward or reverse primers of the EZ-Tn5 transposon. The resulting sequences were compared against the SA190 reference genome to precisely characterize the disrupted genes.
Materials and reagents
Biological materials
1. Pseudomonas argentinensis SA190 (isolated from surface-sterilized root nodules of the plant Indigofera argentea in the Jizan area (16°56.475′N, 42°36.694′E) of Saudi Arabia [13], kindly provided by Heribert Hirt Lab at King Abdullah University of Science and Technology (KAUST), Jeddah, Saudi Arabia).
Reagents
1. Kanamycin sulfate (Fisher Bioreagents, catalog number: 25389-94-0)
2. BactoTM tryptone (Gibco, catalog number: 211705)
3. Sodium chloride (NaCl) (Merck, catalog number: 1064049025)
4. BactoTM yeast extract (Gibco, catalog number: 212750)
5. Sodium hydroxide (NaOH) (Merck, catalog number: 71687-500G)
6. LB broth with agar (Sigma-Aldrich, catalog number: L2897-250G)
7. Potassium chloride (KCI) (Merck, catalog number: P9541-500G)
8. Magnesium chloride (MgCI2) (Merck, catalog number: 208337-100G)
9. Magnesium sulfate (MgSO4) (Merck, catalog number: M7506-500G)
10. Glucose (Merck, catalog number: G8270-100G)
11. Absolute glycerol (Sigma-Aldrich, catalog number: G5516)
12. Nuclease-free water (New England Biolabs, catalog number: B1500S)
13. Phusion® High-Fidelity DNA polymerase (New England Biolabs, catalog number: M0530L)
14. ExoSAP-IT PCR product cleanup reagent (Thermo Fisher Scientific, catalog number: 78-201-1ML)
15. 10 mM dNTPs (Thermo Fisher Scientific, catalog number: 18427089)
Solutions
1. Luria-Bertani (LB) agar (see Recipes)
2. Luria-Bertani (LB) media (see Recipes)
3. Super optimal broth with catabolite repression (SOC) media (see Recipes)
4. Kanamycin sulfate stock solution (50 mg/mL) (see Recipes)
Recipes
1. Luria-Bertani (LB) agar
| Reagent | Final concentration | Quantity |
|---|---|---|
| Tryptone | 1% (w/v) | 10 g |
| Yeast extract | 0.5% (w/v) | 5 g |
| NaCl | 1% (w/v) | 10 g |
| Agar | 0.9% | 9 g |
| Milli-Q H2O | variable | Fill to 1 L |
| Total | n/a | 1 L |
a. Adjust pH to 7.0 using 5 M NaOH
b. Autoclave (121 °C, 15 min, 100.8 kPa).
2. Luria-Bertani (LB) media
| Reagent | Final concentration | Quantity |
|---|---|---|
| Tryptone | 1% (w/v) | 10 g |
| Yeast extract | 0.5% (w/v) | 5 g |
| NaCl | 1% (w/v) | 10 g |
| Milli-Q H2O | variable | Fill to 1 L |
| Total (optional) | n/a | 1 L |
a. Adjust pH to 7.0 using 5 M NaOH
b. Autoclave (121 °C, 15 min, 100.8 kPa).
3. Super optimal broth with catabolite repression (SOC) media
| Reagent | Final concentration | Quantity |
|---|---|---|
| Tryptone | 2% (w/v) | 20 g |
| Yeast extract | 0.5% (w/v) | 5 g |
| NaCl | 10 mM | 2 mL |
| KCl | 2.5 mM | 2.5 mL |
| MgCl2 | 10 mM | 10 mL |
| MgSO4 | 10 mM | 10 mL |
| Milli-Q H2O | variable | Fill to 980 mL |
| Total | n/a | 980 mL |
a. Autoclave (121 °C, 15 min, 100.8 kPa).
b. After autoclaving, add 20 mL of filter-sterilized 1 M glucose.
4. Kanamycin sulfate stock solution (50 mg/mL)
| Reagent | Final concentration | Quantity |
|---|---|---|
| Kanamycin sulfate | 50 mg/mL | 0.5 g |
| Milli-Q H2O | variable | 10 mL |
| Total | n/a | 10 mL |
a. Filter sterilize the solution using a 0.2 μm filter and 10 mL syringe.
b. Aliquot 500 μL stocks and store at -20 °C.
Laboratory supplies
1. 1.5 mL Eppendorf tubes (Thermo Fisher Scientific, catalog number: AM12450)
2. 2 mL Eppendorf tubes (Thermo Fisher Scientific, catalog number: AM12475)
3. NuncTM Petri dishes (Thermo Fisher Scientific, catalog number: 263991)
4. NuncTM square BioAssay dishes (Thermo Fisher Scientific, catalog number: 240845)
5. Inoculating loop (VWR®, catalog number: 612-7277)
6. L-shaped spreader (VWR®, catalog number: 612-1561)
7. 50 mL conical tubes (Thermo Fisher Scientific, catalog number: AM12502)
8. 0.1 cm electroporation cuvette (Thermo Fisher Scientific, catalog number: P41050)
9. NalgeneTM polycarbonate Erlenmeyer flask (Thermo Fisher Scientific, catalog number: 4103-0250PK)
10. Serological pipette (Thermo Fisher Scientific, catalog number: 170358T)
11. EZ-Tn5TM <KAN-2>
Note: This product is no longer available; as an alternative, we suggest using EZ-Tn5 Transposase (LGC Biosearch Technologies, catalog number: TNP92110)
12. CorningTM CostarTM 96-well plate, sterile (Fisher Scientific, catalog number: 10695951)
13. PCR tubes (Thermo Fisher Scientific, catalog number: AB0490)
14. 0.2 μm FisherbrandTM syringe filters, sterile (Thermo Fisher Scientific, catalog number: 09-719C)
15. 10 mL BD Luer-LokTM disposable syringes without needles (Thermo Fisher Scientific, catalog number: 14-823-2A)
16. Toothpick (local purchase)
Equipment
1. MicroPulser Electroporator (Bio-Rad Laboratories, catalog number: 1652100)
2. Centrifuge 5430 (Eppendorf, catalog number: 5427000610)
3. New Brunswick Innova® 40/40R Benchtop Orbital Shaker (Eppendorf, catalog number: M1299-0090)
4. HerathermTM General Protocol Microbiological Incubator (Thermo Fisher Scientific, catalog number: 51028063)
5. SimpliAmpTM Thermal Cycler (Thermo Fisher Scientific, catalog number: A24811)
6. 96-Pin Microplate Replicator (Boekel Scientific, catalog number: 140500)
7. EppendorfTM ResearchTM plus, mechanical single-channel pipettes multipacks (Thermo Fisher Scientific, catalog number: 13-684-250)
8. Autoclave (any manufacturer)
9. Thermo ScientificTM RevcoTM ExF -86 °C upright ultra-low temperature freezer (Thermo Fisher Scientific, catalog number: EXF24086A)
10. Liquid nitrogen (N2) tank (any manufacturer)
Software and datasets
1. SnapGene® (SnapGene, Version 6.2.1, https://www.snapgene.com/)
2. NCBI Blastn program, free use, web-based platform (https://blast.ncbi.nlm.nih.gov/Blast.cgi)
Procedure
A. Preparation of competent cells of Pseudomonas argentinensis SA190
1. Freshly streak the glycerol stock of Pseudomonas argentinensis SA190 onto an LB agar plate (Recipe 1) using a loop and incubate at 28 °C for overnight (16 h) incubation.
Note: The genome size of SA190 is 5,076,637 bp, and approximately 20,000 bacterial colonies are required to achieve 4× full genome coverage.
2. The next day, select a single colony from the plate and transfer to a flask containing 40 mL of LB media (Recipe 2).
3. Incubate the flask at 28 °C and 220 rpm.
4. Transfer 100 μL from the overnight culture to a new flask containing 40 mL of LB broth media.
5. Incubate at 28 °C and 220 rpm until the OD600 reaches 0.4.
Note: The doubling time of SA190 is approximately 60 min at 28 °C in LB broth media.
6. Grow the bacterial culture to an OD600 of 0.4, evenly transfer into two 50 mL tubes, and place on ice.
7. Centrifuge the 50 mL tubes at 5,660× g for 5 min at 4 °C.
8. Discard the supernatant from the 50 mL tubes.
9. Resuspend the pellet in 20 mL of ice-cold H2O by gently shaking the 50 mL tubes.
10. Centrifuge at 5,660× g for 5 min at 4 °C.
11. Discard the supernatant from the 50 mL tubes.
12. Resuspend the pellet in 2 mL of 10% glycerol by gently shaking the 50 mL tubes.
13. Aliquot the sample into 1.5 mL Eppendorf tubes by transferring 100 μL to each.
14. Transfer the 1.5 mL Eppendorf tubes from ice to liquid nitrogen.
15. For long-term storage, store the tubes at -80 °C.
Note: Electrocompetent cells can be stored at -80 °C for up to 6 months without a significant loss of efficiency.
B. Transformation
1. Place two tubes of competent cells from -80 °C on ice.
2. Put electroporation cuvettes and 2 mL Eppendorf tubes on ice.
3. Thaw EZ-Tn5 transposon DNA and competent cells on ice for 15 min.
4. Transfer 100 μL of the competent cells to a 0.1 cm electroporation cuvette® as a control.
5. Wipe around the cuvette with tissue paper to prevent arcing.
6. Use the Ec1 program (V = 1.8 kV, 2.5 ms) for electroporation.
7. Insert the cuvette into the electroporation device and press the button to apply the pulse.
8. Immediately add 900 μL of prewarmed SOC media (room temperature or 28 °C; Recipe 3) to the cuvette.
9. Transfer all the liquid from the electroporation cuvette to a 2 mL Eppendorf tube and place it on ice.
10. Add 0.5 μL of EZ-Tn5 transposon DNA to the second tube of competent cells.
Note: EZ-Tn5 DNA is diluted in sterile water, and typically 0.5–1.0 μg is used per electroporation.
11. Incubate on ice for approximately 10 min.
12. Transfer 100 μL of the competent cells containing EZ-Tn5 transposon DNA to an electroporation cuvette.
13. Wipe around the cuvette with tissue paper to prevent arcing.
14. Use the Ec1 program for electroporation.
15. Insert the cuvette into the electroporation device and press the button to apply the pulse.
16. Immediately add 900 μL of SOC media to the cuvette.
17. Transfer all the liquid from the electroporation cuvette to a 2 mL Eppendorf tube.
18. Place both 2 mL Eppendorf tubes in the incubator at 28 °C and 220 rpm for 1.5 h.
19. Remove the Eppendorf tubes from the incubator and work on a clean bench.
20. Prepare LBkan plates using the kanamycin sulfate stock (50 mg/mL) (Recipe 4).
21. Using an L-shaped loop, plate 100 μL from each tube onto an LBkan plate (final concentration: 50 μg/mL kanamycin).
22. Centrifuge the remaining culture in the tube at 5,660× g for 3 min.
23. Discard the supernatant.
24. Resuspend the pellet in 200 μL of SOC media.
25. Spread 50 μL of the suspension on LBkan plates (50 μg/mL).
26. Incubate the plates at 28 °C for 1.5 days.
27. Following incubation on LBkan plates, transformants appear as distinct colonies.
Note: Typically, 300–500 colonies are obtained per electroporation, but this number may vary depending on the bacterial species. PCR screening or sequencing confirms that 100% of the colonies contain unique insertions.
C. Preparation of the EZ-Tn5 transposon library
1. Check the control and sample plates in the incubator.
2. Take a 96-well plate.
3. Pour LBkan (50 μg/mL) into a reservoir.
4. Transfer 200 μL of LBkan to each well of the 96-well plate using a multichannel pipette.
5. Pick a bacterial colony from the sample plate with a sterile toothpick.
Note: The toothpick is sterilized using an autoclave.
6. Place the colony into a well of the 96-well plate. After finishing each row, remove the toothpicks and continue with the next row.
7. Complete filling the whole plate and cover it.
8. Place the plate in a 28 °C incubator overnight without shaking.
9. The next day, take a new 96-well plate.
10. Add 75 μL of 20% glycerol to each well of the new 96-well plate using a multichannel pipette.
11. Remove the overnight 96-well plate from the incubator and transfer 75 μL from each well to the new plate for preparing glycerol stock.
12. Cover the new plate and store it at -80 °C.
D. Reviving the transposon library
1. Prepare LBkan agar plates in square BioAssay plates (final concentration on the plate: 50 μg/mL kanamycin).
2. Remove the 96-well plate from -80 °C and place it on ice.
3. Revive the bacterial cells from the glycerol stock by placing them on the LBkan agar plate using a 96-pin microplate replicator.
Note: The 96-pin replicator is sterilized by dipping it in 70% ethanol and flaming.
4. Incubate at 28 °C overnight.
E. Preparation of bacterial mutants for RATE-PCR
1. Prepare autoclaved Milli-Q water, toothpicks, and sterile PCR tubes.
2. Label the PCR tubes according to the position of the bacterial colonies on the 96-well plate.
3. Add 50 μL of autoclaved Milli-Q water to the sterile PCR tubes.
4. Take a small colony from the LBkan plate, starting from position A1 to H12, and transferring it to the PCR tubes using a toothpick.
5. Heat the samples at 95 °C for 10 min using a thermocycler.
6. Use the crude cell extract as a DNA template for the RATE-PCR protocol.
F. RATE-PCR to map the insertion site
1. Prepare the master mix for the RATE-PCR reaction (per sample: 1 μL of DNA, 0.2 μL of Phusion polymerase, 4 μL of 5× HF reaction buffer, 1 μL of 10 μM Inv1 or Inv2 primer (Table 1), 0.4 μL of 10 mM dNTPs, and 13.4 μL of nuclease-free water).
Table 1. Primers used for the RATE-PCR reaction
| Primer name | Sequence (5′ to 3′) |
|---|---|
| Inv-1 | ATGGCTCATAACACCCCTTGTATTA |
| Inv-2 | GAACTTTTGCTGAGTTGAAGGATCA |
2. Set the thermocycler for the RATE-PCR conditions: Initial denaturation at 94 °C for 1 min, 20 cycles of 30 s at 94 °C, 30 s at 55 °C, 3 min at 72 °C, 30 cycles of 30 s at 94 °C, 30 s at 30 °C, 2 min at 72 °C, 30 cycles of 30 s at 94 °C, 30 s at 55 °C, 2 min at 72 °C, final extension at 72 °C for 5 min, and hold at 4 °C.
G. Preparation of RATE-PCR products for sequencing
1. Take 5 μL of RATE-PCR products and place them in a 96-well microplate using a multichannel pipette.
2. Add 2 μL of ExoSap-IT to the PCR products and put the 96-well microplate on ice.
3. Incubate at 37 °C for 15 min using the thermocycler.
4. Incubate at 80 °C for 15 min using the thermocycler.
5. Put the 96-well microplate on ice.
6. Add 5 μL of 10 μM KAN-2 forward or reverse primer (Table 2).
7. Add 3 μL of nuclease-free water.
8. Cover the 96-well microplate and send for Sanger sequencing.
Table 2. Primers used for Sanger sequencing
| Primer name | Sequence (5′ to 3′) |
|---|---|
| KAN-2-F | ACCTACAACAAAGCTCTCATCAACC |
| KAN-2-R | GCAATGTAACATCAGAGATTTTGAG |
Data analysis
RATE-PCR generates specific and nonspecific PCR products using Inv-1 or Inv-2 primers, resulting in multiple bands on an agarose gel. The Sanger sequencing primer (Kan-2-R or Kan-2-F) was selected based on the inverse primer (Inv-1 or Inv-2) used in the RATE-PCR. The Kan-2 primer specifically binds to the end of the kanamycin resistance cassette in the RATE-PCR product. It is used in Sanger sequencing to read the genomic region adjacent to the transposon insertion site.
The Sanger sequencing data were analyzed using the NCBI BLASTN tool, which compares the query sequence against entries in the nucleotide database to identify regions of similarity. The SA190 (CP056087.1) genome appeared at the top of the BLASTN results, indicating a high-confidence match. By examining the alignment between the query sequence and the SA190 genome, the exact gene or intergenic region disrupted by the transposon can be identified. The aligned sequence is then mapped onto the SA190 reference genome using the SnapGene program (Version 6.2.1), allowing for precise characterization of the insertion site and detailed annotation of the affected gene or genomic region.
Validation of protocol
Insertion site validation was primarily conducted using Sanger sequencing, which remains the standard for confirming the precise genomic location of transposon insertions amplified via RATE-PCR (Table 3). To further assess the robustness and accuracy of the protocol, a subset of mutants was additionally analyzed using Illumina HiSeq sequencing (Table 4). The concordance between both approaches confirmed the reliability of the RATE-PCR method for mapping EZ-Tn5 insertion sites. Detailed insertion site data, including genomic coordinates, gene annotations, and sequencing confirmation methods, are presented in Tables 3 and 4.
Table 3. Identification of the SA190 mutants by Sanger sequencing
| Mutant ID | Insertion Site (bp) | Strand | Disrupted gene | Gene function (NCBI annotation) | Flanking genomic sequence (5'–3') | Confirmation method |
|---|---|---|---|---|---|---|
| BET15-E8 | 2,168,204 | – | SA190_FliD | Flagellar filament capping protein | …GCTCA**[Tn5]**TGGCG… | Sanger sequencing |
| BET18-H5 | 2,717,175 | – | SA190_RpoE | RNA polymerase sigma factor | …CGTGC**[Tn5]**GCGGC… | Sanger sequencing |
| BET13-G8 | 4,458,951 | + | SA190_YejK | Nucleoid-associated protein | …GTCGT**[Tn5]**ATGTA… | Sanger sequencing |
| BET2-A1 | 2,965,440 | + | SA190_OmpA | TCOOP OmpA-OmpF porin OOP family protein | …TTCCG**[Tn5]**GTGAC… | Sanger sequencing |
Table 3 summarizes the identification of insertion sites in individual SA190 mutants through Sanger sequencing of RATE-PCR amplicons. The table includes the Sanger read identifier, corresponding gene name or intergenic region, nucleotide match identity, alignment length, and insertion coordinates on the SA190 genome. All insertions were confirmed by 100% identity to the reference genome (GenBank: GCA_001044605.1), validating the protocol's ability to reliably localize EZ-Tn5 transposon insertions at single-nucleotide resolution.
Table 4. Validation of insertion sites in SA190 mutants by HiSeq Sequencing technology
| Sample ID | Mutant1 | Mutant 2 | Mutant 3 | Mutant 4 | Mutant 5 |
|---|---|---|---|---|---|
| qseqid | Tn5 | Tn5 | Tn5 | Tn5 | Tn5 |
| sseqid | NODE_1_length_680816_cov_194.442609 | NODE_1_length_497665_cov_196.568265 | NODE_19_length_79072_cov_140.97375 | NODE_20_length_79072_cov_144.562517 | NODE_2_length_497665_cov_160.085036 |
| Percentage of identical matches | 100 | 100 | 100 | 100 | 100 |
| Alignment length | 108 | 108 | 108 | 108 | 108 |
| Mismatch | 0 | 0 | 0 | 0 | 0 |
| gapopen | 0 | 0 | 0 | 0 | 0 |
| qstart | 1 | 1 | 1 | 1 | 1 |
| qend | 108 | 108 | 108 | 108 | 108 |
| sstart | 214682 | 317444 | 18276 | 60797 | 34962 |
| send | 214575 | 317551 | 18383 | 60690 | 349728 |
| evalue | 9.2E-52 | 9.2E-52 | 9.2E-52 | 9.2E-52 | 9.2E-52 |
| bitscore | 200 | 200 | 200 | 200 | 200 |
Table 4 summarizes the alignment results of Tn5 insertion site flanking sequences from five representative mutants mapped against the SA190 genome. For each sample, query and subject sequence identifiers (qseqid and sseqid), alignment statistics (percentage identity, alignment length, mismatches, gaps), and mapping coordinates (start and end positions) are shown. All insertions displayed 100% identity with the reference genome, confirming the accuracy of the RATE-PCR amplification and insertion mapping. The e-value and bit score further confirm the significance of each alignment. Sequencing was performed using Illumina HiSeq technology.
General notes and troubleshooting
General notes
1. Sterility: Throughout the entire procedure, ensure that all materials and work surfaces are sterile to prevent contamination. Use aseptic techniques, including working near a flame or in a biosafety cabinet, where applicable.
2. Competent cells: Handle competent cells with care, as they are highly sensitive to temperature changes. Always keep them on ice except during electroporation. Avoid repeated freeze-thaw cycles to maintain cell competency.
3. Electroporation: Ensure that the electroporation cuvettes are dry on the outside before use to prevent arcing, which can lead to the loss of your sample or damage to the electroporator.
4. Incubation: During all incubation steps, maintain consistent temperature and shaking conditions as specified. Variations in these parameters can significantly affect bacterial growth and transformation efficiency.
5. SOC media: SOC media is critical for the recovery of cells post-electroporation. Ensure that the media is prewarmed and that you add it immediately after electroporation to maximize recovery rates.
6. Plating: When plating transformed cells, ensure even spreading of the culture on the agar surface for consistent colony growth. Use freshly prepared LBkan plates to ensure proper antibiotic selection.
7. Glycerol stocks: When preparing glycerol stocks, ensure that the glycerol is thoroughly mixed with the bacterial culture to prevent cell damage during freezing. Store the glycerol stocks promptly at -80 °C to preserve viability.
8. Labeling: Properly label all tubes, plates, and 96-well plates throughout the procedure. Mislabeling can lead to confusion and loss of valuable data, especially when dealing with large numbers of samples.
9. RATE-PCR preparation: When preparing the RATE-PCR master mix, ensure accurate pipetting of all reagents to maintain the correct concentrations. Use nuclease-free water and freshly prepared dNTPs to avoid degradation of the PCR reaction components.
10. PCR cycling conditions: Double-check the thermocycler settings before starting the PCR run. The specific cycling conditions are crucial for the success of the RATE-PCR, particularly the multiple cycling steps and temperature variations.
11. Product handling: After PCR amplification, handle the PCR products carefully to avoid contamination or loss. When using ExoSap-IT for cleanup, ensure that the incubation times and temperatures are precisely followed for optimal results.
12. Sequencing preparation: Properly prepare the samples for sequencing by ensuring accurate addition of primers and water. Seal the 96-well plate securely before sending it to the core lab to prevent any contamination or evaporation during transport.
Troubleshooting
Problem 1: No colony growth on plates.
Possible cause: Inefficient electroporation or damaged competent cells.
Solution: Verify that the electroporation conditions (voltage, pulse length) are optimal and that cuvettes are clean and dry. Ensure competent cells were handled properly and remain on ice.
Problem 2: Low transformation efficiency.
Possible cause: Suboptimal cell concentration or incorrect handling during electroporation.
Solution: Ensure that the OD600 of the bacterial culture is correctly measured and falls within the range of 0.4–0.6 before proceeding with electroporation. Confirm that the DNA concentration and quality are adequate.
Problem 3: Arcing during electroporation.
Possible cause: Residual moisture on the outside of the cuvette or high salt concentration in the sample.
Solution: Thoroughly dry the outside of the cuvette before electroporation. Check the composition of the sample and buffer to ensure low ionic strength.
Problem 4: Inconsistent colony growth.
Possible cause: Uneven spreading of bacteria on agar plates or inconsistency in antibiotic concentration.
Solution: Use sterile technique to evenly spread the bacterial culture. Prepare fresh LBkan plates with the correct antibiotic concentration and ensure they are evenly poured.
Problem 5: Contamination.
Possible cause: Non-sterile equipment or materials.
Solution: Double-check the sterility of all materials and equipment. Always use sterile pipette tips, and work in a clean environment. Consider using a biosafety cabinet if contamination persists.
Problem 6: Poor glycerol stock viability.
Possible cause: Inadequate mixing of glycerol or improper freezing.
Solution: Ensure that the glycerol is thoroughly mixed with the culture before aliquoting. Immediately transfer the aliquots to -80 °C after preparation.
Problem 7: Difficulty in colony picking.
Possible cause: Dense or small colonies on the agar plate.
Solution: Adjust the plating density to prevent overcrowding. If colonies are too small, consider extending the incubation time to allow for better growth.
Problem 8: Failed or low-yield RATE-PCR.
Possible cause: Incorrect PCR setup or degraded reagents.
Solution: Ensure that all PCR components are fresh and properly stored. Double-check the accuracy of pipetting and the thermocycler settings.
Problem 9: ExoSap-IT cleanup issues.
Possible cause: Incorrect incubation times or temperatures.
Solution: Follow the exact protocol for ExoSap-IT cleanup. Ensure that the incubator or thermocycler is properly calibrated.
Problem 10: Sequencing problems.
Possible cause: Incorrect primer or low DNA concentration.
Solution: Verify the primer sequences and concentrations before adding them to the PCR products. Ensure that the PCR products are at an appropriate concentration for sequencing.
Acknowledgments
The authors would like to thank all members of Hirt lab, the CDA management team, and the Bioscience Core Labs in KAUST for the technical assistance and their help in many aspects of this work. The work was funded by KAUST fund BAS/1/1062-01-01 to H.H. as part of the DARWIN21 desert initiative (http://www.darwin21.org/). S.K. and B.E. are supported by the Deutsche Forschungsgemeinschaft (DFG) under Germany’s Excellence Strategy – EXC 2048/1 – project 390686111 and under Priority Programme "2125 Deconstruction and Reconstruction of Plant Microbiota (DECRyPT)," project 401836049.
Graphical overview was created with BioRender.com: Elkatmis, B. (2025) https://BioRender.com/3a2mg1e
Author contributions
B.E. data curation, investigation, validation, methodology, writing—original draft, and writing—review and editing; B.H. methodology, data curation, S.P. methodology, data curation; S.K. conceptualization, supervision and review and editing. H.H. funding acquisition, supervision, and writing—review and editing; M.M.S. methodology establishment, conceptualization, supervision, validation, and writing—review and editing.
Competing interests
The authors declare that they have no conflicts of interest.
References
- Freed, N. E. (2017). Creation of a Dense Transposon Insertion Library Using Bacterial Conjugation in Enterobacterial Strains Such As Escherichia Coli or Shigella flexneri. J Visualized Exp. e3791/56216. https://doi.org/10.3791/56216
- Hayes, F. (2003). Transposon-Based Strategies for Microbial Functional Genomics and Proteomics. Annu Rev Genet. 37(1): 3–29. https://doi.org/10.1146/annurev.genet.37.110801.142807
- Naorem, S. S., Han, J., Zhang, S. Y., Zhang, J., Graham, L. B., Song, A., Smith, C. V., Rashid, F. and Guo, H. (2018). Efficient transposon mutagenesis mediated by an IPTG-controlled conditional suicide plasmid. BMC Microbiol. 18(1): e1186/s12866–018–1319–0. https://doi.org/10.1186/s12866-018-1319-0
- Nlebedim, V. U., Chaudhuri, R. R. and Walters, K. (2021). Probabilistic identification of bacterial essential genes via insertion density using TraDIS data with Tn5 libraries. Bioinformatics. 37(23): 4343–4349. https://doi.org/10.1093/bioinformatics/btab508
- Filloux, A., Ramos, J. L. (2014). Preface. Pseudomonas methods and protocols. Methods Mol Biol. 1149:v. https://doi.org/10.1007/978-1-4939-0473-0
- Capadona, J. and Jarvis, B. Kits and Reagents for Nucleic Acid Extraction and Purification Research. https://www.genetargetsolutions.com.au/wp-content/uploads/2014/06/Epicentre-Mutagenesis-Tn5-Transposomics.pdf
- Xu, T., Bharucha, N. and Kumar, A. (2011). Genome-Wide Transposon Mutagenesis in Saccharomyces cerevisiae and Candida albicans. Methods Mol Biol. : 207–224. https://doi.org/10.1007/978-1-61779-197-0_13
- Vidal, J. E., Chen, J., Li, J. and McClane, B. A. (2009). Use of an EZ-Tn5-Based Random Mutagenesis System to Identify a Novel Toxin Regulatory Locus in Clostridium perfringens Strain 13. PLoS One. 4(7): e6232. https://doi.org/10.1371/journal.pone.0006232
- Epicentre Biotechnologies EZ-Tn5TM Insertion Kit. Cold Spring Harb Protoc. 2006(17): pdb.kit13–pdb.kit13. https://www.yumpu.com/en/document/view/37026513/protocol-for-ez-tn5tm-insertion-kit
- Alwutayd, K. M., Rawat, A. A., Sheikh, A. H., Almeida‐Trapp, M., Veluchamy, A., Jalal, R., Karampelias, M., Froehlich, K., Alzaed, W., Tabassum, N., et al. (2023). Microbe‐induced drought tolerance by ABA‐mediated root architecture and epigenetic reprogramming. EMBO Rep. 24(8): e202256754. https://doi.org/10.15252/embr.202256754
- Karlyshev, A. V., Pallen, M. J. and Wren, B. W. (2000). Single-Primer PCR Procedure for Rapid Identification of Transposon Insertion Sites. Biotechniques. 28(6): 1078–1082. https://doi.org/10.2144/00286bm05
- Ribot, E. M., Quinn, F. D., Bai, X. and Murtagh, J. J. (1998). Rapid Amplification of Transposon Ends for the Isolation, Cloning and Sequencing of Transposon-Disrupted Chromosomal Genes. Biotechniques. 24(1): 16–22. https://doi.org/10.2144/98241bm01
- Lafi, F. F., Alam, I., Geurts, R., Bisseling, T., Bajic, V. B., Hirt, H. and Saad, M. M. (2016). Draft Genome Sequence of the Phosphate-Solubilizing Bacterium Pseudomonas argentinensis Strain SA190 Isolated from the Desert Plant Indigofera argentea Genome Announc. 4(6): e01431–16. https://doi.org/10.1128/genomea.01431-16
Article Information
Publication history
Received: Apr 9, 2025
Accepted: Jun 19, 2025
Available online: Jul 9, 2025
Published: Jul 20, 2025
Copyright
© 2025 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Elkatmis, B., Han, B., Parween, S., Kopriva, S., Hirt, H. and Saad, M. M. (2025). Comprehensive Mapping of EZ-Tn5 Transposon Insertion Sites in Pseudomonas argentinensis SA190 Using RATE-PCR. Bio-protocol 15(14): e5389. DOI: 10.21769/BioProtoc.5389.
Category
Microbiology > Microbial genetics > Mutagenesis
Molecular Biology > DNA > Mutagenesis
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.