- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Protocol for Generation of Single-Gene Knockout in Hard-to-Transfect THP1 Cell Lines Using CRISPR/Cas9
Published: Vol 15, Iss 13, Jul 5, 2025 DOI: 10.21769/BioProtoc.5365 Views: 4382
Reviewed by: Pooja MukherjeeAnonymous reviewer(s)

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






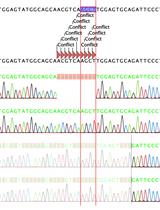
Abstract
This protocol provides a step-by-step approach for generating single-gene knockout in hard-to-transfect suspension immune cell lines like THP1, specifically demonstrated by knocking out the GSDMD gene. By employing CRISPR-Cas9 system delivered via lentivirus, this protocol enables precise gene disruption through targeted single-guide RNAs (sgRNAs). Key steps include designing specific sgRNAs, cloning them into a CRISPR vector, viral packaging, and transducing the target cells, followed by selection and validation. This optimized protocol is particularly useful for functional studies in immune cells, allowing researchers to reliably explore gene function in complex cellular pathways.
Key features
• CRISPR-Cas9-based knockout strategy tailored for hard-to-transfect THP1 cells using lentiviral delivery.
• Lentiviral transduction ensures stable gene delivery with high efficiency compared to other traditional methods like transfection and electroporation.
• Stepwise validation through colony PCR, sequencing, and western blotting to confirm successful knockout.
• Scalable approach, applicable to various cell models for functional genomic studies.
Keywords: CRISPR/Cas9Graphical overview
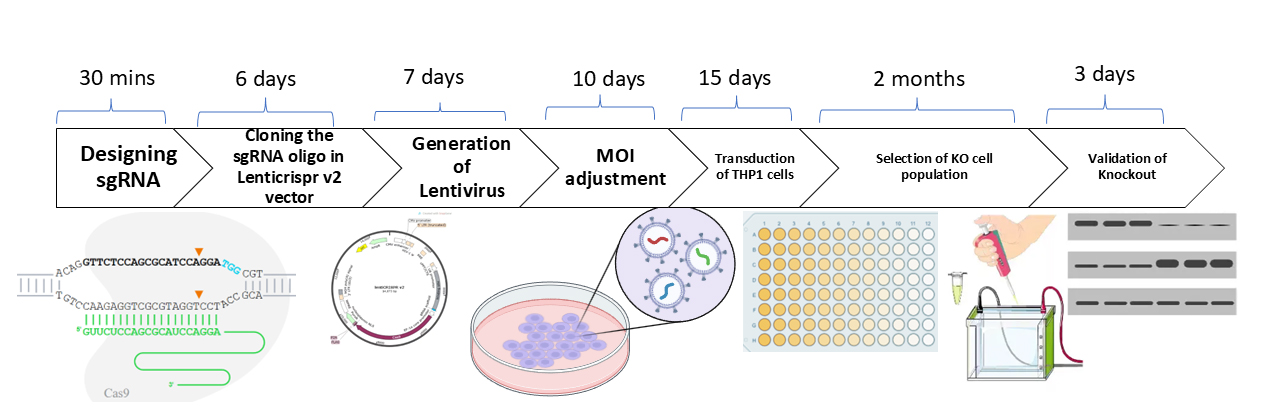
Background
The advent of CRISPR-Cas9 genome editing has revolutionized genetic research by providing a versatile and precise tool for targeted genetic modifications. CRISPR (clustered regularly interspaced short palindromic repeats) and its associated protein Cas9 function together as a programmable nuclease system. The Cas9 enzyme is guided by a single-guide RNA (sgRNA) to introduce double-strand breaks (DSBs) at specific genomic loci. These breaks are repaired by the cell through two major endogenous pathways: non-homologous end joining (NHEJ), an error-prone process that can introduce insertions or deletions (indels), and homology-directed repair (HDR), which can incorporate a donor DNA template for precise edits. However, the effective delivery of CRISPR components in hard-to-transfect suspension cells remains a challenge. Conventional transfection techniques, such as electroporation and chemical transfection, are inefficient with high levels of cytotoxicity, rendering them unsuitable for functional genomic applications [1,2].
To overcome these limitations, lentiviral transduction offers a robust alternative, ensuring stable and efficient gene delivery into immune cell models. This protocol optimizes lentivirus-based CRISPR-Cas9 knockout strategies using sgRNA to achieve high knockout efficiency in THP1 cells, particularly targeting GSDMD, a key regulator in pyroptotic cell death pathways. This protocol builds upon and optimizes previously published protocols for creating single-gene knockouts in THP1 cells. This protocol provides improvements over earlier methods, namely the incorporation of LentiX cells, which are subcloned for high viral titer, and the involvement of GoStix-based viral titration, which offers a rapid and user-friendly alternative to more complex techniques like qPCR or ELISA [3]. In addition, the protocol streamlines puromycin selection and validation steps through a combination of colony PCR, Sanger sequencing, and western blotting. The approach is highly adaptable, offering a scalable method for gene knockout studies in immune cells and other hard-to-transfect models.
Materials and reagents
Biological materials
1. THP1 cells (ATCC, catalog number: TIB-202)
2. Lenti-X cells (Takara, catalog number: 632180)
3. LentiCRISPRv2 (Addgene, catalog number: 52961)
4. E. coli Stbl3 competent cells
5. PsPAX2 (Addgene, catalog number: 12260)
6. pMD2.G (Addgene, catalog number: 12259)
Reagents
1. T4 PNK (New England Biolabs, catalog number: M0201S)
2. T4 DNA ligase (New England Biolabs, catalog number: M0202)
3. Puromycin (Invitrogen, catalog number: A1113803)
4. Ampicillin (HiMedia, catalog number: MB104)
5. 10× PCR buffer (Sigma, catalog number: D4545)
6. Taq polymerase (Sigma, catalog number: D4545)
7. MgCl2 (Sigma, catalog number: D4545)
8. dNTPs (Thermo Fisher Scientific, catalog number: R0191)
9. T4 ligation buffer (New England Biolabs, catalog number: B0202S)
10. Polybrene (Sigma, catalog number: TR-1003-G)
11. Lipofectamine 2000 (Thermo Fisher Scientific, catalog number: 11668500)
12. PLUS reagent (Thermo Fisher Scientific, catalog number: 11514015)
13. Penstrep (Gibco, catalog number: 15140122)
14. Agarose (Invitrogen, catalog number: 16500500)
15. LentiX concentrator (Takara, catalog number: 631231)
16. Lenti Go stix (Takara, catalog number: 631280)
17. Methanol (SRL, catalog number: 65524)
18. 30% acrylamide:bisacrylamide (Bio-Rad, catalog number: 1610156)
19. Bovine serum albumin (BSA) (MP Biomedicals, catalog number: 0215240150)
20. APS (Bio-Rad, catalog number: 1610700)
21. BsmbI-v2 (New England Biolabs, catalog number: R0739S)
22. r3.1 buffer (New England Biolabs, catalog number: B6003S)
23. TEMED (Sigma-Aldrich, catalog number: 1107320100)
24. cOmplete Mini (Sigma-Aldrich, catalog number: 4693159001)
25. Polyvinylidene fluoride (PVDF) membrane (Merck, catalog number: IPVH00010)
26. Luminescent Cell Viability Assay kit (Promega, catalog number: G7571)
27. Clarity western ECL substrate (Bio-Rad, catalog number: 1705061)
28. LB broth (HiMedia, catalog number: M1245)
29. Opti-MEM reduced serum media (Gibco, catalog number: 31985062)
30. RPMI 1640 (HiMedia, catalog number: AL162A)
31. Trypsin-EDTA (0.25%) (Gibco, catalog number: 15400054)
32. DMEM (HiMedia, catalog number: AL0662A)
33. 1× PBS (HiMedia, catalog number: TL1006)
34. FBS (Gibco, catalog number: 16140071)
Solutions
1. 10× TBE buffer (see Recipes)
Recipes
1. 10× TBE buffer (pH 8.3)
| Component | Final concentration in 10× | Quantity |
|---|---|---|
| Tris base | 0.89 M | 108 g |
| Boric acid | 0.89 M | 55 g |
| 0.5 M EDTA (pH 8.0) | 20 mM | 40 mL |
| Distilled water | Up to 1 L |
Laboratory supplies
1. 96-well plates (Corning, catalog number: CLS3360)
2. 12-well plates (Corning, catalog number: CLS3513)
3. 6-well plates (Corning, catalog number: CLS3516)
4. 15 mL centrifuge tubes (Corning, catalog number: CLS430791)
5. 50 mL centrifuge tubes (Corning, catalog number: CLS430829)
6. 10 mL steripipettes (Corning, catalog number: CLS4488)
7. 90 mm Petri dishes (Corning, catalog number: CLS430166)
8. T75 flasks (Corning, catalog number: 430641U)
9. T25 flasks (Corning, catalog number: CLS431463)
10. 0.45 μm vacuum filter (Corning, catalog number: 430314)
Equipment
1. Thermal cycler (VeritiTM Thermal Cycler, 96-well Fast, catalog number: 4375305)
2. CO2 incubator (Thermo Fisher Scientific, FormaTM Steri-CycleTM CO2 Incubator, model: 371)
3. Centrifuge (Thermo Fisher Scientific, Sorvall legend X1R, catalog number: 75004241)
4. Dry bath incubator (Benchmark, catalog number: BSH1004)
5. Electrophoresis power supply (Bio-Rad, Mini-Sub Cell GT System, catalog number: 1704486EDU)
6. Gel documentation system (Bio-Rad, model: Gel Doc EZ Gel Documentation System)
7. Biosafety cabinet (Thermo Fisher Scientific, 1300 Series A2 Class II, model: 1386)
Software and datasets
1. Synthego CRISPR Design Tool (https://www.synthego.com)
2. BioEdit (for Sanger sequencing result visualization)
Procedure
A. sgRNA designing
Time: 30 min
A wide variety of tools are available online to design sgRNAs, like CRISPOR, Synthego, and CHOPCHOP. In this study, we have used Synthego to design the sgRNA according to instructions on the website (https://www.synthego.com/products/bioinformatics/crispr-design-tool) for the GSDMD gene (Figure 1). For choosing a suitable guide RNA sequence, alternative splice variants have to be considered. We suggest picking two different guide sequences for testing editing efficiency. Synthego prioritizes sgRNA based on its on-target score, which is calculated using Azimuth 2.0 model and off-target score.
Note: In this protocol, we have selected the most common exon present in all isoforms for sgRNA design; in case researchers are interested in functional differences between different isoforms, targeting an isoform-specific exon would be a better choice.
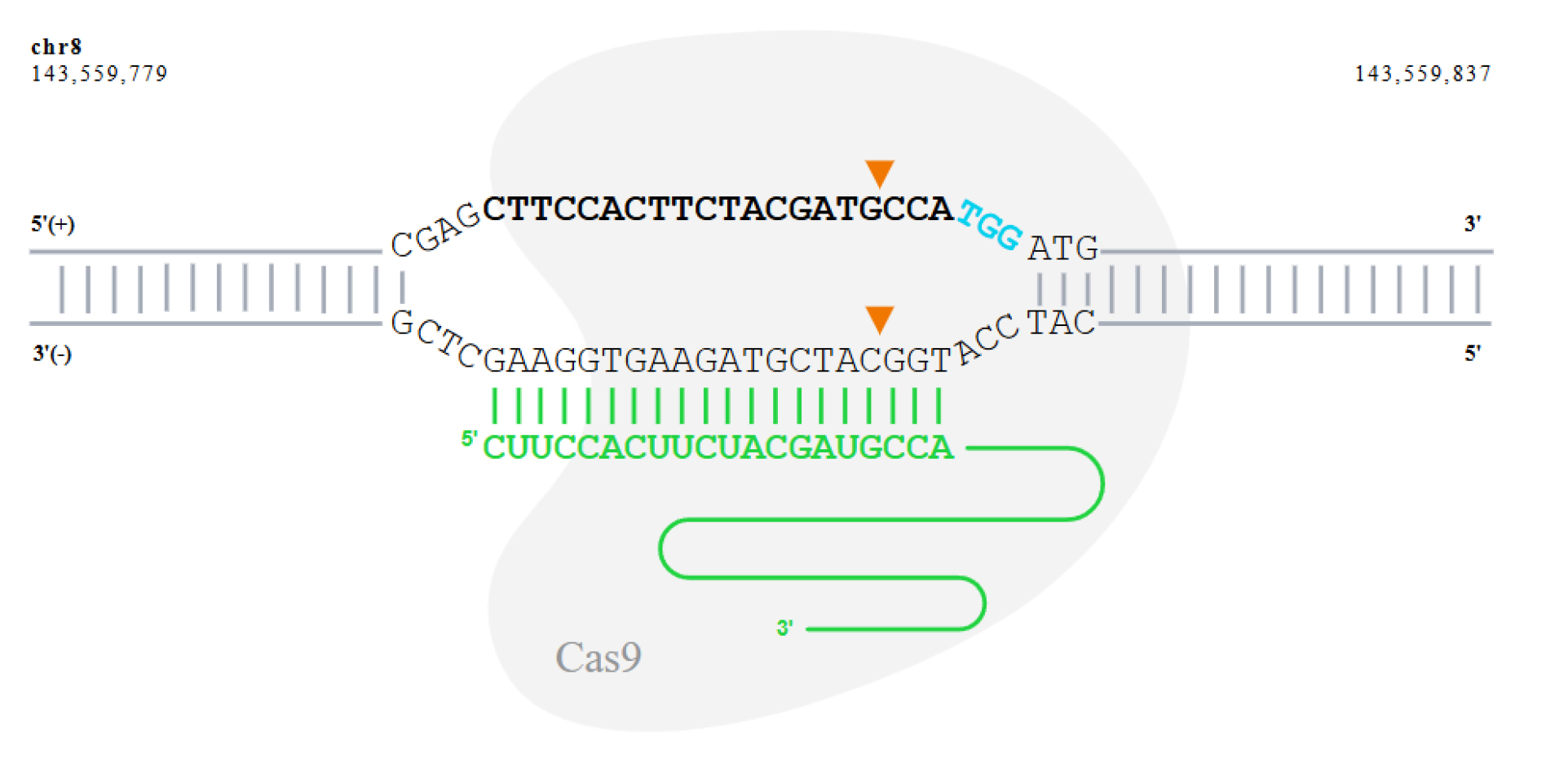
Figure 1. Schematic illustration of the CRISPR-Cas9 cleavage site on the GSDMD gene targeted by sgRNA sequence 5'-CTTCCACTTCTACGATGCCA-3'. The sgRNA binds to the target DNA strand. The PAM sequence (NGG) on the target strand is highlighted in blue. Cas9 cleaves both DNA strands three base pairs upstream of the PAM sequence, as indicated by the arrowheads, resulting in a double-strand break at the cleavage site.
B. sgRNA synthesis
Time: 15 days
1. Design the sgRNA oligonucleotide primers as follows:
sgRNA Forward: 5'- CACCGCTTCCACTTCTACGATGCCA -3'
sgRNA Reverse: 5'- AAACTGGCATCGTAGAAGTGGAAGC -3'
Note: The underlined region represents the 20-nt sgRNA sequence; the forward primer is 20-nt upstream of 5'-NGG, and the reverse primer reverse complements the 20 bp highlighted nucleotides of the forward primer. The overhangs are required for cloning by Golden Gate assembly (https://www.neb.com/en/golden-gate/golden-gate) in the LentiCRISPRv2 vector.
2. Order the designed oligos from IDT.
Note: It costs approximately INR25 per base for each oligo ordered from IDT.
C. Preparation of lentiCRISPR-sgRNA vector
Time: 6 days
C1. Preparation of sgRNA oligonucleotide inserts
1. Dissolve the sgRNA oligos in ddH2O at a final concentration of 100 μM. Set up the following reaction (Table 1) for each pair of sgRNA oligonucleotides:
Table 1. Preparation of sgRNA oligo annealing reaction
| Component | Stock concentration | Final concentration | Volume (μL) |
|---|---|---|---|
| Oligo 1 | 100 μM | 10 μM | 1 |
| Oligo 2 | 100 μM | 10 μM | 1 |
| T4 ligation buffer | 10× | 1× | 1 |
| T4 PNK | 10,000 U/mL | 500 U/mL | 0.5 |
| ddH2O | — | — | 6.5 |
| Total | 10 |
2. Anneal in a thermocycler using the following parameters (Table 2):
Table 2. Annealing and ramp-down parameters for sgRNA oligos
| Temperature | Time |
|---|---|
| 37 °C | 30 min |
| 95 °C | 5 min and |
| ramp down to 25 °C at 5 °C/min |
3. The annealed oligos are verified by running a non-denaturing 20% PAGE native gel (Table 3):
Table 3. Composition of 20% native PAGE gel for verification of annealed oligos
| Components (conc.) | Volume |
|---|---|
| 30% acrylamide:bisacrylamide | 8 mL |
| ddH2O | 1.39 mL |
| TBE (5×) | 2.4 mL |
| APS (10%) | 200 μL |
| TEMED | 10 μL |
4. Pre-run the gel for 1 h in 0.5× TBE buffer, then load 1:10 times diluted annealed sgRNA oligos to the wells and run the gel at 100 V for 2 h. Stain the gel with ethidium bromide for 30 min and take the image in a gel doc.
CAUTION: It is necessary to pre-run the native gel to remove excess ions and unpolymerized acrylamide from the gel.
C2. Digestion of LentiCRISPRv2
1. The lentiCRISPRv2 plasmid was a gift from Feng Zhang. Digest the lentiCRISPRv2 plasmid (1 μg) with 0.5 μL of BsmBI enzyme at 55 °C for 1 h. Run samples on a 1% agarose gel to verify the appropriately sized fragment (12,994-nt DNA fragment). Successful digestion should yield two fragments (~13 kb and ~2 kb). Prepare the reaction mixture as mentioned in Table 4.
Table 4. Digestion reaction components for LentiCRISPRv2 plasmid
| Components (conc.) | Volume |
|---|---|
| LentiCRISPRv2 | 1 μg |
| BsmBI | 0.5 μL |
| r3.1 buffer (10×) | 5 μL |
| ddH2O | Up to 50 μL |
2. The digested vector (~13 kb) fragment is purified using the Gel Purification kit.
C3. Cloning of sgRNA into the lentiCRISPRv2 vector
1. Set up the ligation reaction for each sgRNA as shown in Table 5.
Table 5. Ligation reaction setup for inserting sgRNA into LentiCRISPRv2
| Components (conc.) | Volume |
|---|---|
| Annealed sgRNA (1:10 diluted) | 1 μL |
| Digested plasmid | 50 ng |
| T4 DNA ligase | 1 μL |
| 10× ligation buffer | 2 μL |
| ddH2O | Up to 20 μL |
2. Incubate the ligation reaction at 15 °C for 6 h.
3. Proceed for transformation: Transform the total ligation reaction mixture into a Stbl3 chemically competent cell. Incubate the mixture on ice for 30 min, followed by heat shock at 42 °C for 30 s, and immediately re-incubate on ice for 2 min.
4. Add 100 μL of LB broth without antibiotics to the bacteria from the previous step and incubate the mixture at 37 °C for 1 h in a shaking incubator.
5. Plate the mixture from the previous step onto an LB agar plate with 100 μg/mL ampicillin. Incubate the LB agar plate at 37 °C for 12–16 h. Prepare a negative control plate.
Note: Negative control plates were prepared as follows: 1) plate with only Stbl3 bacteria without plasmid DNA; 2) Stbl3 cells transformed with BsmBI-digested and re-ligated LentiCRISPRv2 plasmid.
6. Assess the plate for colony growth. Typically, multiple colonies should grow on the lentiCRISPR-sgRNA (sgRNA oligo inserted into lentiCRISPRv2 plasmid) plate, and the negative control plate should be free of any colonies.
7. Select 4–5 colonies from the lentiCRISPR-sgRNA plate to confirm the insertion of sgRNA into the lentiCRISPRv2 vector. Grow each single colony in 1 mL of LB broth with 100 μg/mL ampicillin and shake the LB broth at 37 °C for 2 h.
8. To verify the positive clones, colonies can be screened by colony PCR (Table 6). Heat 2 μL of grown culture from the previous step with 4.33 μL of ddH2O for 5 min at 95 °C and add 3.67 μL of PCR master mix (Table 7). The forward primer used here is the hU6 primer, and the reverse primer is the reverse oligo of sgRNA (Table 8).
Table 6. PCR cycling conditions
| Steps | Temperature | Time | Cycles |
| Initial denaturation | 95 °C | 3 mins | 1 |
| Denaturation | 95 °C | 30 s | 35 cycles |
| Annealing | 55 °C | 30 s | |
| Extension | 72 °C | 30 s | |
| Final extension | 72 °C | 7 min | 1 |
| Hold | 4 °C | Hold | |
Table 7. Master mix
| Component | Stock concentration | Final concentration | Volume (μL) |
|---|---|---|---|
| PCR buffer | 10× | 1× | 1 |
| MgCl2 | 25 mM | 1.5 mM | 0.6 |
| dNTPs | 2.5 mM | 200 μM | 0.8 |
| Forward primer | 10 μM | 0.5 μM | 0.5 |
| Reverse primer | 10 μM | 0.5 μM | 0.5 |
| Taq polymerase | 5 U/μL | 0.135 U/μL | 0.27 |
| ddH2O | 6.33 | ||
| Total | 10 |
Table 8. Oligos
| Name | Sequence (5' → 3') |
|---|---|
| sgRNA forward oligo (Oligo 1) | 5'-CACCGCTTCCACTTCTACGATGCCA-3' |
| sgRNA reverse oligo (Oligo 2) | 5'-AAACTGGCATCGTAGAAGTGGAAGC-3' |
| hU6 primer | 5'-GAGGGCCTATTTCCCATGATT-3' |
9. Grow the positive clones in 5 mL of LB media supplemented with 100 µg/mL ampicillin at 37 °C for 12–16 h and isolate the plasmid using the Plasmid Mini Kit.
10. Verify the isolated plasmid by PCR using the abovementioned primers (Table 6) and PCR conditions (Table 8). Visualize the fragments on agarose gel. Positive clones will have an amplification of a ~200 bp fragment. Confirm the sequence of each plasmid from the hU6 promoter using the hU6 forward primer by Sanger sequencing. The 20-bp sgRNA sequence should be inserted between the U6 promoter and the remainder of the sgRNA scaffold.
Pause point: The sequenced verified plasmid can be stored and used later.
D. Lentivirus packaging
Time: 7 days
Here, we use the third-generation lentivirus packaging system plasmid psPAX2, a gift from Didier Trono (Addgene plasmid #12260), with a third-generation transfer plasmid pMD2.G, a gift from Didier Trono (Addgene plasmid #12259).
D1. LentiX cell culture
1. Culture LentiX cells in DMEM supplemented with 10% FBS at 37 °C and 5% CO2. LentiX cells are HEK293T cells subcloned for high transfectability and high titer virus production.
Note: LentiX cells were maintained for two passages before transfection after reviving.
D2. Transfection
1. To prepare cells for transfection, plate LentiX cells onto 10 cm plates in DMEM supplemented with 10% FBS for 16–24 h prior to transfection. Typically, plate ∼4 × 106 cells in a 10 cm plate (∼5 × 104 cells/cm2). It is recommended to transfect the plasmid mix when cells are 80%–90% confluent.
2. On the day of transfection, replace the growth medium with 6–8 mL of serum-free DMEM 2–4 h before transfection. Prepare Lipofectamine 2000-PLUS mixture as mentioned below (Tables 9 and 10):
Table 9. Solution 1
| Components | Volume |
|---|---|
| OptiMEM | 900 μL |
| LentiCRISPR-sgRNA | 12 μg |
| PsPAX2 | 9 μg |
| pMD2.G | 6 μg |
| PLUS reagent | 48 μL |
Mix all reagents and incubate for 5 min at room temperature.
Table 10. Solution 2
| Components | Volume |
| Lipofectamine 2000 | 60 μL |
| OptiMEM | 900 μL |
3. Mix solution 1 and 2 and incubate for 30 min. Add the solution dropwise onto the cells.
4. After 4–6 h, replace DMEM with serum-containing medium.
5. After transfection, incubate the cells for 48 h. The viral particles will be released in the cell supernatants. To collect the virus-containing cell supernatant, filter the medium with a 0.45 μm vacuum filter.
D3. Concentration of lentivirus
1. Add one-third volume of LentiX concentrator to the supernatant collected from the previous step and incubate at 4 °C for 14 h.
2. Centrifuge the samples at 1,500× g for 45 min at 4 °C. After centrifugation, an off-white pellet will be visible.
3. Carefully remove supernatant, taking care not to disturb the pellet. Residual supernatant can be removed with a pipette.
4. Gently resuspend the pellet in 2 mL of complete DMEM.
5. Immediately titrate the sample or store at -70 °C in single-use aliquots of 50 μL.
Pause point: At this stage, the virus can be stored at -70 °C for a year.
E. Titration of lentivirus
Time: 1 day
There are various methods to titrate the virus, such as p24 ELISA, limiting dilution, quantitative PCR, and reporter gene expression. Although these methods are efficient, they can be tedious and time-consuming. Thus, a commercially available kit, Lenti-X GoStix, was used in this study. We need to download GoStix Plus app on our phone for scoring.
1. Enter the lot number by scanning the QR code on the Lenti-X GoStix Plus foil pouch. (Press the QR code icon to activate the scanner.) The lot number can also be entered manually.
2. Enter the number of tests to be scanned.
3. Enter the sample names and the appropriate dilutions. Click Start.
4. Take 20 μL of lentivirus suspended in DMEM and apply it to the sample well of the Lenti-X GoStix Plus cassette.
5. Add 80 μL of chase buffer to the sample well and allow the chase buffer front to appear in the cassette window.
6. Press Start to activate the timer on the app.
7. Allow the test to run for the full 10 min. A test band (T) will start to appear within 5 min and reach maximum intensity at 10 min.
8. After 10 min, scan the GoStix cassette in the app, and the result will be displayed with the GoStix value, i.e., ng/mL of viral protein p24. Empirically, a GoStix value of 50 is equivalent to ~5 × 105 IFU/mL.
F. Multiplicity of infection (MOI) verification
Time: 10–15 days
1. Plate the THP1 cells at confluency in 12-well plates.
2. Add a different concentration of virus (10–100 IFU/mL) in duplicates.
3. Add puromycin (1 μg/mL) in one of the duplicate wells and incubate for 3 days.
4. Measure the cell viability after 3 days using the Luminescent Cell Viability Assay kit.
5. Calculate the ratio of luminescence values from wells without puromycin to those with puromycin. This ratio reflects the proportion of cells that survived selection and is used to estimate the effective MOI (i.e., the number of infectious viral particles per cell). A higher ratio indicates a higher infection efficiency.
G. Lentiviral transduction of THP1 cells
Time: 30–45 days
1. Maintain THP1 cells in high-glucose RPMI 1640 supplemented with 10% FBS at 37 °C and 5% CO2. Passage the cells every 3 days.
2. Plate THP1 in 6-well plates at confluency (1 million cells per well) for lentiviral infection. After approximately 8 h, add lentiviruses (carrying sgRNA) to cells at a MOI of 1–5. Polybrene (8 μg/mL) can be added to increase transduction efficiency.
3. After 24 h of transduction, treat cells with 1 μg/mL puromycin for 3 days to remove un-transduced cells.
4. After 3 days of puromycin treatment, collect cells and plate in a new 12-well plate. Repeat this process twice so that all the non-transduced cells are eliminated.
5. Once the cells proliferate normally, monoclonal cell line generation is performed by the limited dilution method.
6. In this approach, the transduced THP1 cells are serially diluted to very low concentrations (typically 1 cell per 100 μL), and 100 μL of this suspension is plated into each well of a 96-well plate, resulting in an average of ~1 cell/well. After 3–5 days, wells are monitored microscopically to identify and mark those with visible growth from a single cell, which are then expanded into larger wells or flasks.
CAUTION: For essential genes, the knockout might be lethal, with poor cell survival.
Validation of protocol
Colony PCR and Sanger sequencing were performed to confirm the integration of sgRNA into the LentiCRISPRv2 vector (Figure 2A, B).
We extracted DNA from THP1 cells post-lentiviral transduction and performed PCR using primers flanking sgRNA to confirm the integration of the CRISPR-Cas9 construct in genomic DNA (Figure 2C).
Successful gene knockout was confirmed by western blotting, showing the absence of GSDMD protein in edited THP1 cells compared to wild-type (WT-THP1) cells (Figure 2D).
We could knock out a different protein involved in cell death, i.e., NLRP3, using sgRNA 5’GAAGACGTACACCGCGGTGG3’ using the same protocol.
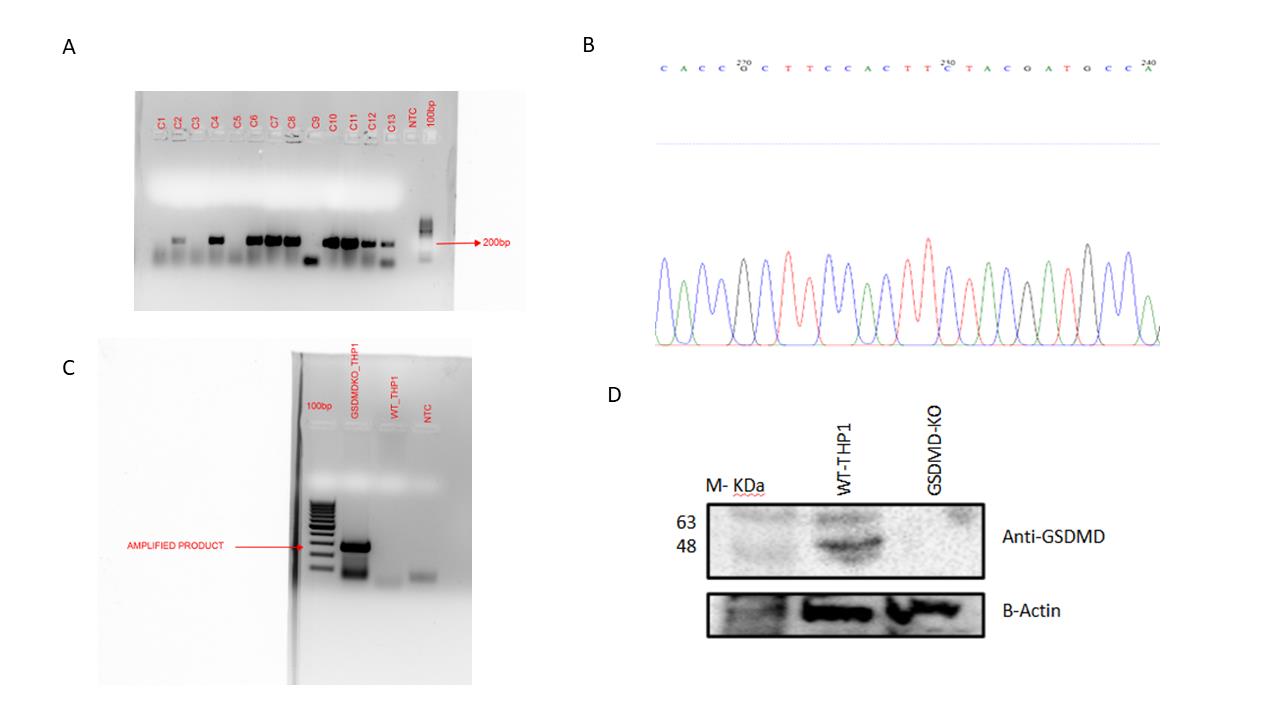
Figure 2. Generation of GSDMD knockout in THP1 cell line. (A) E. coli Stbl3 was transformed with LentiCRISPRv2:sgRNA vector. Preliminary confirmation of clones was performed with colony PCR using hU6 primer as forward primer and reverse sgRNA oligo as reverse primer. Lane 1: 100 bp ladder; lanes 2–14: colony PCR from different colonies; lane 15: no-template control. A product of ~200 bp size was observed for positive transformed colonies. (B) The plasmid DNA was isolated from positive clones, and Sanger sequencing was done using hU6 forward primer. A representative image is shown confirming the presence of sgRNA in the clone. (C) Confirmed positive clones were packaged into lentiviruses. The lentivirus was used to transduce THP1 cells. The integration of the lentiviral vector was confirmed by isolation of genomic DNA from THP1 cells and performing PCR from the region flanking the sgRNA. Lane 1: 100 bp ladder; lane 2: PCR from GSDMD KO THP1 cells; lane 3: PCR from WT-THP1 cells; lane 4: no-template control. The presence of a 300 bp band confirms the integration of lentivirus in the genome. (D) GSDMD KO THP1 cells do not show any detectable production of GSDMD protein compared to WT-THP1 cells, as shown by western blotting. Expression of beta-actin remains unchanged in both GSDMD KO THP1 cells and WT-THP1 cells.
General notes and troubleshooting
Expected outcome
The protocol will reportedly provide successful knockouts of single genes in hard-to-transfect immune cell lines, including THP1. The combination of lentiviral transduction, puromycin selection, and molecular validation makes it an efficient and reproducible methodology to use. This methodology will allow the researcher to examine gene functions across multiple pathways. Additionally, it is scalable and can be applied to other cell types, thus expanding its scope in functional genomics. Thus, the protocol will provide a reliable framework for targeted gene disruption as it aids in further exploration of the responses from immune cells and potential therapeutic targets.
Limitations
This protocol has some drawbacks. The primary challenge is low transfection efficiency in hard-to-transfect cell lines, such as THP1, even with lentiviral delivery. Moreover, sgRNA efficiency varies, and more than one design and optimization are required to ensure successful gene knockout. Off-target effects are a concern because CRISPR-Cas9 can introduce unintended mutations, which necessitates careful validation. The entire workflow is very time-consuming, taking several weeks from the design of sgRNA to clone validation. Cell viability may also be affected by over-selection with puromycin or viral toxicity. In addition, partial knockouts may result in residual gene function in some cells.
Troubleshooting
Traditional transfection methods, such as nucleofection and lipofection, are often ineffective in hard-to-transfect suspension cells like THP1. As shown in Figure 3, nucleofection produced robust GFP expression in adherent MIAPaCa cells, but THP1 cells showed minimal expression. Furthermore, lipofection of THP1 cells resulted in poor survival post-selection. These observations support the use of lentiviral delivery as described in this protocol.
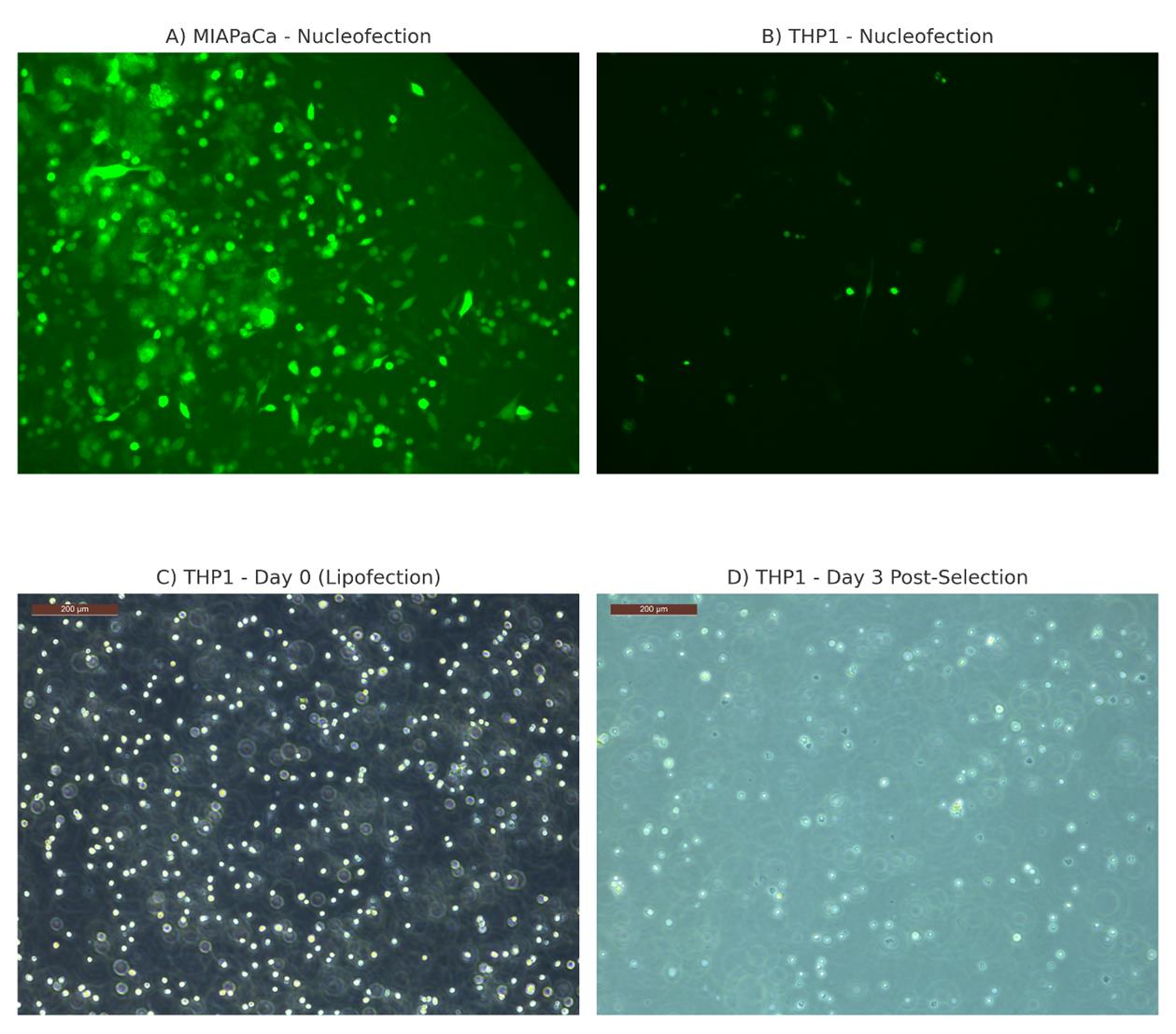
Figure 3. Comparative analysis of gene delivery efficiency and cytotoxicity in THP1 cells using conventional transfection methods. (A) MIAPaCa adherent cells show robust GFP expression following nucleofection with pMAX-GFP, indicating high transfection efficiency. (B) In contrast, THP1 suspension cells show poor GFP expression under the same conditions, demonstrating low efficiency. (C) THP1 cells at the time of transfection using Lipofectamine LTX exhibit healthy morphology. (D) By day 3 post-transfection and puromycin selection, most THP1 cells have died, indicating poor transfection efficiency with lipofection.
These observations support the need for alternative approaches, such as lentiviral transduction, for gene delivery in hard-to-transfect suspension cells like THP1.
Problem 1: Low titers of lentiviruses.
Potential solutions: This might occur due to multiple reasons. First, poor LentiX cell health. It is recommended to use low-passage cells. We usually discard cells that have been passaged more than 15 times. Second, low transfection efficiency of plasmids. Check the quality of transfection reagents or change to other more efficient transfection reagents. Improper cell density (too low or too high) also results in low transfection efficiency. Third, the time for transfection. It is best to transfect plasmids 16–24 h after seeding the cells. This will improve lentivirus titer.
Problem 2: At the end of cell line screening, all of the cells are dead.
Potential solutions: First, we should ensure the THP-1 cells are successfully infected with the virus. Then, ensure that the concentration of puromycin is not too high. The appropriate concentration of puromycin should completely kill wild-type THP-1 cells in 3–7 days, and is normally 1–2 μg/mL for THP-1 cells.
Problem 3: When the screening is completed, the target protein is not reduced at all.
Potential solutions: The most likely cause of this problem is that inappropriate sgRNAs were selected. You can try using new sgRNAs that are frequently used by other researchers. Besides, you should make sure that the concentration of puromycin is not too low. Furthermore, you have to consider that antibodies sometimes recognize non-target proteins, thus giving us an incorrect interpretation.
Acknowledgments
We acknowledge NIBMG Intramural funding(Project code: 60101) for fund support. We thank Dr Anup Majumder for kindlyproviding MIAPaCa cells.
Competing interests
The authors declare no competing interests.
References
- Busch, M., Trabi, M., Cseresnyés, Z., Zagorac, D., Kiemer, A. K. and Hoppstädter, J. (2022). Investigating the role of the NLRP3 inflammasome pathway in acute intestinal inflammation: Use of THP-1 knockout cell lines in an advanced triple culture model. Front Immunol. 13: 898039. https://doi.org/10.3389/fimmu.2022.898039
- Schmidt, T., Schmid-Burgk, J. L., Ebert, T. S., Gaidt, M. M. and Hornung, V. (2016). Designer Nuclease-Mediated Generation of Knockout THP1 Cells. In: Kühn, R., Wurst, W., Wefers, B. (eds) TALENs. Methods in Molecular Biology, vol 1338. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-
- Li, Y., Li, S., Wang, J., Liu, G. and Ma, J. (2022). A simple and efficient protocol for CRISPR/Cas9-mediated gene knockout in human THP-1 monocytes. STAR Protoc. 3(2): 101393. https://doi.org/10.1016/j.xpro.2022.101393
Article Information
Publication history
Received: Mar 26, 2025
Accepted: May 29, 2025
Available online: Jun 10, 2025
Published: Jul 5, 2025
Copyright
© 2025 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Srivastava, K. and Pandit, B. (2025). Protocol for Generation of Single-Gene Knockout in Hard-to-Transfect THP1 Cell Lines Using CRISPR/Cas9. Bio-protocol 15(13): e5365. DOI: 10.21769/BioProtoc.5365.
Category
Molecular Biology > DNA > DNA recombination
Immunology > Immune cell function
Systems Biology > Genomics
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.