- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Synchronized Visualization and Analysis of Intracellular Trafficking and Maturation of Orthoflavivirus Subviral Particles
Published: Vol 15, Iss 10, May 20, 2025 DOI: 10.21769/BioProtoc.5324 Views: 2432
Reviewed by: Keisuke TabataLuis Alberto Sánchez VargasAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Oct 2023


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
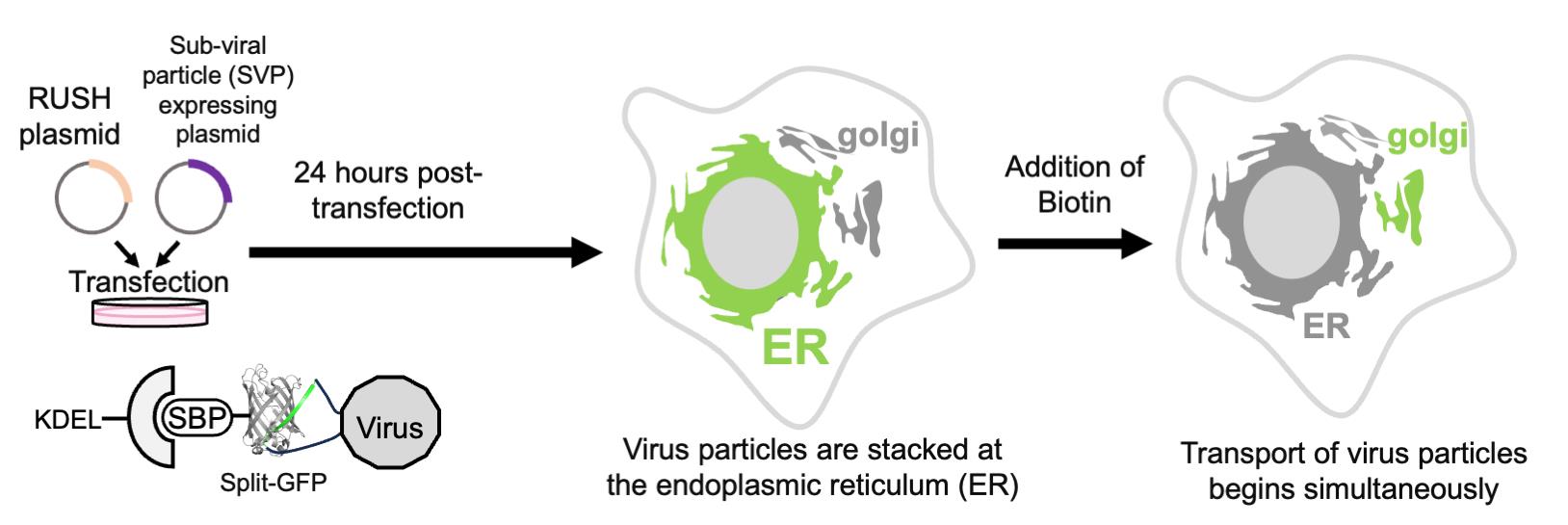
Orthoflavivirus is an enveloped, positive-stranded RNA virus that buds into the endoplasmic reticulum (ER) lumen. The budded virus particles are subsequently transported to the Golgi apparatus and secreted into the extracellular environment via the conventional secretion pathway. In this protocol, we describe a method for monitoring the secretion of Orthoflavivirus particles from the ER. To visualize intracellular membrane trafficking, we combine two distinct imaging techniques: the retention using selective hooks (RUSH) system and the split green fluorescent protein (GFP) system. In this approach, GFP11, a peptide tag fused to prME, the outer coat structural protein of Japanese encephalitis virus particles, was co-expressed in HeLa cells along with two additional components: GFP1-10 fused to a streptavidin-binding peptide and a hook construct consisting of streptavidin fused to the ER retention sequence KDEL. Time-lapse imaging was performed after the addition of biotin, which releases the captured GFP-labeled subviral particles from the ER. This method enables synchronized visualization of intracellular subviral particle trafficking and serves as a valuable tool for analyzing the maturation process of Orthoflavivirus particles within cells.
Key features
• Synchronized intracellular movement of Orthoflavivirus particles is visualized by the retention using selective hooks (RUSH) system.
• Split GFP system is used to label viral particles.
• This protocol has broader applications in investigating the transport of secretory proteins, especially those that are challenging to tag with full-length fluorescent proteins.
Keywords: Retention using selective hooks (RUSH)Graphical overview
Background
Orthoflaviviruses (renamed in 2023, formerly known as flaviviruses) are enveloped, positive-stranded RNA viruses that include several human pathogens, such as Japanese encephalitis virus (JEV), Dengue virus, Zika virus, West Nile virus, and yellow fever virus. The virus particles are approximately 40–60 nm in diameter and consist of genomic RNA encapsulated by capsid proteins within a lipid bilayer. Two proteins are anchored on the outer side of the membrane: precursor membrane protein/membrane protein (prM/M) and envelope protein (E) [1,2]. Virions are considered to bud into the endoplasmic reticulum (ER) lumen, from where they are subsequently transported to the extracellular environment via the conventional secretory pathway [3]. In addition to the virion assembly and secretion pathway, empty particles (subviral particles, SVPs), formed by prM/M and E proteins and lacking the nucleocapsid, are also released from cells through the same secretory pathway as virions [4]. During the budding and secretion of these particles, prM and E undergo several post-translational modifications, including N-linked glycosylation, processing of pr and M proteins, conformational changes, and re-assembly [3,5].
The tracking of membrane vesicle transport within cells has been extensively studied, revealing the dynamics of secretory proteins in living cells. Membrane trafficking, which involves enveloped cargo proteins, primarily begins in the ER lumen and proceeds in an anterograde direction to various Golgi regions (cis, medial, trans, and the trans-Golgi network), which serve as sorting and modification stations [6]. In these sorting zones, the fate of cargo vesicles is determined, directing them to their final destination. Recently, an intriguing approach was developed for tracking protein secretion: the retention using selective hooks (RUSH) system. This elegant methodology enables the study of synchronized cargo trafficking by observing fluorescent-tagged vesicles in cells [7,8]. The RUSH system consists of two key components: (1) a protein of interest (POI) tagged with a fluorophore and a streptavidin-binding peptide (SBP) and (2) a streptavidin molecule bound to a retention molecule (the "hook") in a specific cellular compartment. In the absence of biotin, these two molecules form a complex that is retained in the donor compartment. However, when biotin is added to the culture medium, the POI–fluorophore–SBP complex is released into the next compartment. This transition allows for time-dependent tracking of the POI using fluorescence microscopy [7].
The GFP11 peptide (amino acid sequence: RDHMVLHEYVNAAGIT) is a part of the split-GFP system that can reconstitute the intact superfolder GFP when paired with the GFP1-10 fragment. As a result, the presence of GFP1-10 and excitation light facilitates the detection of the GFP11 peptide [9]. Since GFP11 is a short peptide tag consisting of only 16 amino acids, it minimally affects the function of the fused POI and its complexes. Screening was conducted to identify positions on the viral particle surface where the insertion of a short peptide would not interfere with the folding of prME proteins or SVP assembly, and the 275 aa loop position was found to be suitable [10]. We inserted the GFP11 peptide at this position and demonstrated that the co-expression of the GFP1-10 fragment facilitates GFP labeling of JEV particles [11].
Based on the combination of RUSH and split-GFP methodologies, we present a protocol that enables the quantitative evaluation of viral particle transport as cargo in a time-dependent manner (Figure 1). Using this system, we confirmed that SVPs formed in the ER were transported to the Golgi apparatus within 30 min of biotin addition (Figure 2). This protocol is not only valuable for studying orthoflavivirus particle secretion but also has broader applications in investigating the transport of secretory proteins, especially those that are challenging to tag with full-length fluorescent proteins (e.g., GFP, mCherry).
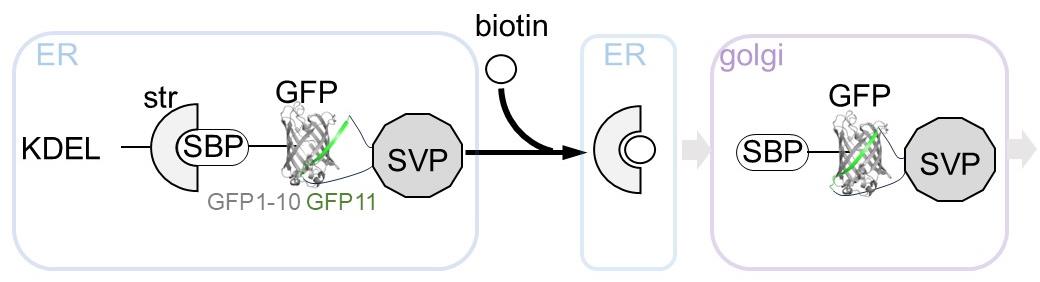
Figure 1. Overview of the retention using selective hooks (RUSH)-based subviral particle (SVP) secretion assay. The schematic illustration represents the RUSH system for SVPs labeled with the split-GFP system. The addition of biotin simultaneously releases the ER-retained GFP-labeled SVPs, enabling synchronized tracking of SVP transport dynamics within cells. Abbreviations used in the figure: ER, endoplasmic reticulum; KDEL, ER retention sequence; Str, streptavidin; SBP, streptavidin-binding peptide; GFP, green fluorescent protein (reconstituted by GFP1-10 and GFP11 peptide); SVP, subviral particle; Golgi, Golgi apparatus.
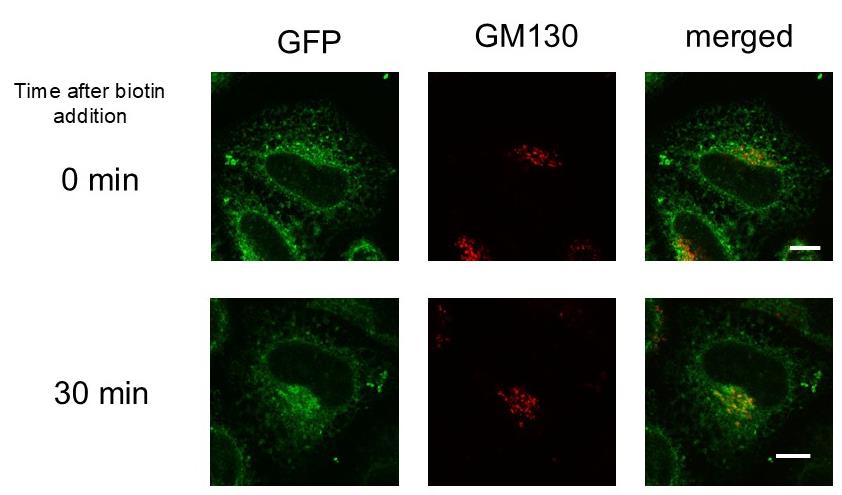
Figure 2. Transport of subviral particles (SVPs) from the endoplasmic reticulum (ER) to the Golgi apparatus. HeLa cells transfected with two plasmids, Str-KDEL_IL-2ss-HiBiT-SBP-GFP1-10 and pCAG-JEprME-S275-GS-GFP11-GS-S276, were treated with 400 μM biotin. Cells were fixed and stained with anti-GM130 either before the addition of biotin (upper panels) or 30 min after its addition (lower panels). Green fluorescent protein (GFP) signals (green, left) and GM130 signals (red, middle) were visualized using confocal laser microscopy. Merged images are shown in the right panels. Scale bar = 10 μm.
Materials and reagents
Note: The materials used in this study can be obtained from the corresponding author, Eiji Morita, upon reasonable request.
Biological materials
1. HeLa cells (ATCC, catalog number: CRM-CCL-2)
Note: HeLa cells, passaged at least 50 times in our laboratory, were used. To prevent mycoplasma contamination, cells were stained with Hoechst to check for intracellular DNA, which indicates contamination. Additionally, cells were periodically treated with BM-Cyclin (Roche, catalog number: 10799050001) to remove mycoplasma contamination.
2. Plasmid Str-KDEL_IL-2ss-HiBiT-SBP-GFP1-10 (V206K)
Notes:
1. This plasmid was constructed by replacing the TNF-SBP-EGFP sequence of Str-KDEL_TNF-SBP-EGFP with an in-frame tandem sequence of interleukin-2 signal peptide, HiBiT tag, SBP, and GFP1-10 (V206K).
2. Str-KDEL_TNF-SBP-EGFP is one of the original RUSH constructs reported by Boncompain et al. [7] and is available from Addgene (#65278). RUSH constructs are mammalian expression constructs that tandemly encode a hook protein (Str-KDEL in this case) and a reporter protein [GOI, interleukin-2 signal peptide-HiBiT-SBP-GFP1-10 (V206K) in this case] linked via an internal ribosome entry site sequence. We selected Str-KDEL as the hook protein based on our experimental requirements (see General note 1).
3. The interleukin-2 signal peptide and HiBiT tag sequences were introduced via overlap polymerase chain reaction (PCR). The interleukin-2 signal peptide and SBP sequence was amplified using Ii-Str_ssSBP-EGFP (Addgene #65277) as a template, and the GFP1-10 (V206K) sequence was amplified using ZipGFP1-10_TEV (Addgene #81242) as a template (see General notes 2 and 3). These fragments were then joined using overlap PCR. The resulting DNA fragment was inserted into the AscI/PacI site of Str-KDEL_TNF-SBP-EGFP (see General note 4).
3. Plasmid pCAG-JEprME-S275-GS-GFP11-GS-S276
Notes:
1. This plasmid was constructed by inserting a sequence encoding a GFP11-containing peptide into the E gene of pCAG-prME.
2. The pCAG-prME plasmid, provided by Dr. Ryosuke Suzuki, was generated in a previous study [12]. This plasmid contains the carboxyl-terminal 22 residues of the JEV Nakayama strain C protein (corresponding to the prM protein signal sequence), the full-length prM protein, and the full-length E protein. For detailed information on the pCAG-prME sequence, please contact Dr. Ryosuke Suzuki (National Institute of Infectious Diseases, Japan).
3. A GFP11 peptide sequence, flanked by GS linker sequences at both ends, was inserted in-frame between amino acid residues S275 and S276 of the JEV E protein. The permissive insertion site within the JEV E protein was previously determined via a Flag-tag insertion screening study [10]. For the full-length amino acid sequence of the E protein, refer to Saga et al. [10] (see General notes 5 and 6).
4. Overlap PCR was performed using pCAG-prME as a template with primers containing this sequence, and the GFP11 peptide coding sequence was inserted into the E gene. The resulting DNA fragment was then inserted into the KpnI/XhoI sites of pCAG-MCS2, a pCAGGS [13] derived plasmid vector containing additional multiple cloning sites (see General note 7).
Reagents
1. DMEM (4.5 g/L glucose) without L-Gln, sodium pyruvate, and phenol red, liquid (Nacalai, catalog number: 08489-45)
2. Fetal bovine serum (FBS) (Thermo, catalog number: 10270-106)
3. 100 mM sodium pyruvate solution (100×) (Nacalai, catalog number: 06977-34)
4. 200 mM L-Glutamine stock solution (Nacalai, catalog number: 16948-04)
5. Phosphate-buffered saline without calcium and magnesium (PBS) (Nacalai, catalog number: 14249-24)
6. Lipofectamine 3000 transfection reagent (Thermo, catalog number: L3000015)
7. OptiMEM (Thermo, catalog number: 31985062)
8. 100 mM biotin stock solution (IBA GmBH, catalog number: 6-6325-001)
9. Sodium hydroxide (Nacalai, catalog number: 31511-05)
10. Hydrochloric acid (Nacalai, catalog number: 18320-15)
11. Paraformaldehyde (Nacalai, catalog number: 26126-25)
12. Triton X-100 (Nacalai, catalog number: 35501-02)
13. Anti-GM130 antibody (BD Biosciences, catalog number: 610822)
14. Anti-mouse IgG antibody, Alexa Fluor 594 conjugated (Jackson ImmunoResearch, catalog number: 115-585-146)
15. Fluoromount-G (Southern Biotechnology Associates, catalog number: 0100-01)
16. MGK-S (Matsunami, catalog number: FX00100)
Solutions
1. Culture media (see Recipes)
2. 1 N NaOH solution (see Recipes)
3. 4% PFA (paraformaldehyde) (see Recipes)
4. 10% TX-100 (w/v) solution (see Recipes)
5. Permeabilizing and blocking solution (see Recipes)
Recipes
1. Culture media
a. 500 mL of DMEM.
b. Add 50 mL of FBS, heat-inactivated by incubation at 56 °C for 45 min.
c. Add 5 mL of 100 mM sodium pyruvate solution (100×).
d. Add 5 mL of 200 mM L-glutamine stock solution.
e. Store at 4 °C.
2. 1 N NaOH solution
a. For 500 mL of 1 N NaOH solution, add 350 mL of Milli-Q water to a glass beaker.
b. Add 20 g of sodium hydroxide drops to Milli-Q water.
c. After completely dissolved, adjust the volume of the solution to 500 mL with water.
d. Transfer the solution to a plastic bottle and store it at room temperature.
3. 4% PFA
a. For 1 L of 4% paraformaldehyde (formaldehyde), add 800 mL of PBS to a glass beaker on a stir plate in a ventilated hood. Heat while stirring to approximately 60 °C. Ensure that the solution does not boil.
b. Add 40 g of paraformaldehyde powder to the heated PBS solution.
c. The powder will not immediately dissolve into the solution. Slowly raise the pH by adding 1 N NaOH dropwise using a pipette until the solution clears.
d. Once the paraformaldehyde is dissolved, cool and filter the solution.
e. Adjust the volume of the solution to 1 L with PBS.
f. Recheck the pH and adjust it to approximately 6.9 with small amounts of diluted hydrochloric acid.
Note: To succeed in the downstream process after cell fixation, please pay attention to the pH of the PFA solution.
g. The solution can be aliquoted and frozen or stored at 2–8 °C for up to one month.
4. 10% TX-100 (w/v) solution
a. For 50 mL of 10% TX-100 solution, add 30 mL of Milli-Q water to a 50 mL tube.
b. Add 5 g of Triton X-100 to Milli-Q water and rotate the tube overnight.
c. After the Triton X-100 is dissolved completely, adjust the volume of the solution to 50 mL with Milli-Q water.
d. Store at room temperature.
5. Permeabilizing and blocking solution
a. Dilute 10% TX-100 solution with PBS by 1:100 (v/v). The final TX-100 concentration is 0.1% (w/v).
b. Dilute FBS with PBS by 1:10 (v/v). The final FBS concentration is 10% (v/v)
Laboratory supplies
1. 35 mm glass-bottom dish (Phoenix Science, catalog number: P35G-0-20-C)
2. Humidified incubator (37 °C, 5% CO2) (PHCbi)
3. 1.5 mL tube (Watson, catalog number: 131-815C)
4. Glass-bottom dish (MatTek Life Science, model: P35G-1.5-14-C)
5. Micro cover glass (Matsunami, thickness: No1S, diameter: 12 mm)
6. White glass, frosted microscope slides (Matsunami, catalog number: S2441)
Equipment
1. Microscope for live imaging based on:
a. EVIDENT IX83 P2ZF inverted fluorescence microscope (Evident, Tokyo, Japan)
b. DP80 CCD digital camera (Evident)
c. IX3-ZDC2 Z-drift compensator (Evident)
d. UPLXAPO 60×/1.35 numerical-aperture oil-immersion objective (Evident, catalog number: UPLXAPO60XO)
e. U-FUNA/U-FBNA/U-FMCHE filter set (Evident)
f. Stage top incubation system (Tokai Hit)
2. Microscope for fixed samples based on:
a. EVIDENT IX83 P2ZF inverted fluorescence microscope (Evident)
b. FV3000 confocal laser scanning microscope (high-speed resonant scanner) (Evident)
c. UPLXAPO 60×/1.35 numerical-aperture oil-immersion objective (Evident, catalog number: UPLXAPO60XO)
d. 5-color laser combiner (Coherent)
Software and datasets
1. cellSens Dimension (Evident)
2. ImageJ/Fiji
Procedure
A. Cell culture and plasmid transfection for live-cell imaging
1. Seed 5 × 104 HeLa cells in a 35 mm glass-bottom dish using 2 mL of culture medium.
2. Incubate the cells at 37 °C and 5% CO2 for 24 h.
3. Set up the transfection reaction:
a. 100 μL of OptiMEM + 0.5 μg of GFP1-10 RUSH construct (hook) + 0.5 μg of GFP11-prME + 1 μL of P3000 reagent.
b. 100 μL of OptiMEM + 2 μL of L3000 reagent
Note: Both P3000 and L3000 reagents are included in Lipofectamine 3000 transfection reagent.
4. Combine the solutions from steps A3a and A3b, vortex the transfection reaction, and incubate it at room temperature for 15 min.
5. Add the transfection reaction to the cells dropwise and incubate at 37 °C and 5% CO2.
6. Six hours post-transfection, exchange the culture media and incubate overnight.
B. RUSH experiment on live-cell imaging (under low light conditions)
1. Set up the microscope and incubation chamber for live-cell imaging at 37 °C and 5% CO2.
Notes:
1. To reduce the flickering of the mercury ARC lamp and laser, we recommend turning on the mercury ARC lamp and laser light source at least 1 h before observation.
2. To minimize XYZ drift caused by temperature differences between the microscope and the CO2 chamber, we recommend pre-heating both before live imaging. Please adjust the room temperature with air conditioning if necessary. To ensure consistent temperature transfer to the cells on the dish, it is recommended to also warm the oil-immersion objective.
2. Bring the dish to the incubation chamber and focus fluorescent cells on multiple fields of view.
3. Add 300 μL of prewarmed culture medium (37 °C), supplemented with 10 μL of 100 mM biotin, to the dish containing the cells, resulting in a final biotin concentration of 400 μM (1:250 dilution) (see Troubleshooting 1).
4. Immediately start acquisition at 3-min time frames for 12 h.
Notes:
1. We typically adjust the camera exposure time between 100 and 500 ms when using the U-FBNA mirror unit. In this time-lapse experiment, we used an exposure time of 200 ms. The gain was set to 1 (default value), the lamp intensity was set to 130, and the ND filter on the mercury lamp side was set to 100. Please note that the ZDC dichroic mirror was inserted into the optical path for multi-point imaging.
2. In phase contrast or brightfield observation, some types of chambers do not provide light-tight cell imaging because room lighting can be a hindrance. Therefore, we kept the lights on in the room while doing overnight live imaging.
3. For live imaging, long-term incubation in the CO2 chamber of the microscope stage can cause the medium to evaporate. We recommend adding more medium when seeding cells or using mineral oil for cell culture.
5. Convert raw files to multi-tiff files using cellSens Dimension.
6. Import the image sequences into the ImageJ/Fiji software and export them.
C. Cell culture and plasmid transfection for fixed cell imaging
1. Seed 3 × 104 HeLa cells onto a micro cover glass using 0.5 mL of culture medium (see Troubleshooting 2).
2. Perform the same steps as described in steps A2–6.
D. RUSH experiment and fixation, indirect immunostaining (under low light conditions)
1. Prepare culture media supplemented with biotin to a final concentration of 400 μM. Keep it warm at 37 °C.
2. Exchange the medium from the cells with prewarmed culture media containing biotin.
3. Incubate cells at 37 °C and 5% CO2.
4. Reach the indicated times, remove media, and treat in 4% PFA in PBS for 10 min.
Note: Generally, the downstream process from this step depends on the target molecule, fixation reagent, antibody, antibody-diluted solution, permeabilizing reagent, blocking compound, mounting reagent, and other conditions. Information about these immunostaining steps should be changed based on previous and upcoming characterization in each laboratory to determine the ideal conditions.
5. Wash three times with 1× PBS.
6. Incubate with permeabilizing and blocking solution for 10 min at room temperature.
7. Wash three times with 1× PBS.
8. Treat with the primary antibody solution, 1× PBS supplemented with anti-GM130 antibody by 1:1,000 (v/v) dilution, for at least 1 h at room temperature.
9. Wash three times with 1× PBS.
10. Treat with the secondary antibody solution, 1× PBS supplemented with anti-mouse IgG antibody, Alexa Fluor 594 conjugated by 1:1,000 (v/v) dilution, for at least 1 h at room temperature.
11. Wash three times with 1× PBS.
12. Mount the glass slides using Fluoromount-G.
13. Carefully store them in a dark place until imaging.
14. Coat the edges of the coverslip with MGK-S to prevent the Fluoromount-G inside from evaporating.
Notes:
1. It is recommended to examine the prepared slides at least 24–48 h after preparation (see General note 8).
2. Pay attention to the time-dependent alternation of the refractive index of Fluoromount-G (see General note 9).
E. Imaging (under low light conditions) and analysis
1. Start the microscope and warm up the required lasers.
Note: In this protocol, we used the 488 and 561 nm lasers. A 60× oil-immersion objective (UPLXAPO60XO) was also used.
2. Focus on single or several clearly defined cells per field of view.
3. Adjust the parameters if necessary.
Notes:
1. Our standard settings were as follows: laser intensity, 0.05%; gain, 1× (default); offset, 0 (default); photo multiplier tube (PMT) voltage, 500 V; pinhole aperture (airy disk), 0.6; sampling speed, 8.0 μs/pixel; scan mode, line (see General note 10).
2. Generally, this process depends on the laser power, voltage on PMT, pinhole aperture, zoom, point spread function, magnification of objective, optical resolution, z-stack series, sample preparation process, and other conditions [14]. Information about these image acquisition steps should be determined based on previous and upcoming characterization, manufacturer’s instructions, and each laboratory’s techniques to determine the ideal conditions (see General note 11).
4. Start imaging.
Note: Check LUT and brightness distribution of samples during XY scanning before image acquisition (see General note 12).
5. Open the image sequence files in Fiji.
Data analysis
1. Open Fiji.
2. In Fiji, open the EzColocalization ImageJ macro available at https://github.com/DrHanLim/EzColocalization [15].
3. Open the microscope (.oir) file in Fiji.
4. Run the macro.
5. Select the two channels to be analyzed (INPUTS tab).
6. Use the manually registered ROI manager for cell identification (INPUTS tab).
Note: A minimum of 20 cells should be selected.
7. In the Analysis tab, check ALL for PCC and check summary.
Note: PCC refers to Pearson's correlation coefficient.
8. Click Analyze to output the analysis results.
9. The output PCC values can be compiled in Excel or visualized in GraphPad Prism.
Validation of protocol
This protocol has been used and validated in the following research article:
• Ishida et al. [11]. N-linked glycosylation of flavivirus E protein contributes to viral particle formation. PLoS Pathog. 11(10): e1011681 (Figure 4A–H).
General notes and troubleshooting
General notes
1. Although the RUSH system is revolutionary, individual researchers must evaluate the applicability of each GOI. We consulted the detailed protocol provided by Boncompain et al. [8] to assess the feasibility of RUSH for our JEV SVP-GFP11. This method encompasses the construction of RUSH constructs for microscopic observation (live imaging and localization analysis), which we recommend as a reference. The RUSH constructs are available from Addgene, and we recommend testing the three available hook constructs (https://www.addgene.org/browse/article/10202/). We purchased constructs encoding the three ER-anchored hook proteins and evaluated the RUSH capabilities of the GOI. Consequently, we selected the Str-KDEL hook construct, which demonstrated effective Golgi localization changes.
2. Although the effect of the V206K mutation on GFP1-10 has not been characterized, the V206K mutation in sfGFP, from which GFP1-10 is derived, contributes to its monomerization (similar to the A206K mutation in EGFP, a well-known monomerization mutation) [16]. Therefore, we introduced the V206K mutation into GFP1-10 to mitigate the potential multimerization due to its overexpression.
Since SVP formation comprising GFP11-inserted (prM)E occurs in the ER lumen, GFP11 and GFP1-10 must be associated within the ER lumen. The ER lumen typically exhibits an oxidizing environment compared with that of the cytosol. Consequently, the expression of proteins not normally found in the ER lumen may lead to unintended intermolecular disulfide bond formation and subsequent accumulation of misfolded proteins [16,17]. Although we did not evaluate ER structural changes or the folding efficiency of ER lumen-expressing fluorescent proteins in this study, introducing mutations that confer resistance to the oxidizing environment may be necessary in some cases.
3. The interleukin-2 signal peptide-HiBiT-SBP-GFP1-10 (V206K) sequence is described as follows:
The interleukin-2 signal peptide is an ER-targeting and secretion signal used to direct the downstream GOI [HiBiT-SBP-GFP1-10 (V206K)] for secretion. This sequence was directly adopted from the original sequence encoded in Ii-Str_ssSBP-EGFP. The HiBiT tag was included as a high-sensitivity detection tag but was not used in this imaging protocol. The SBP is a streptavidin-binding peptide, and this sequence was directly adopted from the original sequence encoded in Str-KDEL_TNF-SBP-EGFP. GFP1-10 corresponds to the GFP1-10 portion of the split-GFP sequence. The V206K mutation was introduced into GFP1-10. (For further details on GFP1-10, refer to General note 2.)
4. The nucleotide sequence encoding the interleukin-2 signal peptide-HiBiT tag-SBP-GFP1-10 (V206K) is as follows: [lowercase, AscI site; uppercase, interleukin-2 signal peptide-HiBiT tag-SBP-GFP1-10 (V206K) sequence; lowercase, PacI site]:
(5') ggcgcgccGCCACCATGTACAGGATGCAACTCCTGTCTTGCATTGCACTAAGTCTTGCACTTGTCACGAATTCCGTGAGCGGCTGGCGGCTGTTCAAGAAGATTAGCGGCGGCGGCGGCAGCGACGAGAAGACCACTGGTTGGCGAGGTGGACACGTTGTTGAAGGACTGGCTGGGGAACTTGAACAACTTCGTGCACGACTGGAGCATCACCCACAAGGTCAACGTGAACCACCTGCAGGTATGTCCAAAGGAGAAGAACTGTTTACTGGTGTTGTGCCAATTTTGGTTGAACTCGATGGTGATGTCAACGGACATAAGTTCTCAGTGAGAGGCGAAGGAGAAGGTGACGCCACCATTGGAAAATTGACTCTTAAATTCATCTGTACTACTGGTAAACTTCCTGTACCATGGCCGACTCTCGTAACAACGCTTACGTACGGAGTTCAGTGCTTTTCGAGATACCCAGACCATATGAAAAGACATGACTTTTTTAAGTCGGCTATGCCTGAAGGTTACGTGCAAGAAAGAACAATTTCGTTCAAAGATGATGGAAAATATAAAACTAGAGCAGTTGTTAAATTTGAAGGAGATACTTTGGTTAACCGCATTGAACTGAAAGGAACAGATTTTAAAGAAGATGGTAATATTCTTGGACACAAACTCGAATACAATTTTAATAGTCATAACGTATACATCACTGCTGATAAGCAAAAGAACGGAATTAAAGCGAATTTCACAGTACGCCATAATGTAGAAGATGGCAGTGTTCAACTTGCCGACCATTACCAACAAAACACCCCTATTGGAGACGGTCCGGTACTTCTTCCTGATAATCACTACCTCTCAACACAAACAAAGCTGAGCAAAGATCCAAATGAAAAATAAttaattaa (3')
5. The GFP11-inserted prME expressed in the pCAG-JEprME-S275-GS-GFP11-GS-S276 construct is expected to display the GFP11 peptide on the SVP particle surface. This expectation is based on a previous study demonstrating that an HCV antigen inserted into the ectodomain of E can react with antibodies [10]. Therefore, in SVPs assembled within the ER lumen, ectodomain-exposed GFP11 should be capable of interacting with (HiBiT tag-SBP-) GFP1-10, which is also present in the ER lumen.
Furthermore, peptide sequence insertion into the orthoflavivirus E protein is challenging, necessitating evaluation of the extent to which the virus particles remain normal [10]. Tag insertion can potentially reduce the efficiency of orthoflavivirus particle formation and secretion. Furthermore, as SVP secretion rates vary among viral species and lineages [11], the impact of tag insertion should be carefully considered when analyzing mutants of different species and multiple lineages of orthoflaviviruses.
Based on these considerations, we performed the following experiments (prior to optimizing the RUSH assay conditions):
a. To determine whether GFP11 peptide insertion affects viral particle formation, we generated GFP11-inserted (prM)E mutants, including those with varying GS linker lengths, expressed them in 293T cells, and assessed intracellular particle formation using sucrose density gradient centrifugation (velocity sedimentation).
b. Using the GFP11-inserted (prM)E mutants selected in Experiment A, we confirmed the generation of a fluorescence signal upon co-expression with ER lumen-localized GFP1-10 in HeLa cells and examined whether the reconstituted GFP fluorescence signal colocalized with the fluorescence of anti-JEV E antibodies.
6. The nucleotide sequence spanning the junction between the E insertion site and the GFP11 peptide coding sequence is as follows (lowercase indicates the E protein's S275 amino acid coding sequence, uppercase indicates the GGGSGGGS linker-GFP11 peptide-GGGSGGGS linker sequence, and lowercase indicates the E protein's S276 amino acid coding sequence):
(5') tcaGGCGGCGGCGGCAGCGGCGGCGGCGGCAGCCGGGACCACATGGTGCTGCACGAGTACGTGAACGCCGCCGGCATCACAGGCGGCGGCGGCAGCGGCGGCGGCGGCAGCagc (3')
7. The nucleotide sequence of the additional multiple cloning site inserted into pCAGGS to generate pCAG-MCS2 is as follows (lowercase, pCAGGS-derived sequence upstream of the inserted sequence; uppercase, additional multiple cloning site sequence containing KpnI and XhoI sites used for JEV prME sequence insertion in this protocol; lowercase, pCAGGS-derived sequence downstream of the inserted sequence):
(5') gcaaagaattATCGATCCGGAGGTACCCGGGCGGCCGCGAGTCTTCGAATTCAAACGCGTCTCGAGAAGCTTGCTAGCAGATCTagatcttttt (3')
8. In standard immunofluorescence staining, it is inevitable that antibodies in the prepared sample dissociate from their antigens over time, leading to diffusion of the fluorescence signal. Therefore, we observe samples as soon as possible after preparation. If a delay is unavoidable, a post-fixation method involving refixation with a chemical fixative after a secondary antibody reaction can be employed as a useful technique.
9. The selection of the mounting medium depends on the type of lens and sample used (sample thickness, transparency, and fluorescent molecules). Given that we observed the cultured cells using an oil-immersion lens, we selected a mounting medium (and coverslip specifications) suitable for this purpose. After mounting the sample, the refractive index of the mounting medium changed as the medium solidified over time. Therefore, it is not recommended to observe the sample immediately after preparation (when the refractive index has not yet reached the required level). We typically observe samples one night after mounting. Conversely, if the samples are left in place for too long, the refractive index of the mounting medium may change because of water evaporation.
10. The parameters are adjusted to achieve an optical resolution close to 60 nm/pixel. To enhance resolution, the pinhole aperture (airy disk) was narrowed, which necessitated an increase in the laser intensity. Unlike Leica systems, which do not employ accumulation, we use an increased pixel scanning speed instead of averaging in the average mode on the FV3000 to reduce noise. It is important to note that this leads to an increased laser exposure of the target fluorophores, resulting in faster photobleaching. Therefore, minimizing the laser power and increasing the PMT voltage during scanning in an image acquisition setup is recommended.
11. For reliable colocalization analysis, it is essential to account for the XYZ shifts between different channels. A plan apochromat objective lens is required as a minimum requirement. To ensure reliable colocalization analysis, it is crucial to minimize the XY shifts between the two channels. For the FV3000, verify the coaxial alignment of the laser paths and the dichroic mirror settings in the optical path of the microscope. Prior to the main experiment, it is recommended to conduct a preliminary experiment using the same molecule labeled with different fluorophores to confirm the absence of fluorescence signal displacement. If an objective lens with a correction collar is used, the Z-axis shifts can be adjusted by using the point spread function as a guide. For FV3000, this adjustment can be performed manually using fluorescent bead samples.
The selection of fluorophores is critical, and it is essential to avoid fluorescence bleed-through between channels, similar to the compensation in flow cytometry. LINE mode is recommended for simultaneous two-color image acquisition.
12. To enhance LUT visualization in the scanned images, we use the Hi-Lo Mode. This allows for parameter adjustment while monitoring the signal saturation. In addition to General note 11, it is crucial to ensure that the LUT during image acquisition falls within the appropriate dynamic range for colocalization analysis. Ideally, prepare a test fluorescent sample that will also be used in the main experiment, and manually determine the dynamic range in which the laser intensity correlates with the fluorescence intensity by observing the LUT as the laser intensity is varied.
Troubleshooting
1. It is important to consider the potential impact of biotin concentration on the RUSH operation. Initially, we attempted to evaluate GOI-fluorophore release using 40 μM biotin, following the recommended RUSH protocol. However, we were unable to observe clear Golgi translocation of fluorescence. Considering that Orthoflavivirus SVPs consist of approximately 60 copies of (prM)E [18] and have a molecular weight of approximately 4.6 MDa, we hypothesized that a higher biotin concentration could be more effective for RUSH release. Consequently, we treated the cells with 400 μM biotin, a 10-fold increase, and observed the translocation. The regulation of biotin concentration is also mentioned in Boncompain and Perez [8].
2. The coating on the cover glass can be removed by washing with water or acid. If cell adhesion efficiency is poor or the background fluorescence is high, the following cleaning procedure is recommended:
a. Incubate the cover glasses in laboratory detergent (SCAT 20X, DKS Co., Ltd.) overnight.
b. Thoroughly rinse with Milli-Q water, followed by sonication in Milli-Q water.
c. Incubate in concentrated sulfuric acid (Nacalai, catalog number: 32520-55) overnight.
d. Thoroughly rinse with Milli-Q water, followed by sonication in Milli-Q water.
e. Replace the Milli-Q water and sterilize by autoclaving.
Acknowledgments
We thank Kaoru Katoh (National Institute of Advanced Industrial Science and Technology) for teaching and suggesting microscopic methodology and a lot of useful tips. We thank Koya Miura (Hirosaki University) for critical suggestions to develop this protocol. This work was supported by JSPS KAKENHI (grant numbers 22H00553, 22H02873, and 22K18378), JST CREST, Japan (grant number JPMJCR17H4), and Japan Agency for Medical Research and Development (AMED) Grant (grant numbers 20333747, 19fk0108168h0001, 20he0622012h0001, and 22fk0108527s0101). The RUSH protocol was adapted from a previous study [8,19]. This protocol is derived from the original research paper (Ishida et al. [11]; DOI: 10.1371/journal.ppat.1011681).
Competing interests
The authors have no competing interests directly relevant to the content of this article.
References
- Wang, X., Li, S. H., Zhu, L., Nian, Q. G., Yuan, S., Gao, Q., Hu, Z., Ye, Q., Li, X. F., Xie, D. Y., et al. (2017). Near-atomic structure of Japanese encephalitis virus reveals critical determinants of virulence and stability. Nat Commun. 8(1): 14. https://doi.org/10.1038/s41467-017-00024-6
- Zhang, X., Ge, P., Yu, X., Brannan, J. M., Bi, G., Zhang, Q., Schein, S. and Zhou, Z. H. (2013). Cryo-EM structure of the mature dengue virus at 3.5-Å resolution. Nat Struct Mol Biol. 20(1): 105–110. https://doi.org/10.1038/nsmb.2463
- Sager, G., Gabaglio, S., Sztul, E. and Belov, G. (2018). Role of Host Cell Secretory Machinery in Zika Virus Life Cycle. Viruses. 10(10): 559. https://doi.org/10.3390/v10100559
- Krol, E., Brzuska, G. and Szewczyk, B. (2019). Production and Biomedical Application of Flavivirus-like Particles. Trends Biotechnol. 37(11): 1202–1216. https://doi.org/10.1016/j.tibtech.2019.03.013
- Barnard, T. R., Abram, Q. H., Lin, Q. F., Wang, A. B. and Sagan, S. M. (2021). Molecular Determinants of Flavivirus Virion Assembly. Trends Biochem Sci. 46(5): 378–390. https://doi.org/10.1016/j.tibs.2020.12.007
- Stalder, D. and Gershlick, D. C. (2020). Direct trafficking pathways from the Golgi apparatus to the plasma membrane. Semin Cell Dev Biol. 107: 112–125. https://doi.org/10.1016/j.semcdb.2020.04.001
- Boncompain, G., Divoux, S., Gareil, N., de Forges, H., Lescure, A., Latreche, L., Mercanti, V., Jollivet, F., Raposo, G., Perez, F., et al. (2012). Synchronization of secretory protein traffic in populations of cells. Nat Methods. 9(5): 493–498. https://doi.org/10.1038/nmeth.1928
- Boncompain, G. and Perez, F. (2013). Fluorescence-Based Analysis of Trafficking in Mammalian Cells. In: Methods for Analysis of Golgi Complex Function. 118: 179–194. https://doi.org/10.1016/b978-0-12-417164-0.00011-2
- Cabantous, S., Terwilliger, T. C. and Waldo, G. S. (2005). Protein tagging and detection with engineered self-assembling fragments of green fluorescent protein. Nat Biotechnol. 23(1): 102–107. https://doi.org/10.1038/nbt1044
- Saga, R., Fujimoto, A., Watanabe, N., Matsuda, M., Hasegawa, M., Watashi, K., Aizaki, H., Nakamura, N., Tajima, S., Takasaki, T., et al. (2016). Bivalent vaccine platform based on Japanese encephalitis virus (JEV) elicits neutralizing antibodies against JEV and hepatitis C virus. Sci Rep. 6(1): 28688. https://doi.org/10.1038/srep28688
- Ishida, K., Yagi, H., Kato, Y. and Morita, E. (2023). N-linked glycosylation of flavivirus E protein contributes to viral particle formation. PLoS Pathog. 19(10): e1011681. https://doi.org/10.1371/journal.ppat.1011681
- Suzuki, R., Ishikawa, T., Konishi, E., Matsuda, M., Watashi, K., Aizaki, H., Takasaki, T. and Wakita, T. (2014). Production of single-round infectious chimeric flaviviruses with DNA-based Japanese encephalitis virus replicon. J Gen Virol. 95(1): 60–65. https://doi.org/10.1099/vir.0.058008-0
- Hitoshi, N., Ken-ichi, Y. and Jun-ichi, M. (1991). Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 108(2): 193–199. https://doi.org/10.1016/0378-1119(91)90434-d
- Montero Llopis, P., Senft, R. A., Ross-Elliott, T. J., Stephansky, R., Keeley, D. P., Koshar, P., Marqués, G., Gao, Y. S., Carlson, B. R., Pengo, T., et al. (2021). Best practices and tools for reporting reproducible fluorescence microscopy methods. Nat Methods. 18(12): 1463–1476. https://doi.org/10.1038/s41592-021-01156-w
- Stauffer, W., Sheng, H. and Lim, H. N. (2018). EzColocalization: An ImageJ plugin for visualizing and measuring colocalization in cells and organisms. Sci Rep. 8(1): 15764. https://doi.org/10.1038/s41598-018-33592-8
- Costantini, L. M., Fossati, M., Francolini, M. and Snapp, E. L. (2012). Assessing the Tendency of Fluorescent Proteins to Oligomerize Under Physiologic Conditions. Traffic. 13(5): 643–649. https://doi.org/10.1111/j.1600-0854.2012.01336.x
- Costantini, L. M., Baloban, M., Markwardt, M. L., Rizzo, M. A., Guo, F., Verkhusha, V. V. and Snapp, E. L. (2015). A palette of fluorescent proteins optimized for diverse cellular environments. Nat Commun. 6(1): 7670. https://doi.org/10.1038/ncomms8670
- Ferlenghi, I., Clarke, M., Ruttan, T., Allison, S. L., Schalich, J., Heinz, F. X., Harrison, S. C., Rey, F. A. and Fuller, S. D. (2001). Molecular Organization of a Recombinant Subviral Particle from Tick-Borne Encephalitis Virus. Mol Cell. 7(3): 593–602. https://doi.org/10.1016/s1097-2765(01)00206-4
- Pacheco-Fernandez, N., Pakdel, M. and von Blume, J. (2021). Retention Using Selective Hooks (RUSH) Cargo Sorting Assay for Protein Vesicle Tracking in HeLa Cells. Bio Protoc. 11(5): e3936. https://doi.org/10.21769/bioprotoc.3936
Article Information
Publication history
Received: Feb 12, 2025
Accepted: Apr 27, 2025
Available online: May 8, 2025
Published: May 20, 2025
Copyright
© 2025 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Ishida, K. and Morita, E. (2025). Synchronized Visualization and Analysis of Intracellular Trafficking and Maturation of Orthoflavivirus Subviral Particles. Bio-protocol 15(10): e5324. DOI: 10.21769/BioProtoc.5324.
Category
Microbiology > Microbial cell biology > Cell staining
Cell Biology > Cell imaging > Live-cell imaging
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.