- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Mycobacterium smegmatis Ribosome Purification, Co-sedimentation, and Subunit Association Assay
Published: Vol 15, Iss 10, May 20, 2025 DOI: 10.21769/BioProtoc.5318 Views: 1845
Reviewed by: Ritu GuptaAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Nov 2024
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
The ribosome, a complex macromolecular machine, plays a vital role in cellular translation. To investigate its structure and conduct in vitro experiments, isolating the ribosomes from cells is the first step. While isolating ribosomes from bacterial cells is routine, obtaining them from mycobacteria proves challenging due to the protective mycolic acid layer, which hinders cell lysis. In this study, we present a straightforward and efficient protocol for isolating ribosomes from Mycobacterium smegmatis. Additionally, we introduce a co-sedimentation assay using density gradient ultracentrifugation, providing a simple yet powerful method for studying ribosome–protein interactions. The re-association assay also offers a practical approach for obtaining tRNA-free 70S ribosomes and evaluating the anti-association properties of potential ligands. While these assays are commonly used, our protocol stands out for its simplicity, requiring limited specialized instruments. These methods can also be scaled up or down per requirement. By employing sonication for cell rupture and utilizing basic lab equipment for ultracentrifugation-based assays, our method greatly simplifies ribosome isolation and related research.
Key features
• Cellular ribosome isolation from Mycobacterium smegmatis using an optimized sonication cycle.
• Detection of ribosome–protein interactions through density gradient ultracentrifugation.
• Simple steps to obtain tRNA-free 70S ribosomes that are crucial for several biochemical experiments.
• Screening of proteins or ligands for their ribosomal anti-association activity.
Keywords: 70S ribosomeGraphical overview
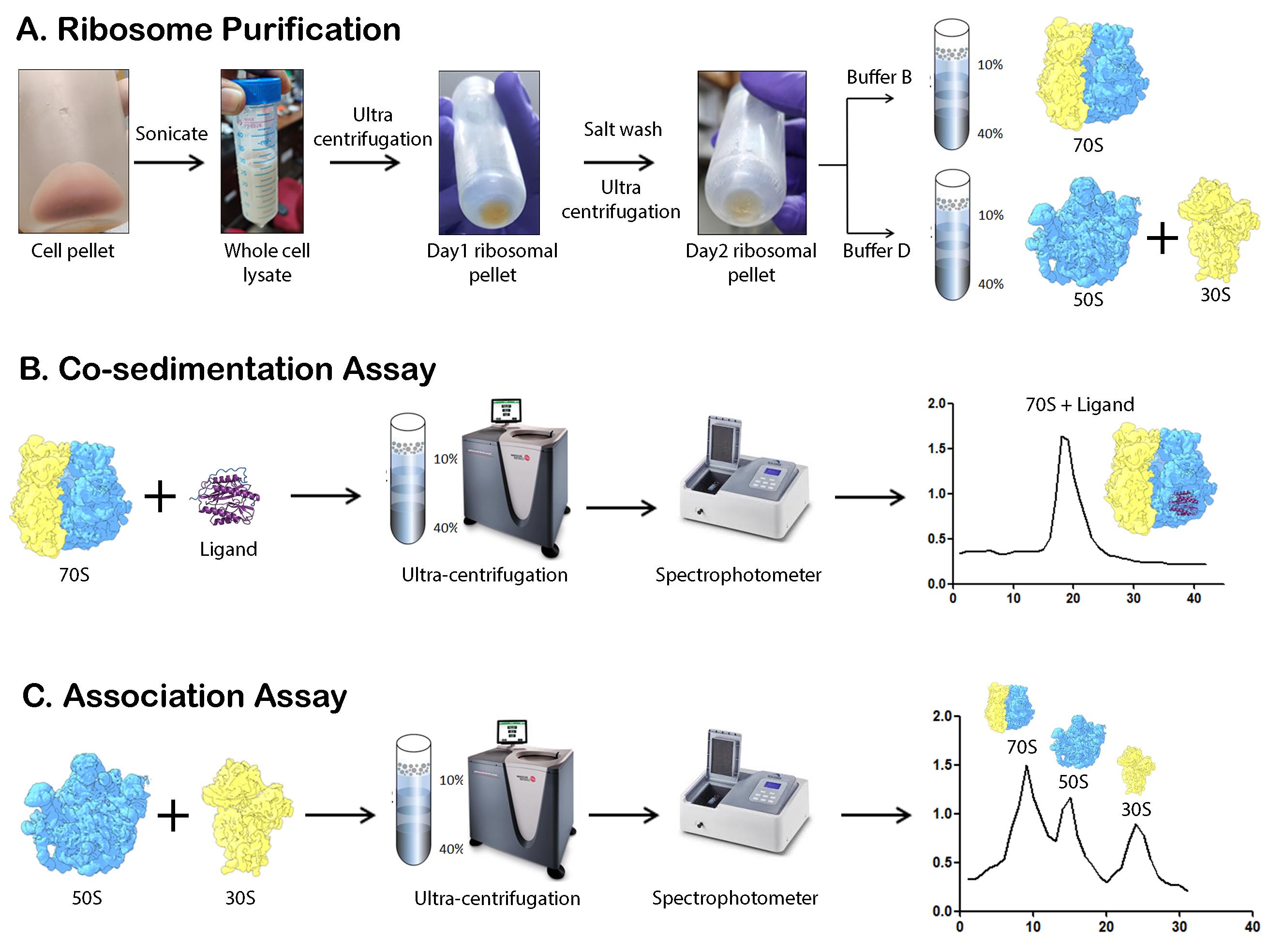
Schematic overview of the major steps involved in M. smegmatis ribosome purification, co-sedimentation, and association studies. (A) The schematic representation highlights the major steps involved in 70S, 50S, and 30S subunit isolation. Cells are lysed by sonication followed by two rounds of ultracentrifugation to obtain the crude ribosome. Depending on the requirement, the pellet is either dissolved in Buffer B or Buffer D, followed by sucrose density gradient ultracentrifugation. (B) Co-sedimentation assay involves the incubation of the ligand with the ribosome and analysis by ultracentrifugation. (C) Association assay involves the incubation of the 50S and 30S ribosomal subunits, followed by ultracentrifugation to analyze the amount of 70S ribosome formed.
Background
In all living cells, protein synthesis is carried out by the ribosome, a large complex composed of RNA and proteins [1–3]. Bacterial ribosomes are composed of a small 30S subunit (SSU) and a large 50S subunit (LSU). Structural studies of ribosomes from both Gram-negative and Gram-positive species have shown that the overall structure of the bacterial ribosome is largely conserved [4]. In E. coli, the 30S subunit consists of 21 ribosomal proteins (r-proteins) and a 16S ribosomal RNA (rRNA), while the 50S subunit contains 36 r-proteins along with 23S and 5S rRNAs. While the core of the ribosome is almost fully conserved, there are subtle variations in ribosome structure. These include species-specific insertions and deletions in various domains of rRNA and r-proteins, as well as novel r-proteins in certain bacterial species [5].
Ribosome isolation is a fundamental step in studying translation, ribosome structure, and ribosome-associated interactions [6]. Mycobacterium is an important bacterial genus that encompasses several prominent pathogens, and its translation machinery is of immense interest due to its implications in antibiotic approaches. While ribosome purification from bacterial cells is well-established, mycobacteria present unique challenges due to their thick, lipid-rich mycolic acid layer, which complicates cell lysis and downstream isolation procedures [7, 8]. Traditional methods often rely on harsh chemical and physical treatments (freeze-thaw), which can be time-consuming and costly, may compromise ribosome integrity, and may result in low yields. Previous studies have utilized various approaches, including mechanical disruption, detergent-based lysis, and enzymatic treatments [9,10], to extract ribosomes from mycobacteria. However, these methods often require specialized reagents, prolonged processing times, or multiple purification steps, making them less accessible for routine laboratory use.
Our protocol offers a simplified yet efficient approach, using sonication for cell disruption and basic ultracentrifugation techniques without the need for alcohol-based precipitation or specialized equipment. This method may be adapted to other mycobacterium species, provided proper safety precautions are taken. The method enables the isolation of high-quality ribosomes suitable for structural and functional studies, including ribosome–protein interaction assays via co-sedimentation as well as functional assays using re-association assays. This protocol has broader applications, such as screening ribosome-targeting antibiotics, studying translation regulation, and investigating ribosome-associated stress responses in mycobacteria. These methods may be scaled up or scaled down as per requirement. Its simplicity and accessibility make it a valuable tool for researchers exploring bacterial ribosome dynamics in both fundamental and applied sciences.
Materials and reagents
Biological materials
1. Mycobacterium smegmatis MC2 155 (gift from Prof. Sujoy Dasgupta, Bose Institute, Kolkata)
Reagents
1. Tris base (Fisher Scientific, catalog number: BP152-1)
2. Magnesium acetate (C4H6MgO4·4H2O) (Sigma-Aldrich, catalog number: M5661-250G)
3. Ammonium chloride (NH4Cl) (Sigma-Aldrich, catalog number: A9434-1KG)
4. 2-Mercaptoethanol (C2H6OS) (Sigma-Aldrich, catalog number: M-7154)
5. Sucrose (C12H22O11) (Calbiochem, catalog number: 8510)
6. HEPES (Fisher Scientific, catalog number: BP310-1)
7. Middlebrook 7H9 broth base (Himedia, catalog number: M198-500G)
8. Bovine serum albumin (SRL, catalog number: 9048-46-8)
9. Polysorbate 80 (SRL, catalog number: 9005-65-6)
10. Glycerol (Sigma-Aldrich, catalog number: G5516-1L)
11. HCl (Rankem, catalog number: H0100)
12. Puromycin (Sigma-Aldrich, catalog number: P8833)
13. Lysozyme (Roche, catalog number: 10837059001)
14. Bis-acrylamide (30%) (Himedia, catalog number: ML037)
15. TEMED (SRL, catalog number: 52145)
16. Ammonium persulphate (SRL, catalog number: 0144142)
17. Sodium lauryl sulphate (SRL, catalog number: 32096)
18. Agarose (SRL, catalog number: 23287)
19. 50× TAE (G2P, catalog number: BUF-07-500ml)
20. Sodium hypochlorite (Sigma-Aldrich, catalog number: S-1898)
Solutions
1. Buffer A (see Recipes)
2. Buffer B (see Recipes)
3. Buffer C (see Recipes)
4. Buffer D (see Recipes)
5. HMA-10 (see Recipes)
6. Mycobacterium smegmatis MC2 155 culture media (see Recipes)
7. Resolving and stacking gel for SDS-PAGE (see Recipes)
8. Agarose gel (see Recipes)
9. Sucrose solutions (see Recipes)
10. Puromycin stock (see Recipes)
Recipes
Note: The different types of buffers used in the experiments are mentioned in the tables below. All stocks are prepared using distilled water, and the final volume is adjusted using the same water. Buffers are then filtered using 0.22 μm membrane syringe filters. The filtered buffers can be stored at 4 °C for a few weeks, although it is ideal to use them within a week. Culture media is prepared using distilled water and autoclaved.
1. Buffer A
| Reagent | Stock concentration | Final concentration | Volume for 500 mL |
|---|---|---|---|
| Tris-HCl (pH 7.6) | 1 M | 20 mM | 10 mL |
| Ammonium chloride | 2 M | 100 mM | 25 mL |
| Magnesium acetate | 1 M | 10 mM | 5 mL |
| 2-Mercaptoethanol | 14.3 M | 5 mM | 175 μL |
2. Buffer B
| Reagent | Stock concentration | Final concentration | Volume for 500 mL |
|---|---|---|---|
| Tris-HCl (pH 7.6) | 1 M | 20 mM | 10 mL |
| Ammonium chloride | 2 M | 30 mM | 7.5 mL |
| Magnesium acetate | 1 M | 10 mM | 5 mL |
| 2-Mercaptoethanol | 14.3 M | 5 mM | 175 μL |
3. Buffer C
| Reagent | Stock concentration | Final concentration | Volume for 500 mL |
|---|---|---|---|
| Tris-HCl (pH 7.6) | 1 M | 20 mM | 10 mL |
| Ammonium chloride | 2 M | 1 M | 250 mL |
| Magnesium acetate | 1 M | 10 mM | 5 mL |
| 2-Mercaptoethanol | 14.3 M | 5 mM | 175 μL |
4. Buffer D
| Reagent | Stock concentration | Final concentration | Volume for 500 mL |
|---|---|---|---|
| Tris-HCl (pH 7.6) | 1 M | 20 mM | 10 mL |
| Ammonium chloride | 2 M | 30 mM | 7.5 mL |
| Magnesium acetate | 1 M | 1 mM | 0.5 mL |
| 2-Mercaptoethanol | 14.3 M | 5 mM | 175 μL |
5. HMA-10
| Reagent | Stock concentration | Final concentration | Volume for 500 mL |
|---|---|---|---|
| HEPES (pH 7.6) | 1 M | 20 mM | 10 mL |
| Magnesium acetate | 1 M | 10 mM | 5 mL |
| Ammonium chloride | 2 M | 60 mM | 15 mL |
| 2-Mercaptoethanol | 14.3 M | 5 mM | 175 μL |
6. Mycobacterium smegmatis MC2 155 culture media
| Reagent | For 1,000 mL |
|---|---|
| Middlebrook 7H9 broth base | 5.3 g |
| Bovine serum albumin | 1 g |
| Polysorbate 80 | 200 μL |
| Glycerol | 2 mL |
7. Resolving and stacking gel for SDS-PAGE
| Reagents for 15% resolving gel | Amount (for 10 mL) |
|---|---|
| Distilled water | 2.2 mL |
| Bis-acrylamide (30%) | 5 mL |
| 1.5 M Tris (PH 8.8) | 2.6 mL |
| 10% SDS | 100 μL |
| 10% APS | 100 μL |
| TEMED | 10 μL |
| Reagents for 5% stacking gel | Amount (for 5 mL) |
| Distilled water | 2.975 mL |
| Bis-acrylamide (30%) | 0.67 mL |
| 1 M Tris (pH 6.8) | 1.25 mL |
| 10% SDS | 50 μL |
| 10% APS | 50 μL |
| TEMED | 5 μL |
8. Agarose gel
| Reagent | Amount (for 100 mL) |
|---|---|
| Agarose | 1.2 g |
| 50× TAE | 2 mL |
| Sodium hypochlorite | 1 mL |
| Distilled water | 97 mL |
9. Sucrose solutions
| Amount of sucrose | Volume of buffer | Final % of sucrose |
|---|---|---|
| 4.8 g | 12 mL | 40% |
| 2.4 g | 8 mL | 30% |
| 1.4 g | 7 mL | 20% |
| 0.6 g | 6 mL | 10% |
10. Puromycin stock
| Puromycin | Buffer D | Final concentration |
|---|---|---|
| 0.0013 g | 100 µL | 13 mg/mL |
Laboratory supplies
1. Pipette (1 mL, 100 μL, 10 μL, and 2.5 μL) (Eppendorf, catalog number: 05-403-151)
2. Microcentrifuge tubes (1.5 mL) (Abdos, catalog number: P10202)
3. 0.22 μm syringe filters (Merck, catalog number: SLGB033RS)
4. 2 L Erlenmeyer narrow mouth conical flask (Borosil, catalog number: 4980030)
5. 50 mL centrifuge tubes (Abdos, catalog number: P10431)
6. 500 mL culture harvest bottles (Nalgene® bottles) (Sigma-Aldrich, catalog number: B9282)
7. Glass homogenizer (Potter-Elvehjem PTFE pestle and glass tube) (Sigma-Aldrich, catalog number: P7984)
8. Oakridge tubes (Nalgene® centrifuge tubes) (Sigma-Aldrich, catalog number: T1418)
9. Centrifuge tubes for ultracentrifugation (Beckman, catalog number: 344058)
10. Amicon® Ultra-4 centrifugal filters, 100 kDa and 10 kDa (Merck, catalog numbers: UFC810024 and UFC801024)
11. Measuring cylinder (Tarsons, catalog number: 343070)
12. Non-absorbent cotton (Himedia, catalog number: LA1017)
13. Quartz cuvette 1 mL (BR Biochem, catalog number: BQ-244)
14. 1 mL pipette tips (Abdos, catalog number: P10114)
15. 200 μL pipette tips (Abdos, catalog number: P10149)
16. 10 μL pipette tips (Abdos, catalog number: P10117)
17. Parafilm (Amcor, catalog number: PM-996)
Equipment
1. Crude weighing balance (Hexane, India, model: DJ602A)
2. Fine weighing balance (Mettler Toledo, model: ME204)
3. Magnetic stirrer (Spinot)
4. Autoclave (Indfos, Autoclave)
5. Class I laminar airflow system (custom assembled)
6. Incubator shaker (Remi, model: CIS-24 Plus)
7. Spectrophotometer (Shimadzu Corp, model: A114546)
8. 4 °C centrifuge (Sigma, model: 330k)
9. Sonicator (Qsonica Sonicators, model: Q125)
10. Ultracentrifuge with AH-629 swing-out rotor (Thermo Scientific, model: Sorval Max Ultra Series)
11. Vortex (Spinix, model: 3737)
12. Peristaltic pump (GE Healthcare Biosciences AV, model: PumpP-1)
13. 4 °C refrigerator (Vestfrost Solutions)
14. -20 °C freezer (Vestfrost)
Software and datasets
1. Prism (v5.00) software (GraphPad, San Diego, CA)
Note: License is required and can be purchased from the website. Graphs can also be prepared using Microsoft Excel.
2. UVProbe 2.42
Note: The license comes with the spectrophotometer.
Procedure
A. 70S ribosome isolation from Mycobacterium smegmatis MC2 155
70S, 50S, and 30S isolation from Mycobacterium smegmatis is a three-day protocol that requires buffer A, buffer B, buffer C, and buffer D. The detailed step-by-step protocol is mentioned below. The volumes of buffer mentioned in the protocol are for a cell pellet obtained from 2 L of culture.
Day 1 of 70S ribosome isolation
1. Prepare a seed culture of Mycobacterium smegmatis by inoculating 100 μL of glycerol stock in 25 mL of media and growing for 24 h. Add 5 mL of seed culture to 500 mL of media and grow until late log phase (OD600 value 1.2); then, harvest the cells in 500 mL of culture harvest bottles using a 4 °C centrifuge at 5,000× g for 10 min.
2. Wash the cell pellet twice using 20 mL of ice-cold buffer A by vortexing. Finally, transfer the pellet to a 50 mL centrifuge tube, dissolve in 25 mL of buffer A, and keep on ice. To ensure that the pellet dissolves properly, vortex the mixture intermittently and mix it by pipetting.
3. Sonicate the dissolved cell pellet (about 25 mL) for 10 cycles (30 s ON; 55 s OFF; amp 90%). Always keep the solution on ice to prevent heating.
4. Add 25 mg of lysozyme to the sonicated solution (final concentration becomes 1 mg/mL) and incubate on ice for 30 min.
5. Repeat the sonication cycle.
6. Transfer the solution to an Oak Ridge tube and centrifuge at 13,000× g for 45 min at 4 °C.
7. Repeat the centrifugation step using a fresh Oak Ridge tube. These two rounds of high-speed centrifugation remove the debris formed after cell lysis.
8. Carefully collect the whole cell lysate and transfer to a 33 mL centrifuge tube. Subject this supernatant to ultracentrifugation at 150,000× g for 2 h and 30 min at 4 °C using a swing-out rotor.
9. Decant the supernatant and store the ribosomal pellet in the centrifuge tube at 4 °C by adding 500 μL of buffer B (Figure 1A) without resuspending. Cover the tube with parafilm to prevent contamination.

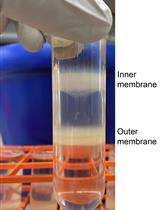
Figure 1. Pellets obtained after Day 1 and Day 2 of ribosome purification. (A) Yellowish pellet obtained at the end of Day 1. (B) More transparent crude ribosome pellet obtained at the end of Day 2 after high salt wash. A black arrow marks the pellet in both panels. (C) Representation of the various layers of the sucrose density gradient.
Day 2 of 70S ribosome isolation
1. Add 19.5 mL of buffer C (high salt wash buffer) to the overnight pellet and pipette it repeatedly to ensure proper dissolving.
2. Transfer the mixture into a glass homogenizer and homogenize the solution for 1 h at every 5 min interval. This step is crucial for the removal of weakly interacting proteins from the ribosome.
3. Transfer the solution to an Oak Ridge tube and centrifuge at 13,000× g for 45 min at 4 °C. Collect the supernatant and repeat the centrifugation step using a fresh Oak Ridge tube.
4. Carefully collect the supernatant in a centrifuge tube and perform ultracentrifugation at 150,000× g for 2 h and 30 min at 4 °C using a swing-out rotor.
5. Decant the supernatant and store the pellet in the centrifuge tube at 4 °C by adding 500 μL of buffer B (Figure 1B). Cover the tube with parafilm to prevent contamination.
Pause point: This crude ribosome pellet can be stored safely in buffer B for 2–3 days at 4 °C without any protease inhibitor.
6. In a fresh centrifuge tube, prepare a sucrose density gradient using buffer B. The bottom layer consists of 40% sucrose (12 mL), followed by 30% (w/v) sucrose (8 mL), 20% (w/v) sucrose (7 mL), and a top-most layer containing 10% (w/v) sucrose (6 mL). Keep this density gradient inside a 4 °C refrigerator overnight for next-day use (Figure 1C).
Critical: Accurate density gradient preparation ensures distinct separation of ribosomes, which results in a well-resolved ribosome profile.
Day 3 of 70S ribosome isolation
1. Dissolve the overnight stored ribosomal pellet in 500 μL of buffer B by pipetting and overlay it on top of the sucrose density gradient.
2. Gently subject this gradient to ultracentrifugation at 150,000× g for 4 h and 30 min at 4 °C using a swing-out rotor.
CAUTION: Ultracentrifuge tubes must be accurately balanced.
3. After completion of ultracentrifugation, collect 750 μL fractions from the gradient into 1.5 mL microcentrifuge tubes using a peristaltic pump. The peristaltic pump collects fractions from bottom to top, with the 40% layer coming out first and the 10% layer last.
4. Measure the OD260 of all fractions to detect the amount of rRNA and plot in Prism (v5.00) software (GraphPad, San Diego, CA) to obtain the profile (Figure 2A). Buffer B is used as a blank.
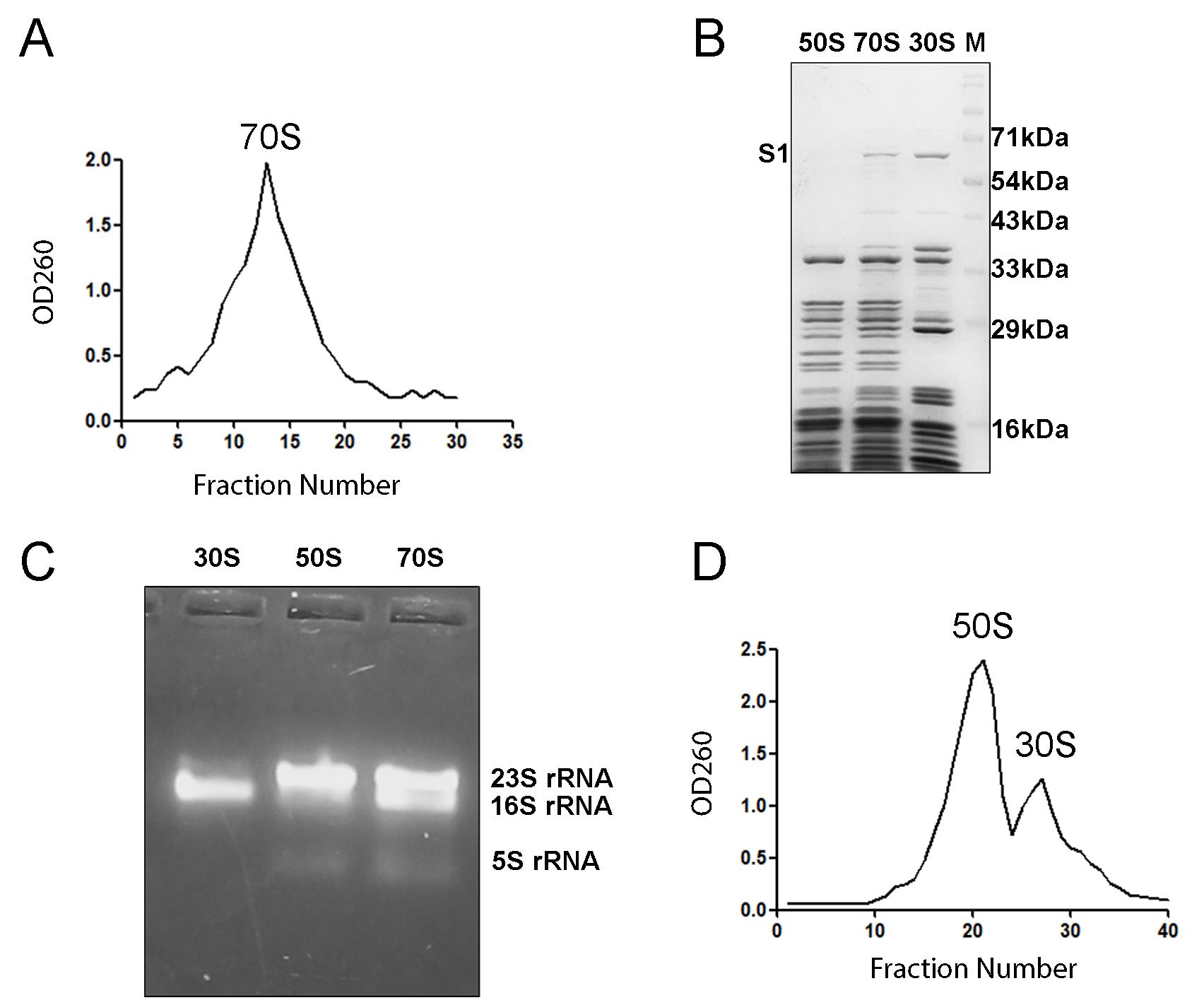
Figure 2. Ribosome isolation and validation by gel electrophoresis. (A) 70S ribosome profile obtained by measuring OD at 260 nm. (B) SDS-PAGE showing the characteristic ribosomal protein bands present in the 70S ribosome, 50S subunit, and 30S subunit. The ribosomal protein S1 is used to identify the presence of the small ribosomal subunit. (C) Agarose gel electrophoresis highlights the 23S, 16S, and 5S rRNA present in the ribosome. The distinct difference in the bands indicates proper purification of the 70S ribosome and its subunits. (D) OD260 profile clearly shows two distinct peaks for 50S and 30S fractions.
5. Pool the fractions (12–18) showing high RNA signal and concentrate them using Amicon® Ultra-4 centrifugal filters (100 kDa). The concentrated ribosome (500 μL) is then diluted with four times the excess of buffer B (2 mL) and again concentrated with Amicon® Ultra-4 centrifugal filters (100 kDa) to approximately 500 μL. This should be repeated at least five times to eliminate sucrose, which would hamper downstream experiments.
6. Calculate the final concentration using the following formula:
Concentration (μM) = (OD260 × Excitation coefficient × Dilution Factor)/Molecular Weight
For the prokaryotic ribosome, the following values are taken:
Extinction coefficient = 67
Molecular weight of 70S ribosome = 2,700
Molecular weight of 50S subunit = 1,800
Molecular weight of 30S subunit = 900
7. Run 10 μmol of sample in 15% SDS-PAGE and 5 μmol of sample in 1% agarose gel to visualize the 70S ribosomal proteins and the ribosomal RNA bands, respectively (Figure 2B, C). Intact rRNA bands and r-proteins indicate that the ribosomal preparation is of proper quality. The quality may further be analyzed by microfluidic capillary electrophoresis and calculation of RNA integrity number (RIN) [11], but from our experience, clear rRNA bands and the absence of a smear on the agarose gel should suffice.
B. 50S and 30S ribosomal subunit isolation from Mycobacterium smegmatis MC2 155
1. For the isolation of ribosomal subunits, follow similar steps until the end of Day 2. The difference comes during the storage of the pellet obtained after the second-day ultracentrifugation. For subunit isolation, it is stored at 4 °C overnight in 500 μL of buffer D. The low magnesium enables the dissociation of the 70S ribosome into 50S and 30S subunits.
2. Prepare the 40%–10% sucrose density gradient using buffer D and keep it inside a 4 °C refrigerator overnight for next-day use.
3. On Day 3, dissolve the pellet in 500 μL of buffer D.
4. Prepare a fresh puromycin solution by adding 0.0013 g of puromycin in 100 μL of buffer D. Add this antibiotic solution (dropwise) to the dissolved pellet and mix by vortexing. Puromycin facilitates the release of peptidyl-tRNA from the 70S ribosome and hence aids in the dissociation of the ribosome into 50S and 30S subunits.
5. Incubate at 37 °C for 20 min and then transfer the tube to ice and incubate for 10 min.
6. Overlay the mixture on top of the density gradient and run ultracentrifugation at 150,000× g for 9 h and 30 min at 4 °C using a swing-out rotor.
7. After completion of the ultracentrifugation, collect the fractions in the same way as done with the 70S ribosome.
8. Measure OD260 and plot in Prism (v5.00) software to obtain the 50S and 30S subunit profiles (Figure 2D).
9. Separately concentrate the two subunits using centrifugal filters, buffer exchange with buffer B (to restore the magnesium level), and finally run 10 μmol of sample in 15% SDS-PAGE and 5 μmol of sample in 1% agarose gel electrophoresis to confirm the ribosomal protein bands and ribosomal rRNA bands of 50S and 30S subunits, respectively (Figure 2B, C).
C. Ribosome co-sedimentation assay
The co-sedimentation assay relies on the principle that if a ligand (protein) interacts with a large molecule (ribosome), then the ligand will migrate along with the larger molecule when subjected to ultracentrifugation.
Note: For our co-sedimentation assay, we used buffer B. The buffer will vary from protein to protein and requires optimization. The incubation temperature and all other parameters are standardized according to our protein of interest, and these need to be optimized when a different protein is used. This protocol will provide an idea of how to check if a protein is binding to the ribosome.
1. Add 15-fold excess protein to 70S ribosome or the 50S/30S subunits in a 1.5 mL microcentrifuge tube.
Note: For running a 33 mL sucrose density gradient, use at least 0.6 μM ribosome. Add 9 μM protein to 0.6 μM ribosome in a final reaction volume of 300 μL. Again, optimization of the buffer, concentration of protein, and other parameters varies for different samples.
2. Incubate the reaction mixture at 37 °C for 15 min, followed by ice for another 15 min.
3. Overlay the reaction mixture on top of a 40%–10% sucrose density gradient prepared using buffer B.
4. Run ultracentrifugation at 150,000× g for 5 h and 30 min at 4 °C using a swing-out rotor.
5. Collect the fractions similarly using a peristaltic pump and measure OD260 to obtain the profile (Figure 3A–C).
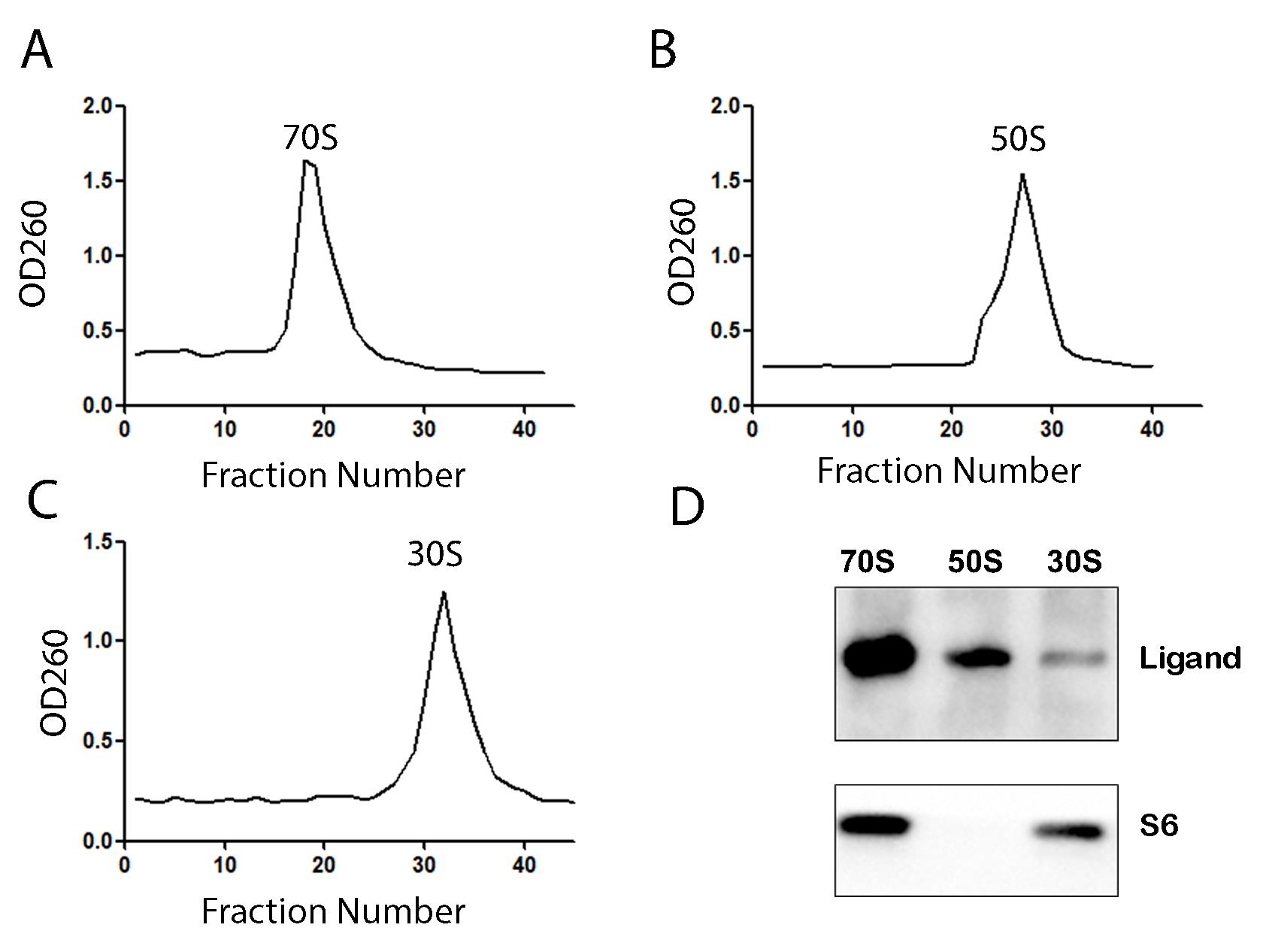
Figure 3. Co-sedimentation of 70S ribosome, 50S subunit, and 30S subunit with a co-translational protein. (A) OD260 profile of the 70S ribosome. (B) OD260 profile of the 50S ribosomal subunit. (C) OD260 profile of the 30S ribosomal subunit. (D) Western blot with protein-specific antibody to detect the binding of the co-translational factor with 70S ribosome and the subunits. Ribosomal protein S6 is used as a loading control.
6. Concentrate the ribosomal fractions using centrifugal filters, remove excess sucrose by exchanging with buffer B, and calculate the final ribosomal concentration using the previously mentioned formula.
Critical: Select the molecular weight cutoff of the centrifugal filters carefully and make sure it is significantly lower than the molecular weight of your protein of interest.
7. To detect if your protein of interest interacts with the ribosome, run SDS-PAGE by loading 0.6 μM 70S ribosome or the subunits. Check if the additional band of the protein is visible on the gel by comparing it with the control ribosome sample without the protein.
8. Sometimes, binding is weak, or the protein band overlaps with ribosomal protein bands. If so, perform western blot with protein-specific antibody to detect the binding (Figure 3D).
D. Ribosomal subunit association assay
Under normal circumstances, the 50S ribosomal subunit joins with the 30S subunit to form the active 70S ribosome. Using sucrose density gradient ultracentrifugation, you can obtain the associated 70S ribosome and calculate the area under the curve from the ribosomal profile to determine the efficiency of association. You can also check if your ligand of interest is an anti-associating agent. We standardized the assay using HMA-10 buffer. The step-by-step protocol to perform the association assay is mentioned below:
1. Add 1 μM 50S subunit and 1 μM 30S subunit in a 1.5 mL microcentrifuge tube. Adjust the total reaction volume to 500 μL with HMA-10 buffer.
Note: If you want to check whether your ligand of interest acts as an anti-associating agent, add the ligand while adding the ribosomal subunits.
2. Incubate the reaction mixture at 37 °C for 20 min and then transfer to ice for another 10 min.
3. Overlay the reaction mixture on top of a 40%–10% sucrose density gradient prepared using HMA-10 buffer, followed by ultracentrifugation at 150,000× g for 5 h and 30 min at 4 °C using a swing-out rotor.
4. Collect the fractions (750 μL in each vial) using a peristaltic pump and measure OD260 of each vial. Use the OD260 values to obtain the ribosomal re-association profile in Prism (v5.00) software (Figure 4A, B). A well-resolved profile will show three distinct peaks corresponding to the 70S ribosome, the 50S subunit, and the 30S subunit.
Note: If you are analyzing whether a ligand is an anti-associating agent, compare the profile with a control set and calculate the area under the curve to quantify the efficiency of anti-association.
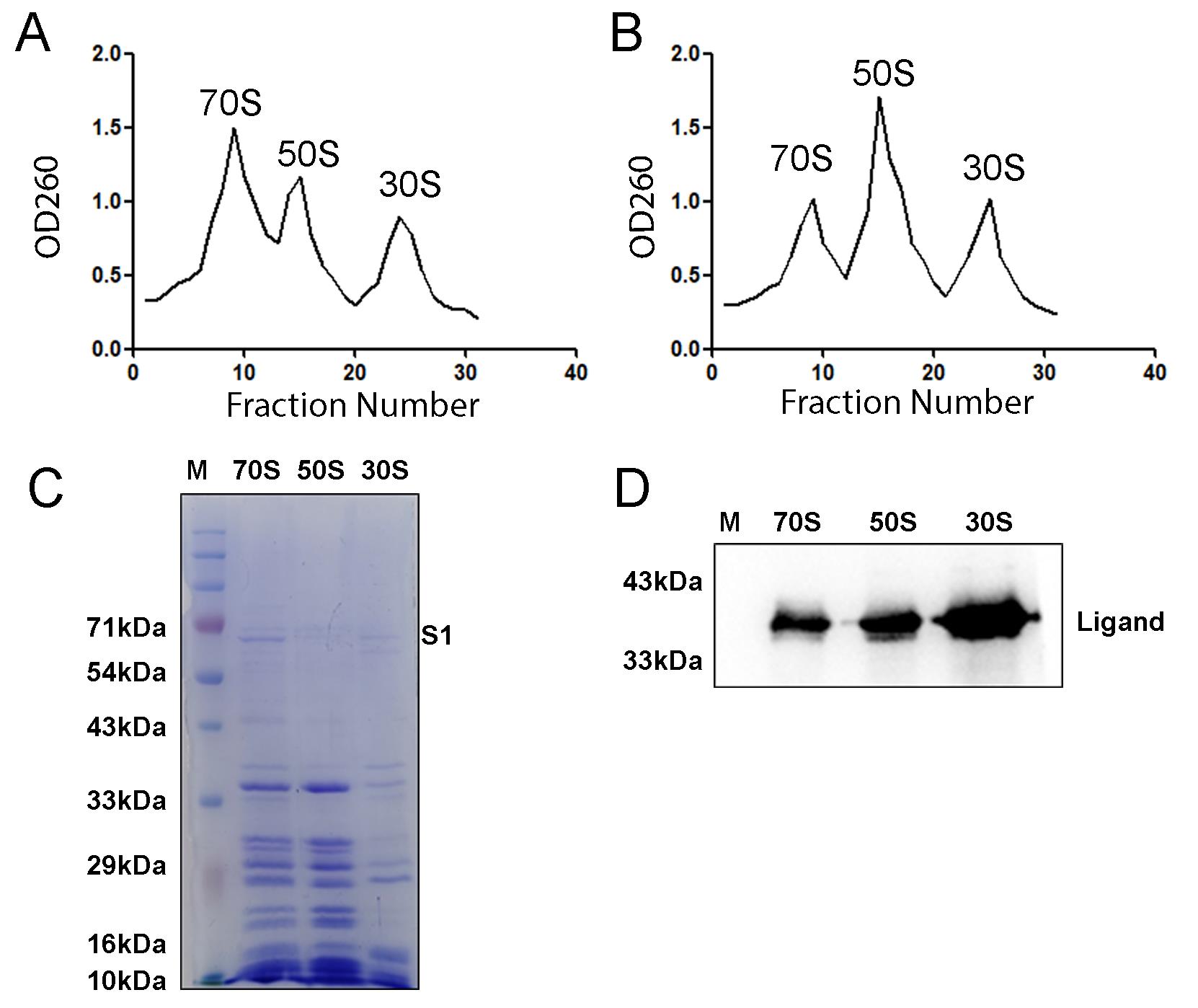
Figure 4. Ribosomal subunit association studies with and without an anti-association agent. (A) OD260 profile indicates the formation of 70S ribosome after the addition of 50S and 30S subunits in the absence of any anti-association agent. (B) In the presence of an anti-association agent, the 70S population is diminished, signifying poor association of the 50S and 30S subunits. It must be noted here that OD260 readings indicate the presence of RNA and cannot alone distinguish between fully assembled 70S ribosomes, ribosomal subunits, or aggregates. The samples must then be verified using 1% agarose gels or SDS-PAGE. (C) SDS-PAGE profile shows the characteristic protein bands of the 70S ribosome, 50S subunit, and 30S subunit. The 5 μM 30S subunit was loaded, which led to lower band intensity. (D) Western blot with protein-specific antibody detects the binding of the anti-association agent on the 70S ribosome and its subunits.
5. Separately concentrate the 70S ribosome, 50S subunit, and 30S subunit using centrifugal filters and remove the excess sucrose by exchanging with HMA-10 buffer.
6. Determine the final concentration of 70S ribosome and the subunits using the previously mentioned formula and run on SDS-PAGE to validate the formation of 70S ribosome (Figure 4C).
Note: The 70S ribosome obtained by re-associating the 50S and 30S subunits is largely free from any tRNA; however, additional validation may be necessary. So, to carry out any experiment requiring tRNA-free 70S ribosome, this re-association is essential.
7. If you used any ligand (protein) for this assay and want to detect the occupancy of that on the ribosomal subunits, run a western blot with a specific antibody (Figure 4D).
Validation of protocol
This protocol or parts of it has been used and validated in the following research article(s):
• Srinivasan et al. [12] Cryo-EM structures reveal the molecular mechanism of HflX-mediated erythromycin resistance in mycobacteria. Structure. (Figure 3A, B, D–I; Figure 4B–F; Figure 5D–F)
• Banerjee et al. [13] Mycobacterial methionine aminopeptidase type 1c moonlights as an anti-association factor on the 30S ribosomal subunit. bioRxiv. (Figure 1I; Figure 4B–D, Figure F–K; Figure 5C–J)
• Baid et al. [14] Cryo-EM captures a unique conformational rearrangement in 23S rRNA helices of the Mycobacterium 50S subunit. International Journal of Biological Macromolecules. (Figure 6C, D)
General notes and troubleshooting
General notes
1. Prepare the buffers the day before use and store at 4 °C.
2. Accurate gradient preparation is critical for obtaining proper ribosomal profiles.
3. The ribosome isolation protocol can vary slightly for other prokaryotic organisms.
4. The ribosome co-sedimentation assay can also be done using a sucrose cushion, but it has some limitations: i) If the protein has an aggregation tendency, it will co-precipitate with the ribosome. ii) Some proteins have the property to split 70S ribosomes into their subunits. Performing a cushion will result in co-precipitation of all the subunits together, and you will miss the splitting indication.
Troubleshooting
Problem 1: Little or no ribosome pellet after ultracentrifugation on Day 2 of ribosome isolation.
Possible cause: Incomplete homogenization of crude ribosomes.
Solution: Ensure proper and thorough homogenization to ensure that the pellet is well dissolved. Ensure that the solution is free from visible particulates.
Problem 2: Obtaining a high OD260 signal in the initial fractions (1–6) of 50S and 30S isolation profiles.
Possible cause: 70S contamination due to incomplete dissociation into its subunits.
Solution: i) Do not take those fractions for subsequent assays; ii) increase the 37 °C incubation time from 20 to 30 min to enhance the splitting.
Acknowledgments
Conceptualization J.S.; Investigation, A.B., S.D., K.S., A.D., P.B.; Writing–Original Draft, A.B., S.D., A.D.; Writing–Review & Editing, K.S., A.B., J.S.
Funding sources that supported the work: SERB, DST (India) sponsored project (SPF/2021/000141), CSIR Niche Creating Project (NCP) MLP-139 and CSIR-Indian Institute of Chemical Biology (OLP120), Kolkata, India. A.B., S.D., K.S., A.D., and P.B. thank DBT, ANRF-NPDF, CSIR, UGC, and CSIR, India, respectively, for their fellowship.
This protocol was described and validated in Baid et al. [14], Srinivasan et al. [12], Banerjee et al. [13]. The protocol was developed, modified, or derived from Dey et al. [15].
Competing interests
The authors declare no conflicts of interest.
Ethical considerations
The work uses non-pathogenic bacteria, and all safety guidelines were followed. The authors have no ethical declarations to declare.
References
- Green, R. and Noller, H. F. (1997). RIBOSOMES AND TRANSLATION. Annu Rev Biochem. 66(1): 679–716. https://doi.org/10.1146/annurev.biochem.66.1.679
- Steitz, T. A. (2008). A structural understanding of the dynamic ribosome machine. Nat Rev Mol Cell Biol. 9(3): 242–253. https://doi.org/10.1038/nrm2352
- Ramakrishnan, V. (2014). The Ribosome Emerges from a Black Box. Cell. 159(5): 979–984. https://doi.org/10.1016/j.cell.2014.10.052
- Khusainov, I., Vicens, Q., Bochler, A., Grosse, F., Myasnikov, A., Ménétret, J. F., Chicher, J., Marzi, S., Romby, P., Yusupova, G., et al. (2016). Structure of the 70S ribosome from human pathogen Staphylococcus aureus. Nucleic Acids Res. 44(21): 10491–10504. https://doi.org/10.1093/nar/gkw933
- Melnikov, S., Ben-Shem, A., Garreau de Loubresse, N., Jenner, L., Yusupova, G. and Yusupov, M. (2012). One core, two shells: bacterial and eukaryotic ribosomes. Nat Struct Mol Biol. 19(6): 560–567. https://doi.org/10.1038/nsmb.2313
- Mehta, P., Woo, P., Venkataraman, K. and Karzai, A. W. (2012). Ribosome Purification Approaches for Studying Interactions of Regulatory Proteins and RNAs with the Ribosome. Methods Mol Biol. 905: 273–289. https://doi.org/10.1007/978-1-61779-949-5_18
- Vandeventer, P. E., Weigel, K. M., Salazar, J., Erwin, B., Irvine, B., Doebler, R., Nadim, A., Cangelosi, G. A. and Niemz, A. (2011). Mechanical Disruption of Lysis-Resistant Bacterial Cells by Use of a Miniature, Low-Power, Disposable Device. J Clin Microbiol. 49(7): 2533–2539. https://doi.org/10.1128/jcm.02171-10
- Rabodoarivelo, M. S., Aerts, M., Vandamme, P., Palomino, J. C., Rasolofo, V. and Martin, A. (2016). Optimizing of a protein extraction method for Mycobacterium tuberculosis proteome analysis using mass spectrometry. J Microbiol Methods. 131: 144–147. https://doi.org/10.1016/j.mimet.2016.10.021
- Trauner, A., Bennett, M. H. and Williams, H. D. (2011). Isolation of Bacterial Ribosomes with Monolith Chromatography. PLoS One. 6(2): e16273. https://doi.org/10.1371/journal.pone.0016273
- Mangan, J. (1997). An effective method of RNA extraction from bacteria refractory to disruption, including mycobacteria. Nucleic Acids Res. 25(3): 675–676. https://doi.org/10.1093/nar/25.3.675
- Schroeder, A., Mueller, O., Stocker, S., Salowsky, R., Leiber, M., Gassmann, M., Lightfoot, S., Menzel, W., Granzow, M., Ragg, T., et al. (2006). The RIN: an RNA integrity number for assigning integrity values to RNA measurements. BMC Mol Biol. 7(1): e1186/1471–2199–7–3. https://doi.org/10.1186/1471-2199-7-3
- Srinivasan, K., Banerjee, A. and Sengupta, J. (2024). Cryo-EM structures reveal the molecular mechanism of HflX-mediated erythromycin resistance in mycobacteria. Structure. 32(9): 1443–1453.e4. https://doi.org/10.1016/j.str.2024.06.016
- Banerjee, A., Srinivasan, K. and Sengupta, J. (2024). Mycobacterial methionine aminopeptidase type 1c moonlights as an anti-association factor on the 30S ribosomal subunit. bioRxiv. e625572. https://doi.org/10.1101/2024.11.26.625572
- Baid, P. and Sengupta, J. (2023). Cryo-EM captures a unique conformational rearrangement in 23S rRNA helices of the Mycobacterium 50S subunit. Int J Biol Macromol. 253: 126876. https://doi.org/10.1016/j.ijbiomac.2023.126876
- Dey, S., Biswas, C. and Sengupta, J. (2018). The universally conserved GTPase HflX is an RNA helicase that restores heat-damaged Escherichia coli ribosomes. J Cell Biol. 217(7): 2519–2529. https://doi.org/10.1083/jcb.201711131
Article Information
Publication history
Received: Mar 3, 2025
Accepted: Apr 18, 2025
Available online: May 8, 2025
Published: May 20, 2025
Copyright
© 2025 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Banerjee, A., Dey, S., Srinivasan, K., Dhur, A., Baid, P. and Sengupta, J. (2025). Mycobacterium smegmatis Ribosome Purification, Co-sedimentation, and Subunit Association Assay. Bio-protocol 15(10): e5318. DOI: 10.21769/BioProtoc.5318.
Category
Microbiology > Microbial cell biology > Organelle isolation
Biochemistry > RNA
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.