- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Isolation and Culture of Primary Pericytes from Mouse
Published: Vol 15, Iss 8, Apr 20, 2025 DOI: 10.21769/BioProtoc.5288 Views: 3276
Reviewed by: Pilar Villacampa AlcubierreJosé M. DiasHaixia Xu
Original research article
The authors used this protocol in:

Nov 2024
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Pericytes are essential for tissue homeostasis, functioning to regulate capillary blood flow. Dysfunctional pericytes are implicated in various pathologies, including cancer progression. Despite their important function in both health and disease, pericytes remain understudied due to a lack of robust model systems that accurately reflect their in vivo biology. Here, we present a comprehensive protocol for isolating and culturing primary pericytes from murine lung, brain, bone, and liver tissues, based on NG2 expression using an antibody-conjugated magnetic bead approach. Our protocol emphasizes the importance of physiological oxygen tension during ex vivo culture (10% O2 for lung pericytes and 5% O2 for brain, bone, and liver pericytes). These conditions stabilize the expression of characteristic pericyte markers at both the transcriptional and protein levels. Importantly, we optimized growth conditions to limit the expression of the plasticity factor Klf4 in order to prevent spontaneous phenotypic switching in vitro. This protocol provides a reliable and reproducible method for obtaining pericytes suitable for high-throughput analyses in order to explore pericyte biology in both physiological and pathological contexts.
Key features
• Isolation of primary pericytes from mouse lung, brain, bone, and liver.
• Emphasis on physioxic culturing conditions to better maintain pericyte phenotype.
• Representative of pericyte biology in both health and disease contexts.
Keywords: PericytesGraphical overview
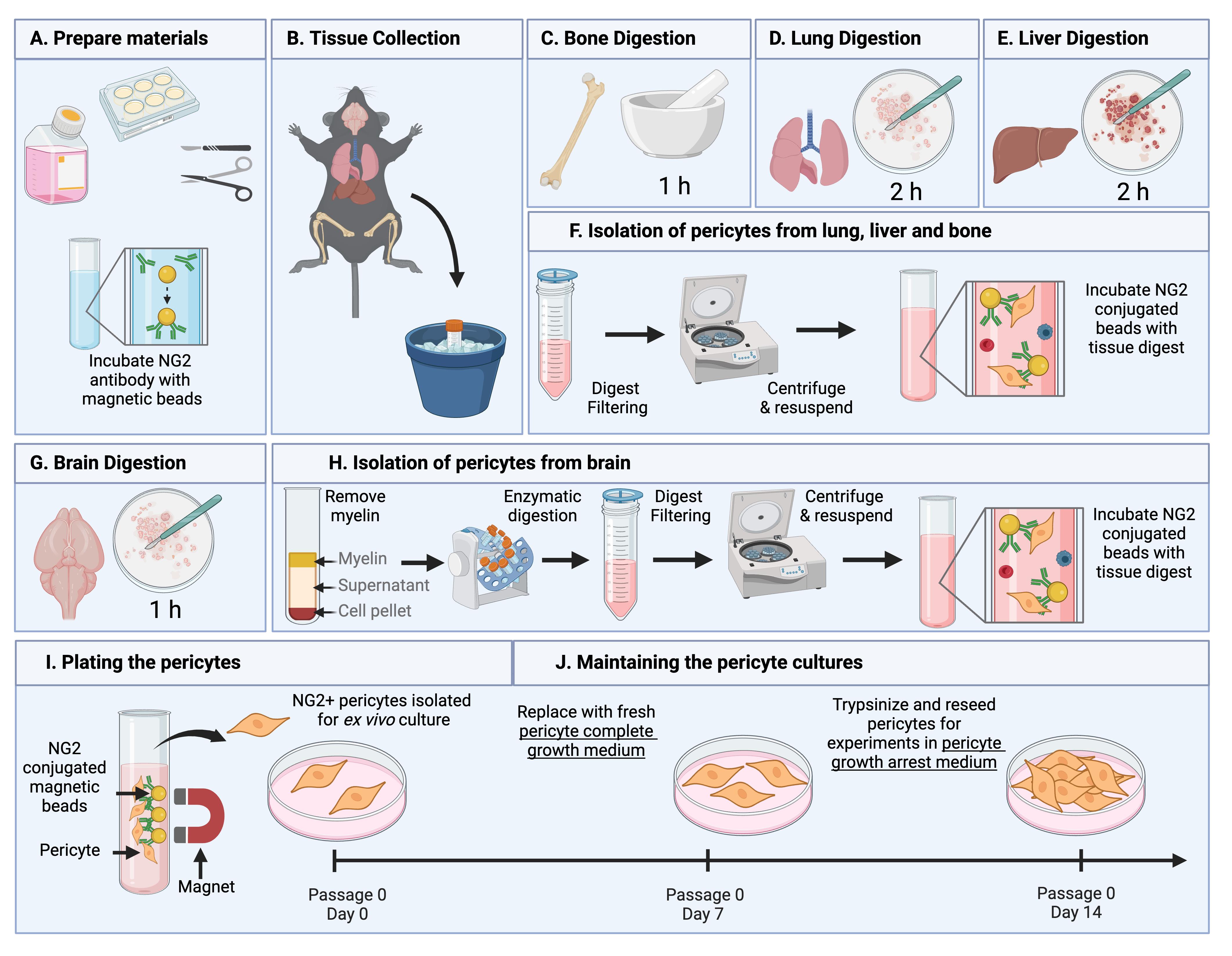
Summary of the pericyte isolation protocol. (A) Before beginning, dissection tools are sterilized, pericyte medium is equilibrated, cell culture plates are coated with collagen, and NG2 antibody–conjugated Dynabeads are prepared. (B) Organs of interest are collected from 12-week-old C57BL/6 mice following euthanasia by isoflurane and cervical dislocation. (D–E) Bone, lung, and liver tissues undergo organ-specific digestion steps prior to isolation described in (F). (F) Digest from the lung, bone, and liver are filtered, centrifuged, and incubated with NG2-conjugated beads. (G) Brain tissues undergo digestion steps. (H) Digest from the brain undergoes myelin removal and further digestion before being filtered, centrifuged, and incubated with NG2 conjugated beads. (I–J) The bead-bound NG2-expressing cells are isolated and washed using a magnet and plated for expansion under optimized pericyte culturing conditions. Figure created in Biorender.com.
Background
Pericytes are integral components of the microvasculature, forming direct interactions with endothelial cells and other mural cells to maintain tissue homeostasis by regulating capillary blood flow and endothelial barrier function [1,2]. Dysfunction or loss of pericytes has been implicated in various pathologies, including aging, ischemic disease, fibrosis, neurodegeneration, and cancer [3–8].
Genetic mouse models have been invaluable tools in elucidating pericyte function during development and disease [9–11]. However, in vitro tools are lacking despite being essential for high throughput and mechanistic studies. This is partly due to the heterogeneity of pericyte origins, functions, and identification markers, as well as their intrinsic plasticity, which can result in undesired lineage differentiation [1,2,7,12–16]. Unfortunately, existing in vitro models present significant limitations. For example, immortalized pericyte cell lines exhibit constitutive proliferation, a hallmark of perivascular activation not typically observed in quiescent pericytes in vivo [7,17,18]. Alternatively, pericytes derived from induced pluripotent stem cells (iPSCs) require complex differentiation protocols and highly specific culturing conditions, making these protocols less accessible [19,20].
Primary pericyte cultures present a more physiologically relevant alternative tool, as they have the potential to retain tissue-specific characteristics and better reflect in vivo biology. While isolation of perivascular cells has been achieved, these methods lack standardization, which hinders reproducibility [21]. A common cell isolation approach involves enzymatic digestion of tissues followed by purification via fluorescence-activated cell sorting (FACS) or immunomagnetic separation. However, despite successful isolation, cell yield remains a challenge. For example, Lee et al. described a protocol for isolating murine cardiac pericytes, but its high cell input requirement and resulting low yield (1.1%) present significant drawbacks [22]. Low pericyte yield during isolation could be overcome with ex vivo expansion; however, their high differentiation capacity poses an additional challenge. In the development of this protocol, we sought to uncover the culture conditions that would support pericyte expansion while retaining their identity and function, a problem that has been overcome in the culture of other plastic cell populations, i.e., iPSCs, by tightly regulating oxygen tension [23–26].
To address the current limitations in pericyte research, we developed a standardized protocol for isolating and culturing primary murine pericytes from the lung, brain, bone, and liver under physiologically relevant oxygen conditions [27–29]. This approach preserved pericyte identity and function in both homeostatic conditions and recapitulated important pericyte phenotypic switching that has been observed in vivo in response to tumors [7]. This protocol uses NG2 as a marker to selectively isolate pericytes, as it is continuously expressed on arteriolar and mid-capillary pericytes, allowing the capture of multiple pericyte subtypes [30]. However, some pericyte populations, such as those associated with post-capillary venules, may not express NG2 [30] and could be underrepresented, which is a limitation of this method. Additionally, NG2 is expressed by other cell types including oligodendrocyte precursor cells in the brain [31], necessitating validation of pericyte purity in culture. Here, we provide a robust protocol for studying pericyte biology in health and disease.
Materials and reagents
Biological materials
1. 12-week-old female C57BL/6 mice used for pericyte isolation
Reagents
1. 0.05% trypsin-EDTA (Thermo Fisher Scientific, catalog number: 25300062)
2. Acetic acid (Millipore Sigma, catalog number: A6283)
3. Bovine serum albumin (BSA) (Millipore Sigma, catalog number: 12657)
4. Collagen from rat tail (Millipore Sigma, catalog number: C7661)
5. Collagenase A (Millipore Sigma, catalog number: 11088793001)
6. Dispase II (Millipore Sigma, catalog number: 4942078001)
7. DNase1 (Millipore Sigma, catalog number: D4263)
8. Dulbecco’s modified Eagle’s medium (DMEM), low glucose (Millipore Sigma, catalog number: D6046)
9. Sheep anti-rat IgG Dynabeads (Thermo Fisher Scientific, catalog number: 11035)
10. Endothelial cell growth supplement (R&D, catalog number: 390599)
11. Fetal bovine serum (FBS) [GeminiBio, catalog number: 900-208-500, lot tested (lot no. A120030)]
12. Ham’s F12 nutrient mix (Thermo Fisher Scientific, catalog number: 11765047)
13. Heparin (Millipore Sigma, catalog number: H3149)
14. HEPES (1 M) (Thermo Fisher Scientific, catalog number: 15630080)
15. MEM non-essential amino acids (100×) (Thermo Fisher Scientific, catalog number: 11140050)
16. CO2-O2-N2 (5-5-90) compressed gas mix (Roberts Oxygen)
17. NG2 monoclonal antibody (546930) (Thermo Fisher Scientific, catalog number: MA5-24247)
18. Compressed nitrogen gas (Roberts Oxygen)
19. PBS, pH7.4 (Thermo Fisher Scientific, catalog number: 10010023)
20. Penicillin-Streptomycin (Thermo Fisher Scientific, catalog number: 15070063)
21. Sodium pyruvate (100 mM) (Thermo Fisher Scientific, catalog number: 11360070)
Solutions
1. NG2 antibody resuspension (see Recipes)
2. Collagenase A stock solution (see Recipes)
3. Dispase II stock solution (see Recipes)
4. DNase1 stock solution (see Recipes)
5. Heparin stock solution (see Recipes)
6. 0.1% BSA/PBS (see Recipes)
7. 20% BSA in DMEM (see Recipes)
8. Collagen stock solution (see Recipes)
9. Collagen working solution (see Recipes)
10. Pericyte basal medium (see Recipes)
11. Pericyte complete growth medium (see Recipes)
12. Pericyte growth arrest medium (see Recipes)
Recipes
1. NG2 antibody resuspension
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| NG2 antibody | 0.5 mg/mL | 100 μg |
| Sterile PBS | n/a | 200 μL |
| Total | n/a | 200 µL |
Once dissolved, store at -20 °C.
2. Collagenase A stock solution
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| Collagenase A | 100 mg/mL | 500 mg |
| Sterile PBS | n/a | 5 mL |
| Total | n/a | 5 mL |
Calcium- and magnesium-free PBS is used to prevent enzyme inhibition. Collagenase may take up to 20 min to fully dissolve. Keep the solution on ice and invert the vial at regular intervals to mix. Aliquot and store at -20 °C for up to 6 months. To produce the working concentration, dilute the stock in PBS or low-glucose DMEM to a final concentration of 2 mg/mL.
3. Dispase II stock solution
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| Dispase II | 10 mg/mL | 1 g |
| HEPES buffered saline (pH 7.4) | 50 mM HEPES, 150 mM NaCl | 100 mL |
| Total | n/a | 100 mL |
Once dissolved, aliquot and store at -20 °C for up to 2 months. To produce the working concentration, dilute the stock in low-glucose DMEM to a final concentration of 1 mg/mL.
4. DNase1 stock solution
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| DNase1 | 1 mg/mL | 1 mg |
| Sterile deionized water | n/a | 1 mL |
| Total | n/a | 1 mL |
Once dissolved, aliquot and store at -20 °C for 1 month. To produce the working concentration, dilute the stock in low-glucose DMEM to a final concentration of 10 μg/mL.
5. Heparin stock solution
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| Heparin | 10 mg/mL | 1 g |
| Sterile deionized water | n/a | 100 mL |
| Total | n/a | 100 mL |
Aliquot and store at -20 °C for up to 2 years. To produce the working concentration, dilute the stock in PBS to a final concentration of 10 µg/mL.
6. 0.1% BSA/PBS
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| BSA | 15 mM | 0.1 g |
| PBS | n/a | 100 mL |
| Total | n/a | 100 mL |
Once dissolved, filter the solution through a 0.2 μm filter and store at 4 °C for up to 1 month.
7. 20% BSA in DMEM
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| BSA | 3 M | 20 g |
| DMEM low glucose | n/a | 100 mL |
| Total | n/a | 100 mL |
For brain digestion, a solution of 20% BSA in low-glucose DMEM is required [29]. BSA will dissolve on a roller at 37 °C. Once dissolved, filter the solution through a 0.2 μm filter. 10 mL is needed per batch of 3–6 organs.
8. Collagen stock solution (for cell culture plate coating)
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| Rat collagen type I | 3 mg/mL | 25 mg |
| 0.1 M acetic acid | 0.1 M | 8,333 μL |
| Total | n/a | 8,333 μL |
Collagen stock should be prepared in 0.1 M acetic acid at a concentration of 3 mg/mL. The collagen in acetic acid should be placed on a roller at room temperature for 2–3 h, followed by overnight rolling at 4 °C. Once dissolved, store the collagen stock at 4 °C.
9. Collagen working solution (for cell culture plate coating)
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| Collagen stock (Recipe 8) | 0.06 mg/mL | 1 mL |
| Sterile deionized water | n/a | 49 mL |
| Total | n/a | 50 mL |
Before coating the plates, 3 mg/mL collagen stock needs to be diluted 1:50 in sterile deionized water to a final concentration of 0.06 mg/mL. Coat tissue culture plates with collagen working solution (enough to cover the entire surface, i.e., 500 μL per well of a 6-well plate) at 37 °C for at least 2 h before seeding pericytes. Plates can be prepared with collagen at a maximum of one day before pericyte isolation and kept at 37 °C.
10. Pericyte basal medium
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| DMEM | n/a | 375 mL |
| Ham’s F12 nutrient mix | n/a | 375 mL |
| Sodium pyruvate (100 mM) | 2 mM | 20 mL |
| HEPES (1 M) | 20 mM | 20 mL |
| MEM non-essential amino acids (100×) | 1× | 10 mL |
| Total | n/a | 800 mL |
Filter sterilize using vacuum filter units and store at 4 °C for up to 1 month. This pericyte basal medium recipe is the same as that reported for endothelial cell isolation and expansion by Reiterer et al. [29] and Sandovici et al. [32].
11. Pericyte complete growth medium
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| Pericyte basal medium (Recipe 10) | n/a | 38 mL |
| FBS | 20% | 10 mL |
| Endothelial cell growth supplement (50×) | 1× | 1 mL |
| Penicillin/Streptomycin | 1% | 0.5 mL |
| Total | n/a | 50 mL |
Prepare the required volume fresh. Ensure the pericyte basal medium is equilibrated to the appropriate oxygen tension 24 h in advance. Endothelial cell growth supplement should be aliquoted and stored at -20 °C; once aliquots are thawed do not refreeze and use within 1 week. The pericyte complete growth medium composition is the same as that reported for endothelial cell isolation and expansion by Reiterer et al. [29] and Sandovici et al. [32] with the exception of heparin, which is not included.
12. Pericyte growth arrest medium
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| Pericyte basal medium (Recipe 10) | n/a | 48.5 mL |
| FBS | 2% | 1 mL |
| Penicillin/Streptomycin | 1% | 0.5 mL |
| Total | n/a | 50 mL |
Prepare the required volume fresh. Ensure the basal medium is equilibrated to the appropriate oxygen tension 24 h in advance.
Laboratory supplies
1. 15 mL sterile conical tubes (Alkali Scientific Inc, catalog number: CN5600-NEST)
2. 50 mL sterile conical tubes (Alkali Scientific Inc, catalog number: CN5602-NEST)
3. 1.7 mL microcentrifuge tubes (Corning, catalog number: 3620)
4. Tissue culture treated 6-well plates (Alkali Scientific Inc, catalog number: TP9006)
5. 70 μm nylon cell strainer (Fisher Scientific, catalog number: 22-363-548)
6. 96-well cell culture plate (Alkali Scientific Inc, catalog number: TPN1096)
7. 96-well clear bottom black cell culture plate (Fisher Scientific, catalog number: 07-200-567)
8. Cell culture dish 100 × 20 mm (Alkali Scientific Inc, catalog number: TD0100)
9. Sterile scalpels (Fisher Scientific, catalog number: 12-460-454)
10. Filter paper (Millipore Sigma, catalog number: WHA1001240)
11. Vacuum filter units (Fisher Scientific, catalog number: FB12566506)
12. 5 mL serological pipettes (Sarstedt, catalog number: 86.1253.001)
13. 10 mL serological pipettes (Sarstedt, catalog number: 86.1254.001)
14. 50 mL serological pipettes (Sarstedt, catalog number: 86.1256.001)
15. 20 μL filter pipette tips (Alkali Scientific Inc, catalog number: FLT-20)
16. 200 μL filter pipette tips (Alkali Scientific Inc, catalog number: FLT-200
17. 1,000 μL filter pipette tips (Alkali Scientific Inc, catalog number: FLT-1000)
18. 25 cm2 cell culture flask with vented cap (Alkali Scientific Inc, catalog number: TV0025)
Equipment
1. Graefe forceps (Roboz Surgical Store, catalog number: RS-5135)
2. Micro dissecting scissors (Roboz Surgical Store, catalog number: RS-5906SC)
3. Rainin pipette kit (Rainin, catalog number: 30456872)
4. Pipette controller (VWR, catalog number: 89009-344)
5. Mortar (Millipore Sigma, catalog number: Z247464)
6. Pestle (Millipore Sigma, catalog number: Z247502)
7. Refrigerator (4 °C)
8. Sorvall X4 Pro centrifuge (Thermo Fisher Scientific, catalog number: 75009505)
9. Water bath (Thermo Fisher Scientific, catalog number: TSGP2S)
10. Automated cell counter (DeNovix, model: DeNovix CellDrop FL)
11. Freezer (-20 °C)
12. BioTek Lionheart FX automated microscope (Agilent, catalog number: LFXW-SN)
13. Physioxia tissue culture incubator (Thermo Fisher Scientific, catalog number: 51033597)
14. Hypoxia cabinet with O2 controller (Coy Lab Products); hypoxia unit is placed inside a standard tissue culture incubator (Thermo Fisher Scientific, catalog number: 51033544)
15. Magnetic separation rack, 1.5 mL tubes (EpiCypher, catalog number: 10-0012)
16. Nutating Mixer (Corning, catalog number: 6720)
17. Rocking oven/incubator (Boekel Scientific, catalog number: 136400)
18. Zeiss Primovert inverted cell culture microscope with Axiocam 208 (Zeiss, catalog number: 415510-1101-000)
Procedure
A. Before you begin
1. Autoclave micro dissecting tools.
2. Prepare NG2 antibody resuspension (see Recipe 1).
3. Prepare and aliquot digestion enzymes (see Recipes 2–4).
4. Prepare and aliquot heparin for perfusion of tissues (see Recipe 5).
5. Prepare 0.1% BSA/PBS (see Recipe 6).
6. If isolating pericytes from the brain, prepare 20% BSA in DMEM (see Recipe 7).
7. Collagen-coat plates for seeding pericytes at least 2 h in advance (see Recipes 8 and 9).
8. Prepare the pericyte basal medium (see Recipe 10).
9. Equilibrate pericyte basal medium by adding the volume needed to a 25 cm2 tissue culture flask with a vented cap and place inside the appropriate tissue culture incubator at 37 °C for at least 24 h. Use the 10% O2 incubator if the medium will be used for lung pericytes and the 5% O2 incubator if the medium will be used for brain, bone, or liver pericytes.
10. On the day of pericyte isolation, use the equilibrated pericyte basal medium to prepare the pericyte complete growth medium (see Recipe 11) and store it in the appropriate oxygen tension tissue culture incubator until pericyte seeding.
B. Preparing Dynabeads
1. Place a 1.5 mL microcentrifuge tube into a non-magnetic holder.
2. Swirl the bottle containing the sheep anti-rat IgG Dynabeads immediately before use to ensure they are in suspension.
Note: Beads are supplied at a concentration of 4 × 108 DynabeadsTM/mL.
3. Pipette 10 µL of beads per organ collected and place this in the microcentrifuge tube in the non-magnetic holder.
4. Add 1 mL of 0.1% BSA/PBS to the aliquot of beads and pipette up and down to ensure sufficient washing.
Note: A volume of 1 mL of 0.1% BSA/PBS is adequate for washing up to 2.4 × 107 beads (e.g., for six lungs, use 60 µL of beads, which is equivalent to 2.4 × 107 beads). Additionally, it is not recommended to prepare more than 2.4 × 107 beads/mL per microcentrifuge tube as beads can get stuck around the lid of the microcentrifuge tube when using a greater volume.
5. Move the microcentrifuge tube to the magnetic holder and allow the solution to turn clear (30–60 s).
6. Gently remove the 0.1% BSA/PBS without disturbing the beads.
7. Repeat this washing step three times; then, leave the beads in the fourth 1 mL of 0.1% BSA/PBS in a non-magnetic holder.
8. Add 1.2 µL of the NG2 monoclonal antibody per organ to the washed bead solution, e.g., for five lungs, add 6 µL of antibody to 50 µL of beads in 1 mL of 0.1% BSA/PBS.
9. Incubate the bead and antibody mix for 1 h at room temperature with gentle agitation and then move to 4 °C with gentle agitation until needed (use within 4 h of preparation).
10. Repeat the wash detailed in steps B4–7 to remove excess antibody.
11. Finally, resuspend the beads in the original volume of 10 µL per organ to be collected (i.e., resuspend the beads for five organs in 50 µL of 0.1% BSA/PBS).
C. Tissue collection
1. For each batch of pericytes, collect the organs from no less than 3 and no more than 6 animals, for optimal cell yield.
2. Anesthetize the mice with isoflurane and then perform cervical dislocation.
Critical: Do not use CO2 to euthanize the animals as this will alter pericyte behavior.
3. Position the mouse in dorsal recumbency on a dissection board and use pins to secure the limbs extended in a diagonal position away from the body. Spray the mouse down with 70% ethanol.
4. Use forceps to grasp the skin over the sternum and make an incision with the scissors at the ventral midline. Extend the incision the length of the ventral midline cutting through the diaphragm, sternum, and ribs.
5. Make a small incision in the right atrium and cut the abdominal aorta below the liver.
6. Perfuse the mouse with 10 mL of ice-cold PBS containing 10 μg/mL heparin through the left ventricle. You should be able to visualize the organs becoming lighter in color.
Optional: Perfusion can be performed with PBS alone or not at all. However, liver pericyte yield is improved following perfusion with PBS containing heparin.
7. To extract the lungs, use the forceps to grab the trachea and cut to release the lungs, gently cutting any connective tissue from behind as you go. Remove the heart from the lung block. Place the lungs in ice-cold low-glucose DMEM. For a visual representation of lung dissection, please refer to D’Agostino et al. [33].
8. To extract the liver, use scissors to cut the liver away from the diaphragm and the abdominal organs. Once the liver is released, place it on a clean paper towel and identify the medial lobe that has a butterfly-like appearance with the gall bladder in the middle. Gently clamp the end of the gall bladder closest to the tissue with your forceps and cut it away with scissors, ensuring the gall bladder contents are not released over the tissue. Place the liver in ice-cold low-glucose DMEM. For a visual representation of liver dissection, please refer to Charni-Natan et al. [34].
9. To extract the femur, extend the midline excision in the lower abdomen above the hip and down the leg to the ankle joint. Use the scissors to cut the anterior quadriceps muscle exposing the femur. Position the scissors posterior to the femur to cut the hamstring at the knee joint. Place the scissor blades on either side of the femur and gently move them up until you reach the femoral head hip joint. With some pressure but without cutting, twist the scissors back and forth gently to dislocate the femur. Use forceps to hold the femur and cut away any remaining muscle. Use the scissors again at the knee joint to gently dislocate the femur from the tibia. Place the femur in ice-cold low-glucose DMEM. For a visual representation of bone dissection, please refer to Amend et al. [35].
10. To extract the brain, decapitate the mouse using scissors. Cut the skin on the dorsal midline and pull away the skin to expose the full skull. Insert one scissor blade into the spinal cord space and carefully cut along the sagittal suture to remove the calvaria (skull cap). Use forceps in a closed position to gently scoop under the brain, releasing any attachments and freeing the brain. Place the brain in ice-cold low-glucose DMEM. For a visual representation of brain dissection, please refer to MacManus et al. [36].
D. Lung digestion
1. Place the lungs into a cell culture dish and wash once with cold PBS.
2. Remove any excess connective tissue including the trachea and bronchi using scissors and place this in a waste dish before washing the remaining lung tissue once more with cold PBS.
3. Mince the lung tissue with scissors followed by a scalpel in a dry cell culture dish until tissue pieces are approximately 1 mm3. Transfer the minced tissue to a 15 mL conical tube.
Note: The tissue mincing step can be performed at room temperature but should be completed as quickly as possible.
4. Add 10 mL of PBS to the minced lungs with collagenase A to a final concentration of 2 mg/mL and parafilm the lid.
Note: Adjust the volume of the digestion solution to the number of organs, i.e., five lungs can be digested in 10 mL.
5. Digest the tissue at 37 °C with gentle rotation for 2 h.
Critical: Do not allow digestion to exceed 2 h; over-digestion will reduce cell yield.
E. Liver digestion
1. Place the livers into a cell culture dish and wash once with cold PBS.
2. Check that the gall bladder has been removed. Then, gently separate the liver lobes and mince with scissors in a dry cell culture dish. Transfer the minced tissue to a 50 mL conical tube.
Note: The tissue mincing step can be performed at room temperature but should be completed as quickly as possible.
3. Add 15 mL of PBS to the minced liver with collagenase A to a final concentration of 2 mg/mL and parafilm the lid.
Note: Adjust the volume of the digestion solution to the number of organs, i.e., five livers can be digested in 15 mL.
4. Digest the tissue at 37 °C with gentle rotation for 2 h.
Critical: Do not allow digestion to exceed 2 h; over-digestion will reduce cell yield.
F. Bone digestion
1. Clean the femurs by removing any remaining muscles or tendons. Place the bones in sterile gauze to carefully rub away any attached soft tissue.
2. Place the bones in a mortar with 1 mL of PBS and gently crush with a pestle to release the bone marrow.
3. Collect the PBS containing bone marrow and place in a clean 15 mL conical tube.
4. Add 1 mL of fresh PBS to the bone fragments remaining in the mortar and repeat the crushing step with increased force. Collect the PBS containing bone marrow into the same collection conical tube.
Notes:
a. PBS containing bone marrow will become lighter in color with each wash/crush.
b. Bone crushing can be performed at room temperature but should be completed as quickly as possible.
5. Repeat step F4 one more time with increased force until only small bone fragments remain.
6. Move the PBS and bone fragments from the mortar into the collection conical tube.
7. Make up the desired digestion volume with PBS.
Note: Adjust the volume of the digestion solution to the number of organs, i.e., five pairs of femurs in 10 mL.
8. Add collagenase A to a final concentration of 2 mg/mL to the collection tube and parafilm the lid.
9. Digest the tissue at 37 °C with gentle rotation for 1 h.
Critical: Do not allow digestion to exceed 1 h; over-digestion will reduce cell yield.
G. Brain digestion
1. Place the brains in a clean, dry cell culture dish at room temperature and use a scalpel to remove the brain stem, cerebellum, and olfactory bulb.
Note: All brain digestion steps are the same as those described by Reiterer et al. [29] used for the isolation of brain endothelial cells.
2. Roll the remaining cortical tissue on sterile blotting paper to remove the meninges.
3. Mince the remaining tissue with a scalpel until no large pieces remain. Transfer the tissue to a 15 mL conical tube and add 10 mL of prewarmed low-glucose DMEM.
Note: Adjust the volume of the digestion solution to the number of organs, i.e., five brains in 10 mL.
4. Break up the tissue further using a 10 mL serological pipette by gently mixing up and down.
5. Add collagenase A to a final concentration of 2 mg/mL and DNase1 to a final concentration of 10 µg/mL and parafilm the lid.
Note: This digestion step is required to break down the complex extracellular matrix associated with the brain microvasculature and to release the microvessels.
6. Digest the tissue at 37 °C with gentle rotation for 1 h.
Critical: Do not allow digestion to exceed 1 h; over-digestion will reduce cell yield.
H. Isolation of pericytes from lung, liver, and bone
1. Collect the digested tissue from the 37 °C digestion incubator.
2. Filter the digest through a 70 µm nylon mesh filter into a new conical tube.
3. Centrifuge the flowthrough at 260× g for 5 min at 4 °C.
Note: This is a good time to perform the bead washing step outlined in steps B4–7.
4. Aspirate the supernatant from the digest carefully as the pellet will be loose.
5. Resuspend the cell pellet in 10 mL of PBS to wash.
6. Centrifuge the washed cell suspension at 260× g for 5 min at 4 °C.
7. Aspirate the supernatant carefully, resuspend in PBS/0.1% BSA, and move to a clean conical tube.
Note: The volume of 0.1% BSA/PBS depends on the number of organs, i.e., 4 mL for five organs.
8. Add the washed pre-coated beads to the cleaned tissue digest, e.g., for five lungs and three livers, the washed beads are resuspended in 80 µL of 0.1% BSA/PBS so that 50 µL of bead suspension will be added to the cleaned lung digest and 30 µL will be added to the cleaned liver digest.
9. Incubate the tissue digest and bead suspension for 1 h at 4 °C with gentle agitation.
I. Isolation of pericytes from brain
1. Collect the digested tissue from the 37 °C digestion incubator.
Note: All the steps for pericyte isolation from the brain are the same as those described by Reiterer et al. [29] used for the isolation of brain endothelial cells.
2. Centrifuge the digest at 280× g for 5 min at 4 °C.
3. Discard the supernatant and resuspend the pellet in 10 mL of 20% BSA in DMEM.
4. Mix the suspension well by pipetting up and down using a 10 mL serological pipette 20 times.
Note: Pipette slowly to reduce bubbles.
5. Centrifuge the suspension at 1,000× g for 10 min at 4 °C. The suspension will separate into three layers: myelin, supernatant, and cell pellet (top to bottom). See Figure 1.
6. Remove the milky myelin layer at the top, taking care to remove the myelin from the sides of the conical tube as well. Then, discard the remaining supernatant.
Notes:
a. It is recommended to use a serological pipette to remove both the myelin layer and the supernatant as the cell pellet can remain loose following centrifugation.
b. It is critical to fully remove the myelin layer prior to removal of the supernatant to prevent contamination of the pellet.
7. Resuspend the pellet in 2 mL of low-glucose DMEM and filter it through a 70 µm nylon mesh strainer into a new conical tube. Wash the strainer with 1 mL of DMEM.
8. Add 13 mL of low-glucose DMEM to the old conical tube to collect any remaining cells. Use a serological pipette to collect the 13 mL suspension and pass this through the same 70 µm strainer into the new tube containing the resuspended cells.
9. Centrifuge the suspension at 280× g for 5 min at 4 °C.
10. Discard the suspension, resuspend the pellet in 2 mL of low-glucose DMEM, and transfer to a new 15 mL conical tube.
11. Wash the old conical tube with 2 mL of low-glucose DMEM and transfer this to the new conical tube.
12. Add dispase II to a final concentration of 1 mg/mL and DNase1 to a final concentration of 10 µg/mL.
Note: This digestion step is required to dissociate the microvascular components and prevent clumping of cells that could lead to contamination in culture.
13. Incubate the suspension at 37 °C for 30 min with gentle rotation.
14. Centrifuge the suspension at 280× g for 5 min at 4 °C.
15. Discard the supernatant and resuspend the pellet in 5 mL of low-glucose DMEM to wash.
16. Centrifuge the suspension at 280× g for 5 min at 4 °C.
Note: This is a good time to perform the bead washing step outlined in steps B4–7.
17. Discard the supernatant, resuspend the pellet in 0.1% BSA/PBS, and move to a new conical tube.
Note: The volume of 0.1% BSA/PBS depends on the number of organs, i.e., 4 mL for five organs.
18. Add the washed pre-coated beads to the cleaned tissue digest.
19. Incubate the suspension for 1 h at 4 °C with gentle agitation.
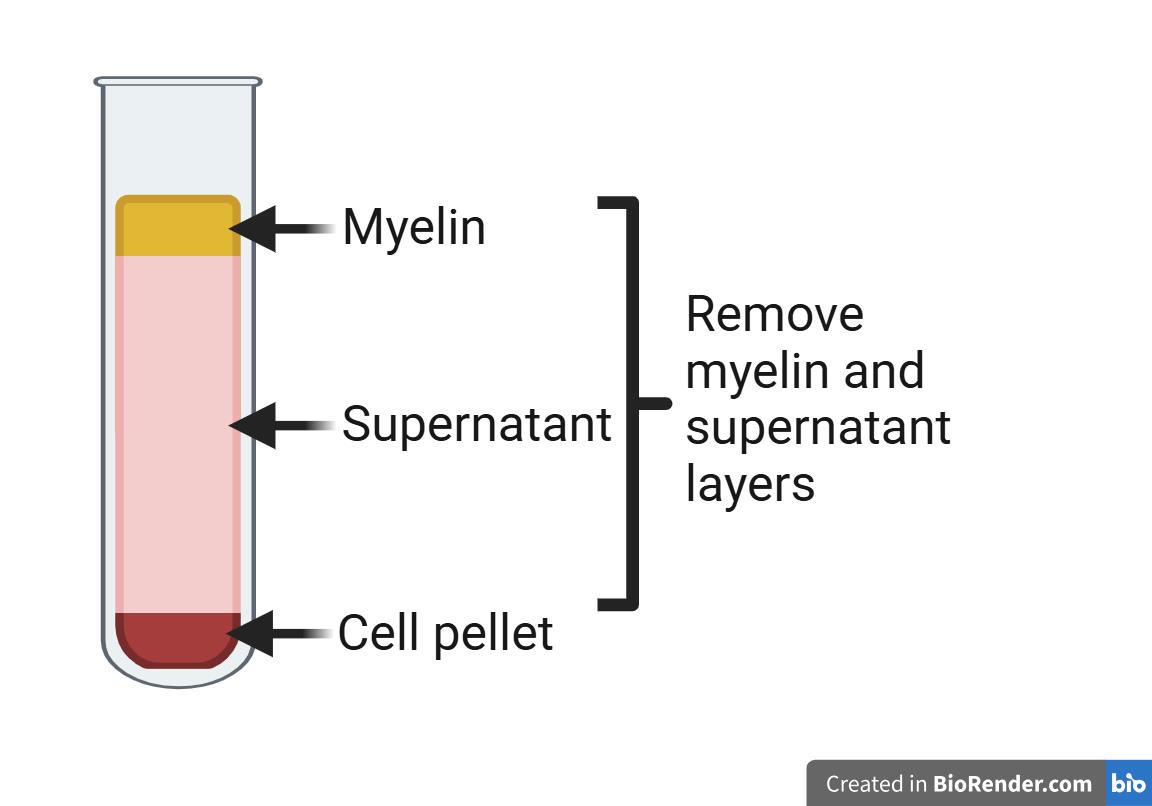
Figure 1. Schematic of myelin separation in brain digestions. Following centrifugation of brain digestions in 20% BSA in DMEM, three layers will form. At the top will be the myelin layer, in the middle the supernatant, and at the bottom a pellet containing dissociated cells and microvessel fragments. Both the myelin layer and the supernatant will be discarded, and the cell pellet will be retained for further digestion as per the protocol. Schematic made with Biorender.com.
J. Plating the pericytes
1. During the 1 h incubation at 4 °C, wash the collagen-coated plates with PBS.
2. Aliquot the bead-cell suspension in 1.5 mL microcentrifuge tubes and place them in the magnetic holder.
3. Wait 30–60 s to allow the beads to separate from the suspension; then, gently remove the suspension without disturbing the beads.
4. Without the magnet, add 1 mL of 0.1% BSA/PBS to one microcentrifuge tube and resuspend the beads.
5. Use this 1 mL volume for subsequent resuspension across the tubes, collecting all the beads into one microcentrifuge tube.
6. Return the microcentrifuge tube containing all the beads to the magnetic holder and perform two more washes with 0.1% BSA/PBS.
7. After washing, resuspend the beads in 1 mL of complete growth medium and do not return the microcentrifuge tube to the magnet.
8. Add 2 mL of complete growth medium per well of a 6-well plate that has been pre-coated with collagen and washed with PBS.
9. Split the 1 mL cell suspension between the wells of the 6-well plate so that one well represents one organ, i.e., for five organs, use five wells on the 6-well plate, adding 200 µL of the 1 mL total cell suspension per well.
Note: 6-well plates are used so that pericytes can easily make contact with neighboring cells while having enough space to expand.
10. Move the plate to the incubator with the appropriate oxygen tension (10% O2 for lung pericytes or 5% O2 for brain, bone, and liver pericytes) and do not disturb for 7 d to ensure adherence and reduce cell loss.
Note: Reference images of pericytes during expansion and after seeding for experiments can be found in the original paper [28].
K. Maintaining the pericyte cultures
1. Seven days after isolation, replace the medium on the pericyte cultures with fresh equilibrated pericyte growth medium and continue to change the medium every 2–3 d until cells are ready to be used.
Notes:
a. Pericyte colonies should be visible at this time and typically will be ready to use in experiments 10–14 d after isolation.
b. While oxygen tension is a critical factor for pericyte growth and retention of phenotype ex vivo, it is difficult to perform medium changes without transiently exposing pericytes to environmental oxygen tensions. When performing any task with pericytes outside the incubator, move as quickly as possible and always use equilibrated medium to limit exposure to non-physiological oxygen levels.
c. Pericytes are used only at passage 1 and are not cryopreserved. This is due to observations of spontaneous phenotypic switching with multiple passages.
L. Setting up experiments with pericytes
1. The pericytes are ready to be used in experiments when they become 70%–80% confluent (usually 10–14 d after isolation). Pericytes may grow in dense colonies, which makes it difficult to estimate confluence, but it would be expected to retrieve between 0.5 and 1 × 106 lung pericytes and 0.1 and 0.5 × 106 brain, liver, and bone pericytes per well of a 6-well plate.
2. Pericytes can be trypsinized with 500 µL of 0.05% trypsin-EDTA for approximately 10 min at 37 °C. Trypsin should be neutralized with 500 µL of pericyte complete growth medium.
Note: With trypsinization, beads will detach from the cells.
3. Collect and centrifuge the cell suspension at 260× g for 5 min at 4 °C.
4. Discard the supernatant and resuspend the cells in pericyte growth arrest medium.
Note: If the pericytes appear clumpy in the resuspension, pass the cell suspension through a 70 µm filter for accurate counting and seeding.
5. Count and seed pericytes onto new collagen-coated plates in pericyte growth arrest medium (Recipe 12) 24 h before experiments begin. See Table 1 for pericyte seeding densities across different experimental plate types.
6. Return the pericytes to the appropriate incubator, i.e., 10% O2 for lungs and 5% O2 for brain, bones, and liver.
Note: Pericytes are seeded in growth arrest medium for 24 h in advance of experiments to model the proliferative quiescence of pericytes in vivo.
Table 1. Pericyte seeding densities
| Plate | Lung pericyte number | Brain/bone/liver pericyte number | Medium volume |
|---|---|---|---|
| 96-well plate | 5 × 103 | 1 × 104 | 100 μL |
| 24-well plate | 5 × 104 | 5 × 104 | 250 μL |
| 12-well plate | 1 × 105 | 1 × 105 | 500 μL |
| 6-well plate | 3 × 105 | 3 × 105 | 1,000 μL |
Validation of protocol
This protocol was developed to generate a primary pericyte cell line that could be used to further interrogate the role of pericytes in both physiology and disease. Validation of the described protocol can be found in the original article [28]. Briefly, it was demonstrated that pericytes isolated from mice and cultured under physiologically relevant oxygen tensions (physioxia) retained important normal and disease-associated pericyte phenotypes. Pericyte phenotypic analysis confirmed that pericytes retained stable transcriptional and protein expression of perivascular markers (see Figures 2 and 4 from reference [28]). Spontaneous pericyte phenotypic switching in vitro was avoided by validating the culture conditions that limited expression of the plasticity factor, Klf4 (see Figure 2 from reference [28]). Pericyte function was validated in tube formation assays, where pericytes were observed to associate with endothelial junctions (see Figure 5 from reference [28]), demonstrating their role in supporting vascular structures and maintaining endothelial barrier function. To confirm that the protocol also resulted in pericytes capable of recapitulating pathological phenotypic switching, similar to that induced by tumor-derived secreted factors in the early metastatic niche, we validated that these primary cell cultures could be induced to undergo phenotypic switching. Primary lung pericytes were observed to increase expression of Klf4 and simultaneously lose expression of Myh11 and Acta2 in response to metastatic stimuli, indicating phenotypic switching representative of the in vivo response (see Figure 6 from reference [28]). Overall, the study concluded that this pericyte isolation protocol resulted in a robust model to study pericyte behavior in both health and disease.
Routine quality control of primary pericytes can be efficiently performed using quantitative real-time PCR for characteristic identity and activation genes. Pericyte identity and function could be assessed using Ng2, Myh11, and Acta2, while activation status could be evaluated using the plasticity marker Klf4. The ideal pericyte preparation would demonstrate the expression of the characteristic marker genes while maintaining low expression of the activation factor. Importantly, a reference for Klf4 should be established as a positive control. This could include pericytes treated with tumor cell–conditioned medium for a cell line such as 4T1, known to induce pericyte phenotypic switching. The details of this methodology, which could be used for quality control, are provided in the methods section of the original publication [28].
General notes and troubleshooting
General notes
1. All experimental validation was performed on pericytes used at passage 1.
2. This protocol has been optimized using 12-week-old female C57BL/6 mice but should be adaptable to variables such as mouse age, sex, or genetic background, as needed.
3. Pooling tissues from 3–6 mice is preferred to processing mice individually as it results in a much greater cell yield, allowing more flexibility in downstream experiments while avoiding the introduction of unnecessary noise.
4. Beads may remain after passaging; persistent beads do not normally interfere with cell growth or downstream experiments.
5. If necessary, collected organs may be stored up to 16 h in low-glucose DMEM at 4 °C.
6. Always use new materials and clean tools between organ digestions to prevent cross-contamination.
7. While this protocol requires controlled oxygen tensions, this can be achieved using either a dedicated tissue culture incubator (e.g., item 13 in the equipment list) or a commercially available hypoxia cabinet (e.g., item 14) that can be filled with a gas mixture and placed inside a standard incubator.
Troubleshooting
Problem 1: Low pericyte yield, i.e., less than two colonies per well.
Possible cause 1: Insufficient removal of connective tissue, indicated by difficulties in the filtering step.
Solution: Spend more time carefully cleaning the tissue to remove excess connective tissue.
Possible cause 2: BSA in solution can create bubbles, which may lead to loss of cells and beads.
Solution: Move gently during cleaning and resuspension steps to limit bubble production.
Acknowledgments
T.M. prepared the manuscript draft. T.M., C.M.B., and M.M. reviewed and edited the manuscript. This protocol was described and validated by McErlain et al. [28]. This protocol was developed and modified from endothelial cell isolation protocols reported by Reiterer et al. [29] and Sandovici et al. [32].
Competing interests
The authors declare no conflicts of interest.
Ethical considerations
All animal experiments were conducted in accordance with the guidelines and regulations outlined in the Guide for the Care and Use of Laboratory Animals and were approved by the NCI-Bethesda Animal Care and Use Committee. All procedures were in accordance with those outlined in the ARRIVE guidelines.
References
- Holm, A., Heumann, T. and Augustin, H. G. (2018). Microvascular Mural Cell Organotypic Heterogeneity and Functional Plasticity. Trends Cell Biol. 28(4): 302–316. https://doi.org/10.1016/j.tcb.2017.12.002
- Armulik, A., Genové, G. and Betsholtz, C. (2011). Pericytes: Developmental, Physiological, and Pathological Perspectives, Problems, and Promises. Dev Cell. 21(2): 193–215. https://doi.org/10.1016/j.devcel.2011.07.001
- van Splunder, H., Villacampa, P., Martínez-Romero, A. and Graupera, M. (2024). Pericytes in the disease spotlight. Trends Cell Biol. 34(1): 58–71. https://doi.org/10.1016/j.tcb.2023.06.001
- Hosaka, K., Yang, Y., Seki, T., Fischer, C., Dubey, O., Fredlund, E., Hartman, J., Religa, P., Morikawa, H., Ishii, Y., et al. (2016). Pericyte–fibroblast transition promotes tumor growth and metastasis. Proc Natl Acad Sci USA. 113(38): e1608384113. https://doi.org/10.1073/pnas.1608384113
- Yamaguchi, M., Hirai, S., Tanaka, Y., Sumi, T., Tada, M., Takahashi, H., Watanabe, A. and Sakuma, Y. (2020). Pericyte-myofibroblast transition in the human lung. Biochem Biophys Res Commun. 528(2): 269–275. https://doi.org/10.1016/j.bbrc.2020.05.091
- Nobre, A. R., Risson, E., Singh, D. K., Di Martino, J. S., Cheung, J. F., Wang, J., Johnson, J., Russnes, H. G., Bravo-Cordero, J. J., Birbrair, A., et al. (2021). Bone marrow NG2+/Nestin+ mesenchymal stem cells drive DTC dormancy via TGF-β2. Nat Cancer. 2(3): 327–339. https://doi.org/10.1038/s43018-021-00179-8
- Murgai, M., Ju, W., Eason, M., Kline, J., Beury, D. W., Kaczanowska, S., Miettinen, M. M., Kruhlak, M., Lei, H., Shern, J. F., et al. (2017). KLF4-dependent perivascular cell plasticity mediates pre-metastatic niche formation and metastasis. Nat Med. 23(10): 1176–1190. https://doi.org/10.1038/nm.4400
- Sengillo, J. D., Winkler, E. A., Walker, C. T., Sullivan, J. S., Johnson, M. and Zlokovic, B. V. (2012). Deficiency in Mural Vascular Cells Coincides with Blood–Brain Barrier Disruption in Alzheimer's Disease. Brain Pathol. 23(3): 303–310. https://doi.org/10.1111/bpa.12004
- Cuervo, H., Pereira, B., Nadeem, T., Lin, M., Lee, F., Kitajewski, J. and Lin, C. S. (2017). PDGFRβ-P2A-CreERT2 mice: a genetic tool to target pericytes in angiogenesis. Angiogenesis. 20(4): 655–662. https://doi.org/10.1007/s10456-017-9570-9
- Guimarães-Camboa, N., Cattaneo, P., Sun, Y., Moore-Morris, T., Gu, Y., Dalton, N. D., Rockenstein, E., Masliah, E., Peterson, K. L., Stallcup, W. B., et al. (2017). Pericytes of Multiple Organs Do Not Behave as Mesenchymal Stem Cells In Vivo. Cell Stem Cell. 20(3): 345–359.e5. https://doi.org/10.1016/j.stem.2016.12.006
- Zhu, X., Hill, R. A., Dietrich, D., Komitova, M., Suzuki, R. and Nishiyama, A. (2011). Age-dependent fate and lineage restriction of single NG2 cells. Development. 138(4): 745–753. https://doi.org/10.1242/dev.047951
- Dias Moura Prazeres, P. H., Sena, I. F. G., Borges, I. d. T., de Azevedo, P. O., Andreotti, J. P., de Paiva, A. E., de Almeida, V. M., de Paula Guerra, D. A., Pinheiro dos Santos, G. S., Mintz, A., et al. (2017). Pericytes are heterogeneous in their origin within the same tissue. Dev Biol. 427(1): 6–11. https://doi.org/10.1016/j.ydbio.2017.05.001
- Hartmann, D. A., Coelho-Santos, V. and Shih, A. Y. (2022). Pericyte Control of Blood Flow Across Microvascular Zones in the Central Nervous System. Annu Rev Physiol. 84(1): 331–354. https://doi.org/10.1146/annurev-physiol-061121-040127
- Crisan, M., Yap, S., Casteilla, L., Chen, C. W., Corselli, M., Park, T. S., Andriolo, G., Sun, B., Zheng, B., Zhang, L., et al. (2008). A Perivascular Origin for Mesenchymal Stem Cells in Multiple Human Organs. Cell Stem Cell. 3(3): 301–313. https://doi.org/10.1016/j.stem.2008.07.003
- Birbrair, A., Borges, I. d. T., Gilson Sena, I. F., Almeida, G. G., da Silva Meirelles, L., Gonçalves, R., Mintz, A. and Delbono, O. (2017). How Plastic Are Pericytes?. Stem Cells Dev. 26(14): 1013–1019. https://doi.org/10.1089/scd.2017.0044
- Birbrair, A., Zhang, T., Wang, Z. M., Messi, M. L., Enikolopov, G. N., Mintz, A. and Delbono, O. (2013). Role of Pericytes in Skeletal Muscle Regeneration and Fat Accumulation. Stem Cells Dev. 22(16): 2298–2314. https://doi.org/10.1089/scd.2012.0647
- Li, P., Wu, Y., Goodwin, A. J., Halushka, P. V., Wilson, C. L., Schnapp, L. M. and Fan, H. (2021). Generation of a new immortalized human lung pericyte cell line: a promising tool for human lung pericyte studies. Lab Invest. 101(5): 625–635. https://doi.org/10.1038/s41374-020-00524-y
- Dzau, V. J., Braun-Dullaeus, R. C. and Sedding, D. G. (2002). Vascular proliferation and atherosclerosis: New perspectives and therapeutic strategies. Nat Med. 8(11): 1249–1256. https://doi.org/10.1038/nm1102-1249
- Faal, T., Phan, D. T., Davtyan, H., Scarfone, V. M., Varady, E., Blurton-Jones, M., Hughes, C. C. and Inlay, M. A. (2019). Induction of Mesoderm and Neural Crest-Derived Pericytes from Human Pluripotent Stem Cells to Study Blood-Brain Barrier Interactions. Stem Cell Rep. 12(3): 451–460. https://doi.org/10.1016/j.stemcr.2019.01.005
- Ghaedi, M., Niklason, L. E. (2016). Organoids, Stem Cells, Structure, and Function. Methods Mol Biol. 1576: 55–92. https://doi.org/10.1007/7651_2016_11
- Alvino, V. V., Mohammed, K. A. K., Gu, Y. and Madeddu, P. (2023). Approaches for the isolation and long-term expansion of pericytes from human and animal tissues. Front Cardiovasc Med. 9: e1095141. https://doi.org/10.3389/fcvm.2022.1095141
- Lee, L. L., Khakoo, A. Y. and Chintalgattu, V. (2019). Isolation and Purification of Murine Cardiac Pericytes. J Visualized Exp. 150: e59571. https://doi.org/10.3791/59571
- Chen, C., Tang, Q., Zhang, Y., Yu, M., Jing, W. and Tian, W. (2018). Physioxia: a more effective approach for culturing human adipose-derived stem cells for cell transplantation. Stem Cell Res Ther 9(1): 148. https://doi.org/10.1186/s13287-018-0891-4
- D'Ippolito, G., Diabira, S., Howard, G. A., Roos, B. A. and Schiller, P. C. (2006). Low oxygen tension inhibits osteogenic differentiation and enhances stemness of human MIAMI cells. Bone. 39(3): 513–522. https://doi.org/10.1016/j.bone.2006.02.061
- Holzwarth, C., Vaegler, M., Gieseke, F., Pfister, S. M., Handgretinger, R., Kerst, G. and Müller, I. (2010). Low physiologic oxygen tensions reduce proliferation and differentiation of human multipotent mesenchymal stromal cells. BMC Cell Biol. 11(1): 11. https://doi.org/10.1186/1471-2121-11-11
- Kim, D. S., Ko, Y. J., Lee, M. W., Park, H. J., Park, Y. J., Kim, D. I., Sung, K. W., Koo, H. H. and Yoo, K. H. (2016). Effect of low oxygen tension on the biological characteristics of human bone marrow mesenchymal stem cells. Cell Stress Chaperones. 21(6): 1089–1099. https://doi.org/10.1007/s12192-016-0733-1
- Ortiz-Prado, E., Dunn, J. F., Vasconez, J., Castillo, D. and Viscor, G. (2018). Partial pressure of oxygen in the human body: a general review. Am J Blood Res. 9:1–14. https://pubmed.ncbi.nlm.nih.gov/30899601/
- McErlain, T., McCulla, E. C., Glass, M. J., Ziemer, L. E., Branco, C. M. and Murgai, M. (2024). Pericytes require physiological oxygen tension to maintain phenotypic fidelity. Sci Rep. 14(1): 29581 https://doi.org/10.1038/s41598-024-80682-x
- Reiterer, M., Eakin, A., Johnson, R. S. and Branco, C. M. (2022). Hyperoxia Reprogrammes Microvascular Endothelial Cell Response to Hypoxia in an Organ-Specific Manner. Cells. 11(16): 2469. https://doi.org/10.3390/cells11162469
- Murfee, W. L., Skalak, T. C. and Peirce, S. M. (2005). Differential Arterial/Venous Expression of NG2 Proteoglycan in Perivascular Cells Along Microvessels: Identifying a Venule‐Specific Phenotype. Microcirculation. 12(2): 151–160. https://doi.org/10.1080/10739680590904955
- Kishida, N., Maki, T., Takagi, Y., Yasuda, K., Kinoshita, H., Ayaki, T., Noro, T., Kinoshita, Y., Ono, Y., Kataoka, H., et al. (2019). Role of Perivascular Oligodendrocyte Precursor Cells in Angiogenesis After Brain Ischemia. J Am Heart Assoc. 8(9): e011824. https://doi.org/10.1161/jaha.118.011824
- Sandovici, I., Reiterer, M., Constância, M. and Branco, C. M. (2022). Protocol to isolate and culture primary mouse feto-placental endothelial cells. STAR Protoc. 3(4): 101721. https://doi.org/10.1016/j.xpro.2022.101721
- D’Agostino, M. R., Afkhami, S., Kang, A., Marzok, A., Miller, M. S. and Xing, Z. (2022). Protocol for isolation and characterization of lung tissue resident memory T cells and airway trained innate immunity after intranasal vaccination in mice. STAR Protoc. 3(3): 101652. https://doi.org/10.1016/j.xpro.2022.101652
- Charni-Natan, M. and Goldstein, I. (2020). Protocol for Primary Mouse Hepatocyte Isolation. STAR Protoc. 1(2): 100086. https://doi.org/10.1016/j.xpro.2020.100086
- Amend, S. R., Valkenburg, K. C. and Pienta, K. J. (2016). Murine Hind Limb Long Bone Dissection and Bone Marrow Isolation. J Visualized Exp. 110: e53936. https://doi.org/10.3791/53936
- MacManus, D. B. (2023). Excision of whole intact mouse brain. MethodsX. 10: 102246. https://doi.org/10.1016/j.mex.2023.102246
Article Information
Publication history
Received: Feb 6, 2025
Accepted: Mar 23, 2025
Available online: Apr 7, 2025
Published: Apr 20, 2025
Copyright
© 2025 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
McErlain, T., Branco, C. M. and Murgai, M. (2025). Isolation and Culture of Primary Pericytes from Mouse. Bio-protocol 15(8): e5288. DOI: 10.21769/BioProtoc.5288.
Category
Cancer Biology > Invasion & metastasis > Cell biology assays > Cell isolation and culture
Cell Biology > Cell isolation and culture > Cell isolation > Dynabead
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.