- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Ex Vivo Measurement of Stable Isotope-Labeled Fatty Acid Incorporation Into Phospholipids in Isolated Mice Muscle
Published: Vol 15, Iss 7, Apr 5, 2025 DOI: 10.21769/BioProtoc.5257 Views: 1905
Reviewed by: Chiara AmbrogioOm Prakash NarayanIstvan Stadler
Original research article
The authors used this protocol in:

May 2023
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
With the advancement of liquid chromatography–mass spectrometry (LC–MS/MS), the quantification of glycerophospholipid (PL) molecules has become more accessible, leading to the discovery of numerous enzymes responsible for determining the acyl groups attached to these molecules. Metabolic tracer experiments using radioisotopes and stable isotopes are powerful tools for defining the function of metabolic enzymes and metabolic flux. We have established an ex vivo muscle experimental system using stable isotope–labeled fatty acids to evaluate fatty acid incorporation into PL molecules. Here, we describe a method to incorporate fatty acids with stable isotope labels into excised skeletal muscle and detect the PL molecules containing labeled acyl chains by LC–MS/MS.
Key features
• Quantify the metabolism of fatty acids into phospholipid acyl chains.
• Enable measurements in excised muscle samples.
• Assess the effects of genetic recombination of acyltransferases.
Keywords: Stable isotope tracerGraphical overview
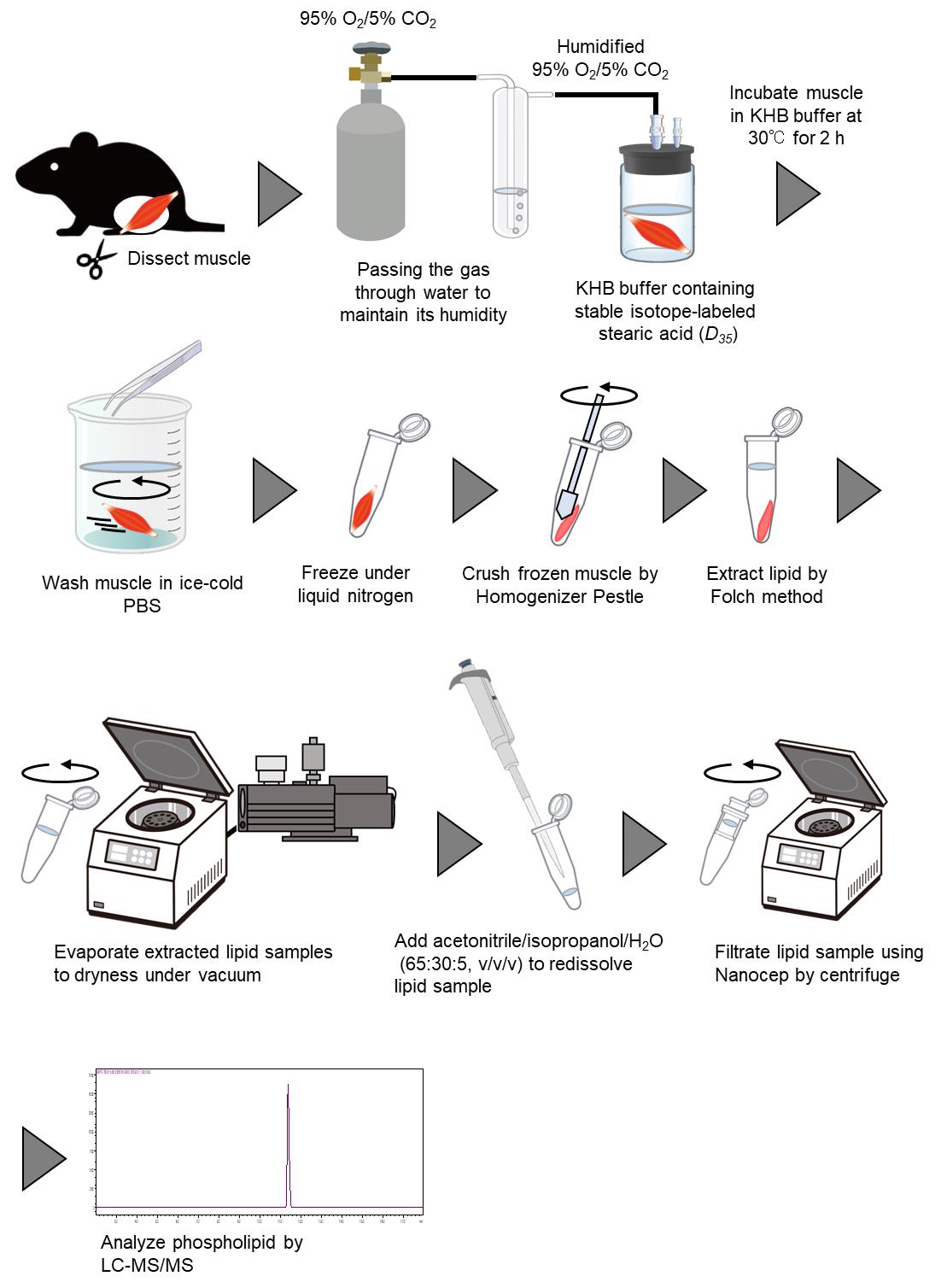
Ex vivo measurement of stable isotope–labeled fatty acid incorporation into phospholipids in isolated mice muscle
Background
Glycerophospholipids (PL) are major constituents of cellular membranes, containing two acyl chains and a polar head. Numerous varieties of PL molecules exist depending on the combination of acyl-chain species and polar heads. Various types of PLs, such as phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidylserine (PS), phosphatidylinositol (PI), and phosphatidylglycerol (PG), are synthesized from glycerol-3-phosphate (G3P) via the de novo pathway (Kennedy pathway) [1]. Acyl-chain species bound to PLs produced in the Kennedy pathway are remodeled by phospholipases and lysophospholipid acyltransferases (LPLATs) [2]. This remodeling pathway, called the Lands’ cycle, is essential for generating PL species diversity and asymmetric distribution of acyl chains [3].
Acyl chain species in PL molecules were traditionally quantified by the measurement of extracted fatty acids using gas chromatography–mass spectrometry (GC–MS) after hydrolyzation of PLs. Thus, measuring individual PL molecules one by one was challenging. In recent years, advancements in lipid molecule separation technology by liquid chromatography (LC) and improved detection sensitivity of MS have made it possible to measure lipid molecules exceeding 5,000 species [4,5]. Mass spectrometers can detect isotope peaks, enabling their use in combination with stable isotopes such as deuterium (2H) and 13C for tracking specific compounds in experiments. Tracking PL remodeling using deuterium-labeled palmitic acid (16:0-D31-FFA) and stearic acid (18:0-D35-FFA) has been reported in cultured cell experiments [6]. However, examples of in vivo or ex vivo PL metabolism tracer experiments are limited, highlighting the need for the development of new methods. Typically, metabolic tracers for carbohydrates and lipids in skeletal muscle are evaluated by incubating isolated skeletal muscle with radiolabeled substrates and measuring the radioactivity of the extracted components [7,8]. However, experiments using radiolabeled compounds cannot measure PL molecules with defined bound acyl group molecular species, as is possible with LC–MS/MS measurements, even though thin-layer chromatography can be used to measure the radioactivity of each PL class, such as PC and PE, with high sensitivity. Therefore, PLs labeled with stable isotopes need to be quantified by mass spectrometry. In addition, ex vivo studies allow organ-specific assessment and reduce the use of stable isotope compounds compared to in vivo studies. Satellite cells isolated from mice differentiate into a mixture of fast and slow muscle fibers, making fast and slow muscle-specific analysis difficult in an in vitro experimental system. An ex vivo experimental system using fast- and slow-twitch muscle excised from mice allows the evaluation of skeletal muscle-specific phospholipid metabolism and overcomes the above problems.
This article provides a detailed method for measuring the incorporation of stable isotope–labeled fatty acids into PLs in isolated skeletal muscle. Although the remodeling of the acyl group in PL measured by this protocol has been validated in a specific LPLAT (KO) model, a limitation of this protocol is that the contribution of Kennedy pathway cannot be ignored. Although the methods described in this article are based on excised skeletal muscle, direct injection of stable-isotope compounds into skeletal muscle may be applicable for in vivo evaluation.
Materials and reagents
Biological materials
1. C57BL/6J mice at 8–12 weeks old
Note: At least five mice are recommended.
Reagents
1. Stearic acid (D35, 98%) (Cambridge Isotope Laboratories, catalog number: DLM-379-1)
2. Fatty acid–free bovine serum albumin (BSA) (Wako, catalog number: 013-25773)
3. Chloroform (Wako, catalog number: 038-02601)
4. Methanol (Wako, catalog number: 134-14523)
5. 1, 2-diheptadecanoyl-sn-glycero-3-phosphocholine (17:0-PC) (Avanti Polar Lipids, catalog number: 850360)
6. 1, 2-dilauroyl-sn-glycero-3-phosphoethanolamine (12:0-PE) (Avanti Polar Lipids, catalog number: 850702)
Note: Dissolve 17:0-PC and 12:0-PE in a chloroform/methanol mixture (2:1) to a final concentration of 10 mg/mL. After dissolution, store the solution at -30 °C or below to prevent volatilization of the organic solvents.
7. Butylhydroxytoluene (BHT) (Wako, catalog number: 029-07392)
8. 2-Propanol (Wako, catalog number: 164-25533)
9. Ultrapure water (Wako, catalog number: 210-01303)
10. Acetonitrile (Wako, catalog number: 018-19853)
11. Ammonium formate (Sigma-Aldrich, catalog number: 70221)
12. Formic acid (Wako, catalog number: 067-04531)
13. Sodium chloride (NaCl) (Wako, catalog number: 191-01665)
14. Potassium chloride (KCl) (Wako, catalog number: 163-03545)
15. Calcium chloride dihydrate (CaCl2·2H2O) (Wako, catalog number: 039-00431)
16. Potassium dihydrogen phosphate (KH2PO4) (Wako, catalog number: 169-04245)
17. Magnesium sulfate heptahydrate (MgSO4·7H2O) (Wako, catalog number: 131-00405)
18. Glucose (Wako, catalog number: 049-31165)
19. Sodium hydrogen carbonate (NaHCO3) (Wako, catalog number: 191-01305)
Solutions
1. Krebs–Henseleit buffer (KHB) (see Recipes)
2. Mobile phase A (see Recipes)
3. Mobile phase B (see Recipes)
Recipes
1. 1 L Krebs–Henseleit buffer (KHB)
| Reagent | Final concentration (mM) | Quantity (g) |
|---|---|---|
| NaCl | 125 | 7.283 |
| KCl | 5.3 | 0.395 |
| CaCl2·2H2O | 1.9 | 0.279 |
| KH2PO4 | 1.3 | 0.171 |
| MgSO4·7H2O | 1.3 | 0.309 |
| Glucose | 5 | 0.9 |
Note: A NaHCO3 stock solution prepared to a final concentration of 1.3% and saturated with CO2 gas (sealed with liquefied CO2 for 1 h) can be used for up to one month when stored at 4 °C in a sealed container.
Caution: As various organic solvents are used for lipid extraction and as LC–MS mobile phase, they should be handled in a safety cabinet to prevent inhalation.
Caution: The KHB buffer should be used within one week. KHB buffer containing NaHCO3, BSA, and stable isotope–labeled stearic acid (D35) should only be used on the day of preparation.
2. Mobile phase A
H2O/acetonitrile (60:40, v/v)
10 mM ammonium formate
0.1% formic acid
3. Mobile phase B
Isopropanol/acetonitrile (90:10, v/v)
10 mM ammonium formate
0.1% formic acid
Laboratory supplies
1. 20 mL glass reaction vial (IWAKI, catalog number: 1890CV20)
2. Homogenizer pestle (BIO-BIK, catalog number: 1005-39)
3. Nanocep with 0.45 μm wwPTFE (PALL, catalog number: ODPTEF04C35)
4. SecurityGuard Guard Cartridge kit (Phenomenex, catalog number: KJ0-4282)
5. Accucore RP-MS column (2.6 μm, 2.1 mm × 50 mm) (Thermo ScientificTM, catalog number: 17626-012105)
Equipment
1. Anesthetic apparatus for small animals (Shinano, model: SN-487-0T AS1)
2. Thermostatic water bath (Thermal Chemical, model: DB-42)
3. Centrifugal evaporator (EYELA, model: CVE-3000)
4. Ultrasonic bath (BRANSON, model: Yamato1510)
5. Triple quadrupole mass spectrometer (Shimadzu, model: LCMS-8040)
6. Binary pump (Shimadzu, model: LC-30AD)
7. Auto sampler (Shimadzu, model: SIL-30AC)
8. Column oven (Shimadzu, model: CTO-20AC)
9. N2 gas supplier (ANEST IWATA, model: 24FD)
10. EM oil-sealed rotary vane pumps (Edwards, model: E2M28)
Software
1. LabSolutions LCMS (Shimadzu, version 5.65)
Procedure
A. Preparation of stable isotope–containing KHB
1. Dissolve stable isotope–labeled stearic acid (D35) stock in 100% EtOH. (Stock fatty acid concentration=250 mM.)
2. Prepare KHB with 4% BSA and 1.25 mM stable isotope–labeled stearic acid (D35).
3. Add 1.3% NaHCO3 solution saturated with CO2 gas to the KHB prepared in step A2. (Add 2 mL of 1.3% NaHCO3 solution to 10.5 mL of KHB.)
4. Dispense 2.5 mL of KHB into a 20 mL glass reaction vial (Figure 1A).
5. Keep the solution at 30 °C in a water bath while ventilating the vial with humidified 95% O2/5% CO2 gas for at least 30 min (Figure 1B, C).

Figure 1. Photographs of experimental equipment. (A) Glass vial with rubber closure with gas inlet and outlet. (B) Overview of the 95% O/5% CO gas supply pathway. (C) Water bath and glass tube that can supply humidified 95% O2/5% CO2 gas into each glass reaction vial.
B. Incubation of muscle sample with KHB under 95% O2/5% CO2 gas
1. Anesthetize the mouse with 2%–5% isoflurane in oxygen.
Caution: When performing inhalation anesthesia, measures should be taken to prevent leakage to the environment by using adsorption devices or draft chambers.
2. To minimize suffering, euthanize mice by cervical dislocation after rendering them comatose under anesthesia.
3. Excise the extensor digitorum longus (EDL) and soleus muscles and weigh them.
Critical: When skeletal muscle is dissected, the tendons are cut to avoid unnecessary incisions in the muscle tissue. Two pieces of EDL and soleus muscle (approximately 20 mg each) are used for analysis.
4. After rinsing in 0.9% sodium chloride solution, place muscles in KHB filled with humidified 95% O2/5% CO2 gas as prepared in section A (Figure 1A).
Critical: Place the skeletal muscle sample as quickly as possible in a vial that has been ventilated with humidified 95% O2/5% CO2 gas (step A5) and immediately resume ventilation.
5. Incubate muscles in KHB for 2 h at 30 °C (Figure 1C).
6. Take the muscles from the KHB and wash them in ice-cold 0.9% sodium chloride solution.
7. Place muscles in 1.5 mL centrifuge tubes.
8. To freeze skeletal muscles, place the 1.5 mL tubes containing the samples in liquid nitrogen until completely frozen (freezing for approximately 1 min is sufficient).
Note: Frozen muscle samples can be stored at -80 °C until the next procedural step.
C. Lipid extraction from muscle
1. Powder frozen muscle using a homogenizer pestle.
Critical: Skeletal muscle is completely pulverized to ensure efficient lipid extraction. If grinding with a pestle homogenizer is difficult, use a mortar and pestle to grind the frozen skeletal muscle.
Caution: Although it is possible to grind skeletal muscle in an organic solvent using a Polytron homogenizer, the heat causes the organic solvent to evaporate, and the skeletal muscle may adhere to the shaft, resulting in sample loss. Therefore, crushing frozen skeletal muscle is the recommended method.
2. To prepare the extraction solvent, mix chloroform and methanol (2:1, v/v) and add butylhydroxytoluene (final concentration 0.2 mg/mL) and 17:0-PC and 12:0-PE (final concentration 0.5 μg/mL).
Note: 17:0-PC and 12:0-PE are used as internal standards to correct the measurement results.
3. Extract total lipid from the powdered muscles overnight with the extraction solvent (use 1 mL of extraction solvent for each two pieces of EDL and soleus muscle sample).
4. Centrifuge at 10,000× g for 10 min at room temperature (RT) to precipitate the skeletal muscle residue and transfer the supernatant to a new 1.5 mL tube.
5. Evaporate the entire lipid extraction solvent to dryness under vacuum.
6. Redissolve with 200 μL of acetonitrile/isopropanol/H2O (65:30:5, v/v/v) as the same volume of the lipid extracted solvent used for evaporation.
7. Dissolve the lipid sample in an ultrasonic bath if necessary.
8. Filtrate reconstituted lipid sample by Nanocep with 0.45 μm wwPTFE centrifuge filter (7,500× g, 10 min, RT).
9. Store filtrated lipid samples at -80 °C until analysis.
D. Quantification of PC- and PE-containing, stable isotope–labeled stearic acid by LC–MS/MS
1. Perform the LC separation with an Accucore RP-MS column (2.6 μm, 2.1 mm × 50 mm) under the specific conditions described below:
Sample injection: 40 μL.
Flow rate: 0.4 mL/min.
Gradient conditions: 10% mobile phase B (see Recipes for the composition of mobile phases A and B) at 0 min, 10% B at 2 min, 100% B at 15 min, 10% B at 15.01 min, and 10% B at 20 min (Table 1).
Column temperature: 40 °C.
Table 1. Gradient run used for LC–MS/MS analyses
| Time (min) | Mobile phase A (%) | Mobile phase B (%) |
| 0 | 90 | 10 |
| 2 | 90 | 10 |
| 15 | 0 | 100 |
| 15.01 | 90 | 10 |
| 20 | 90 | 10 |
2. Set the MS equipment as described below:
Nebulizer gas flow: 3.0 L/min.
Drying gas flow: 15.0 L/min.
Desolvation line temperature: 250 °C.
Heat-block temperature: 400 °C.
Collision-induced dissociation (CID) gas: 230 kPa.
3. Analyze stable isotope–labeled PC and PE using multiple reaction monitoring (MRM) in negative ionization mode. The precursor ion for PC and PE species containing stable isotope–labeled stearic acid (D35) is set to Q1, and the detection of fatty acid fragments is set to Q3. The MRM conditions are listed in Table 2.
Table 2. MRM transitions used for LC–MS/MS analyses for PC and PE species containing stable isotope–labeled stearic acid (D35)
| Species | Q1 m/z | Q3 m/z | Collision energy (CE) |
|---|---|---|---|
| PC (17:0/17:0) | 806.6 | 269.2 | 35 |
| PE (12:0/12:0) | 578.4 | 199.2 | 35 |
| PC (18:0-D35-22:6) | 913.6 | 318.3 | 35 |
| PE (18:0-D35-22:6) | 825.5 | 318.3 | 35 |
4. Quantify the peak area of PC (18:0-D35-22:6) and PE (18:0-D35-22:6) using the software LabSolutions LCMS.
5. Normalize the obtained peak area of PC (18:0-D35-22:6) and PE (18:0-D35-22:6) against the peak area of the internal standard (17:0-PC and 12:0-PE) and muscle weight (Figure 2).
Caution: Prepare a blank sample without incorporation of the stable isotope–labeled stearic acid (D35) to confirm the specificity of the results.
At MS1, [M+HCOO]- ions for PC and [M-H]- ions for PE were selected. At MS3, the m/z values of 318.3 were selected for species containing stable isotope–labeled stearic acid (D35).
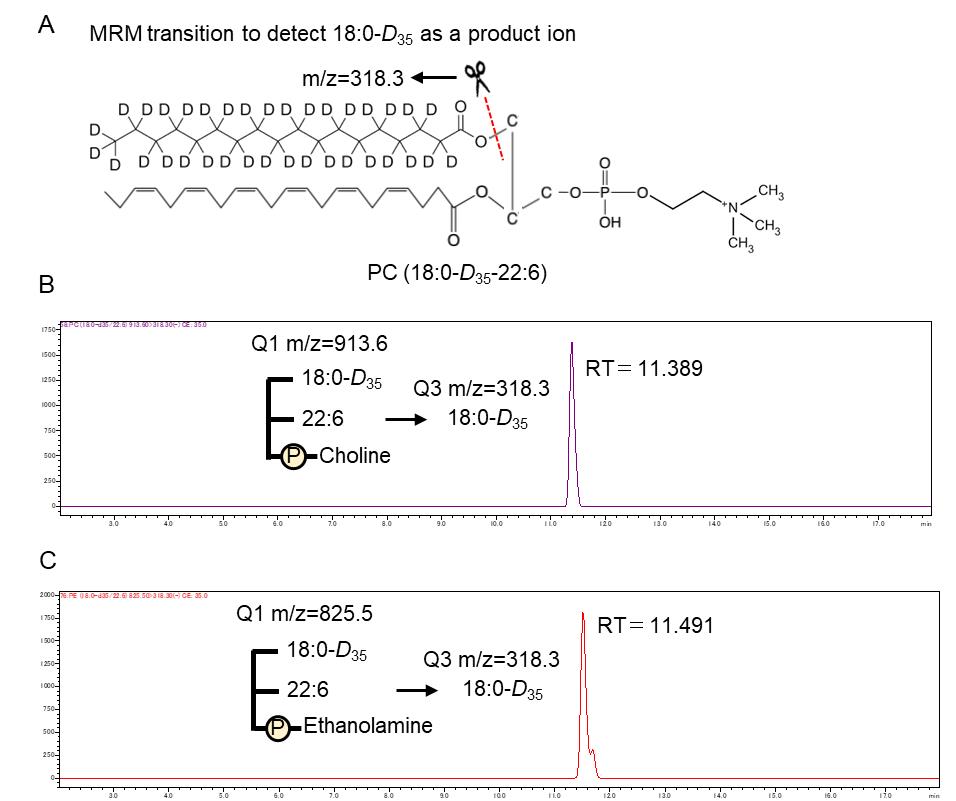
Figure 2. Multiple reaction monitoring (MRM) event of phospholipids containing stable isotope–labeled stearic acid (D35) and chromatograph. (A) Structural formula of phosphatidylcholine (PC) (18:0-D35-22:6). (B and C) Mass chromatogram obtained using MRM (m/z 913.60>318.30) for PC (18:0-D35-22:6) and MRM (m/z 825.50>318.30) for phosphatidylethanolamine (PE) (18:0-D35/22:6).
Validation of protocol
The authors evaluated mice deficient in LPLAT7, which is responsible for incorporating stearoyl-CoA into lysophospholipids in the remodeling pathway, and confirmed a reduction in PLs-containing isotope-labeled stearic acid (D35) in LPLAT7-deficient mice (Figure 3). This protocol was adapted from a previous study [9].
This protocol or parts of it has been used and validated in the following research article(s):
Sato et al. [9] LPGAT1/LPLAT7 regulates acyl chain profiles at the sn-1 position of phospholipids in murine skeletal muscles. J Biol Chem (Figure 5, panels E and F).

Figure 3. Quantification of phosphatidylcholine (PC) (18:0-D35-22:6) and phosphatidylethanolamine (PE) (18:0-D35-22:6). (A) Schematic representation of stearoyl-CoA incorporation into lysophospholipids by lysophospholipid acyltransferase (LPLAT) 7. (B) Extensor digitorum longus (EDL) and soleus (SOL) were treated with 1.25 mM of stable isotope–labeled stearic acid (D35). The levels of PC (18:0-D35-22:6) and PE (18:0-D35-22:6) were measured using LC–MS/MS. fl/fl=LPLAT7flox/flox mice; cKO=LPLAT7 cKO mice.
General notes and troubleshooting
General notes
1. The detection sensitivity in LC–MS analysis is significantly reduced due to ion suppression, as the quantity of PL molecules containing the stable isotope is extremely low compared to intravital PLs. Since the measurement is performed in negative ion mode, the detection peak is notably weaker than in positive ion mode; however, detection with high specificity is still achievable.
2. This method cannot distinguish between the remodeling pathway and the de novo pathway.
3. If you are using an instrument different from the mass spectrometer described here, you will need to optimize settings such as nebulizer gas flow, drying gas flow, desolvation line temperature, heat-block temperature, collision-induced dissociation (CID) gas, and collision energy for accurate measurements.
Troubleshooting
Problem 1: Low signal intensity of PC and PE containing stable isotope–labeled stearic acid (D35) in MS/MS analysis.
Possible cause: Insufficient volume of stable isotope–labeled stearic acid (D35) during sample incubation.
Solution: Increase the amount of stable isotope–labeled stearic acid (D35) used for sample incubation. Detection of PC and PE containing stable isotope–labeled stearic acid (D35) was not achieved at a concentration of 1.25 mM. However, increasing the concentration to 2.5 mM was sufficient for successful detection.
Problem 2: Low efficiency in the uptake of stable isotope–labeled stearic acid (D35) into skeletal muscle during incubation.
Possible cause: Damage to skeletal muscle during dissection and/or insufficient incubation time.
Solution: Handle skeletal muscle samples with care during dissection to avoid unnecessary damage and extend the incubation time. Extending the incubation period by 1 h, compared to the original radioisotope tracer protocol, improved uptake efficiency.
Acknowledgments
This work was supported by the Grant-in-Aid for Scientific Research (KAKENHI) (grant number 21K21224) from the Japanese Ministry of Education; the Uehara Memorial Foundation (Tokyo, Japan) (to TS); Council for Science, Technology and Innovation (CSTI), Cross-Ministerial Strategic Innovation Promotion Program (SIP, No. 14533567); “Technologies for creating next-generation agriculture, forestry and fisheries,” funded by the Bio-oriented Technology Research Advancement Institution, NARO; and a Grant-in-Aid for Scientific Research (KAKENHI) (grant numbers 19K22814 and 21H03362) from the Japanese Ministry of Education, Culture, Sports, Science and Technology (MEXT, Tokyo) (to S.M.). This method was developed by modifying the radioisotope tracer experimental protocol described by Clinton R. Bruce [7].
Specific contributions of each author
Conceptualization, S.M.; Investigation, T.S.; Writing-Original Draft, T.S.; Writing-Review & Editing, S.M.; Funding acquisition, T.S., and S.M.; Supervision, S.M.
Competing interests
The authors declare no conflict of interest.
Ethical considerations
This protocol for animal care and use was approved by the Institutional Animal Care and Use Committee of the University of Shizuoka (number 165122).
References
- Kennedy, E. P. and Weiss, S. B. (1956). The Function of Cytidine Coenzymes in the Biosynthesis of Phospholipides. J Biol Chem. 222(1): 193–214. https://doi.org/10.1016/s0021-9258(19)50785-2
- Shindou, H., Hishikawa, D., Harayama, T., Eto, M. and Shimizu, T. (2013). Generation of membrane diversity by lysophospholipid acyltransferases. J Biochem. 154(1): 21–28. https://doi.org/10.1093/jb/mvt048
- Lands, W. E. (1958). Metabolism of Glycerolipides: A Comparison of Lecithin and Triglyceride Synthesis. J Biol Chem. 231(2): 883–888. https://doi.org/10.1016/s0021-9258(18)70453-5
- Harayama, T. and Riezman, H. (2018). Understanding the diversity of membrane lipid composition. Nat Rev Mol Cell Biol. 19(5): 281–296. https://doi.org/10.1038/nrm.2017.138
- Tsugawa, H., Ikeda, K., Takahashi, M., Satoh, A., Mori, Y., Uchino, H., Okahashi, N., Yamada, Y., Tada, I., Bonini, P., et al. (2020). A lipidome atlas in MS-DIAL 4. Nat Biotechnol. 38(10): 1159–1163. https://doi.org/10.1038/s41587-020-0531-2
- Kawana, H., Ozawa, M., Shibata, T., Onishi, H., Sato, Y., Kano, K., Shindou, H., Shimizu, T., Kono, N., Aoki, J., et al. (2022). Identification and characterization of LPLAT7 as an sn-1-specific lysophospholipid acyltransferase. J Lipid Res. 63(10): 100271. https://doi.org/10.1016/j.jlr.2022.100271
- Bruce, C. R., Brolin, C., Turner, N., Cleasby, M. E., van der Leij, F. R., Cooney, G. J. and Kraegen, E. W. (2007). Overexpression of carnitine palmitoyltransferase I in skeletal muscle in vivo increases fatty acid oxidation and reduces triacylglycerol esterification. Am J Physiol Endocrinol Metab. 292(4): E1231–E1237. https://doi.org/10.1152/ajpendo.00561.2006
- Miura, S., Kai, Y., Kamei, Y., Bruce, C. R., Kubota, N., Febbraio, M. A., Kadowaki, T. and Ezaki, O. (2009). α2-AMPK activity is not essential for an increase in fatty acid oxidation during low-intensity exercise. Am J Physiol Endocrinol Metab. 296(1): E47–E55. https://doi.org/10.1152/ajpendo.90690.2008
- Sato, T., Umebayashi, S., Senoo, N., Akahori, T., Ichida, H., Miyoshi, N., Yoshida, T., Sugiura, Y., Goto-Inoue, N., Kawana, H., et al. (2023). LPGAT1/LPLAT7 regulates acyl chain profiles at the sn-1 position of phospholipids in murine skeletal muscles. J Biol Chem. 299(7): 104848. https://doi.org/10.1016/j.jbc.2023.104848
Article Information
Publication history
Received: Dec 25, 2024
Accepted: Feb 27, 2025
Available online: Mar 13, 2025
Published: Apr 5, 2025
Copyright
© 2025 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Sato, T. and Miura, S. (2025). Ex Vivo Measurement of Stable Isotope-Labeled Fatty Acid Incorporation Into Phospholipids in Isolated Mice Muscle. Bio-protocol 15(7): e5257. DOI: 10.21769/BioProtoc.5257.
- Sato, T., Umebayashi, S., Senoo, N., Akahori, T., Ichida, H., Miyoshi, N., Yoshida, T., Sugiura, Y., Goto-Inoue, N., Kawana, H., et al. (2023). LPGAT1/LPLAT7 regulates acyl chain profiles at the sn-1 position of phospholipids in murine skeletal muscles. J Biol Chem. 299(7): 104848. https://doi.org/10.1016/j.jbc.2023.104848
Category
Biochemistry > Lipid > Membrane lipid
Cell Biology > Cell metabolism > Lipid
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.