- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

A Highly Sensitive and Selective DAMP Assay for the Detection of Bacterial Pathogens Associated With Brain Inflammation
Published: Vol 15, Iss 6, Mar 20, 2025 DOI: 10.21769/BioProtoc.5240 Views: 1887
Reviewed by: Anupama SinghSrestha DasguptaDebashis Dutta
Original research article
The authors used this protocol in:

Jul 2024
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
The early detection of meningitis pathogens—including Haemophilus influenzae, Neisseria meningitidis, Streptococcus pneumoniae, and Klebsiella pneumoniae—through point-of-care (POC) systems is essential for mitigating the risk of neurological damage, enhancing patient outcomes, and facilitating prompt clinical decision-making. Nucleic acid amplification testing (NAAT) is a promising tool for improving the diagnosis process of bacterial pathogens associated with brain inflammation. This is due to its high sensitivity, rapidity, and compatibility with portable diagnostic platforms, making it particularly suitable for POC applications. This protocol introduces an innovative diagnostic approach designed to function effectively without the need for advanced laboratory equipment. By leveraging dual-priming isothermal amplification (DAMP), the assay uses custom internal primers to enhance specificity and minimize false results. Brilliant Green is used in this assay for fluorescence detection due to its availability, high fluorescence level, and optimal sample-to-background (S/B) ratio. The assay demonstrated excellent specificity, absence of false positives, sensitivity comparable to loop-mediated isothermal amplification (LAMP), and a high S/B ratio.
Key features
• A POC-compatible diagnostic assay developed to enable rapid and accurate diagnosis of meningitis without the need for advanced laboratory infrastructure.
• A DAMP-based approach for the identification of pathogens responsible for bacterial meningitis with high sensitivity and specificity while ensuring the absence of false-positive results.
Keywords: DNA quantitative detectionGraphical overview
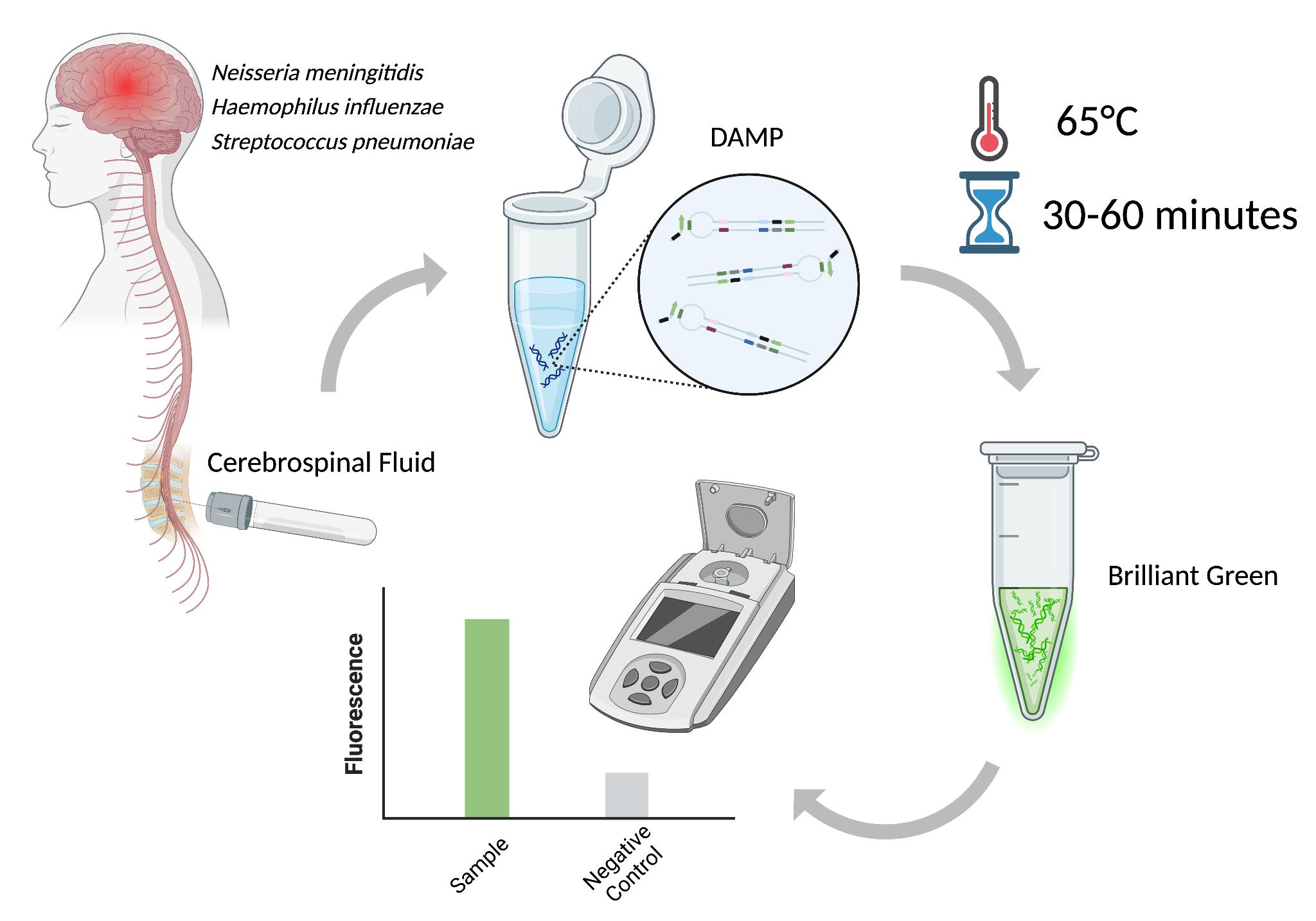
Schematic workflow of this protocol [1]
Background
The rapid and accessible diagnosis of infectious diseases is essential for effective treatment, especially in cases of brain inflammations such as meningitis and encephalitis, where rapid intervention is crucial to preventing complications and decreasing morbidity [2]. For these infections, diagnostic methods need to be highly sensitive, specific, and accessible, ideally matching the point-of-care (POC) parameters: a testing duration of less than 2 h, a cost of less than $10 per test, and suitable for use by untrained personnel. Nucleic acid amplification testing (NAAT) is regarded as the gold standard for molecular diagnostics in this area [3]. PCR-based technologies are among the most commonly used NAAT methods in clinical practice. Recent research indicates that NAATs are essential diagnostic tools [4], particularly in instances where cerebrospinal fluid (CSF) cultures yield negative results [5], as these culturing methods become significantly limited upon antibiotic treatment.
Among NAAT techniques, isothermal amplification has emerged as particularly well-suited for POC diagnostics as it eliminates the need for complex lab equipment [6]. Isothermal amplification eliminates the need for thermal cycling and utilizes specialized enzymes with strand displacement activity to amplify target DNA or RNA sequences at a constant temperature. Of these techniques, loop-mediated isothermal amplification (LAMP) is the most widely used due to its simplicity, rapidity, and high sensitivity. However, despite its advantages, LAMP is prone to nonspecific amplification, which may be acceptable for screening purposes but is unsuitable for guiding therapeutic decisions. Thus, in general practice, LAMP often requires an additional PCR test to confirm primary results [7,8]. A novel method, dual-priming isothermal amplification (DAMP), was developed to overcome the limitations of LAMP. DAMP incorporates a dual-priming mechanism using specialized primers to enhance specificity and reduce false-positive results. While LAMP uses six primers to target eight distinct regions of the target sequence, with inner primers spanning 40–60 nucleotides to facilitate self-priming strand extension, DAMP targets six distinct regions using six primers, with inner primers designed to span less than 40 nucleotides [9]. Additionally, DAMP incorporates pairing-competition primers, which enhance amplification efficiency by balancing self-priming and pairing-priming strand extensions, making it a promising tool for rapid, accurate detection of meningitis in both laboratory and POC settings [9].
A critical aspect of introducing NAAT-based methods into clinical practice is the detection of amplification products. The detection can be achieved through direct or indirect methods. Various visualization techniques can use fluorescent compounds (e.g., DNA intercalating) or naked-eye detection [10]. Although naked eye readouts do not require any specialized tools, their dynamic ranges are often limited, and personnel should have the skills to distinguish subtle changes in turbidity or color. On the other hand, fluorescent detection is preferred for its high sensitivity and quantitative capabilities [11]. Previously, we evaluated three fluorescent dyes for their effectiveness in detecting double-stranded DNA (dsDNA) amplification products: dsGreen, Thioflavin T, and Brilliant Green. Each dye was assessed based on its sample-to-background (S/B) ratio, which is crucial for minimizing background noise and ensuring accurate detection [1]. Brilliant Green was identified as the optimal dye due to its high fluorescence intensity, affordability, and superior S/B ratio. The performance of this dye, combined with the DAMP reaction, provides a highly sensitive and specific assay for detecting the four primary bacterial pathogens responsible for meningitis: Haemophilus influenzae, Neisseria meningitidis, Streptococcus pneumoniae, and Klebsiella pneumoniae. These pathogens were selected as the leading causes of bacterial meningitis in individuals beyond the neonatal stage.
The developed assay was evaluated using bacterial culture and clinical cerebrospinal fluid (CSF) samples. It demonstrated both high specificity and sensitivity. Its versatility allows it to function as a standalone diagnostic tool or to be integrated into POC devices, offering a reliable, rapid, and accessible solution for the diagnosis of meningitis.
Materials and reagents
Biological materials
Thermally inactivated cell cultures of the pathogens Klebsiella pneumoniae, Haemophilus influenzae, Neisseria meningitidis, and Streptococcus pneumoniae were obtained from the Pediatric Research and Clinical Center for Infectious Diseases (PRCCID) under the Russian Federal Medical Biological Agency.
Note: This protocol can also be performed on DNA isolated from CSF samples. Thermally inactivated CSF clinical samples were also provided by the PRCCID.
Reagents
1. Nuclease-free water (Invitrogen, catalog number: 1097704)
2. dNTPs (Evrogen, catalog number: PB032)
3. Fine powder MgSO4 (AppliChem, catalog number: 141404)
4. Ethylene glycol liquid (Lenreactiv, catalog number: 263059)
5. Fine-powder KCl (AppliChem, catalog number: 131494.1210)
6. Fine-powder (NH4)2SO4 (AppliChem, catalog number: A3485,5000)
7. Fine powder Tris-HCl (AppliChem, catalog number: 633654.0415)
8. Tween 20% (AppliChem, catalog number: A4974,0500)
9. Bst DNA polymerase (BioLabMix, catalog number: E-10010)
10. Fine powder Brilliant Green (Lenreactiv, catalog number: 020264)
11. 50 bp+ DNA ladder (Evrogen, catalog number: NL003)
12. Ethidium bromide (BioLabMix, catalog number: EtBr-10)
13. 50× TAE buffer (BioLabMix, catalog number: BE-DNA-1000)
14. Fine powder agarose (Helicon, catalog number: SYS-Q0009-0.1)
15. 4× gel loading dye (Evrogen, catalog number: PB020)
16. Primers (Evrogen, direct order):
For Klebsiella pneumoniae:
Outer forward primer (FO): GCGAAGAAACAGAACGGC
Outer reverse primer (RO): TGATCGGGGTCATCTTGC
Inner forward primer (FI): TACGGTCGGAGCTGGTGTAGTTTTCTATGATTTCGGCGGCAG
Inner reverse primer (RI): GACCGGCCTGAAATATGACGCTTTTTACATGGTCGCCAGGTAG
Paring-competition forward primer (FC): TACGGTCGGAGCTGGTGTAG
Pairing-competition reverse primer (RC): GACCGGCCTGAAATATGACGC
Note: The primer sequences for the other pathogens can be found in the original article [11].
Solutions
1. 1× TAE buffer (see Recipes)
2. 10× Bst buffer (see Recipes)
3. Brilliant Green 100 μM stock (see Recipes)
4. MgSO4 1 M stock (see Recipes)
Recipes
1. 1× TAE buffer
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| 50× TAE buffer | 1× | 40 mL |
| Deionized water | n/a | 1,960 mL |
| Total | 2,000 mL |
2. 10 × Bst buffer
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| (NH4)2SO4 1 M | 100 mM | 1 mL |
| KCl 1 M | 120 mM | 1 mL |
| MgSO4 1 M (Recipe 4) | 20 mM | 0.2 mL |
| Tris-HCl (pH 8.8) 1 M | 200 mM | 2 mL |
| Tween 20% | 2% | 0.2 mL |
| Ethylene glycol | 5% | 0.5 mL |
| Total | 10 mL |
3. Brilliant Green 100 μM stock
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| Brilliant Green | 100 μM | 48.3 ng |
| Deionized water | n/a | 1 mL |
| Total | 1 mL |
4. MgSO4 1 M stock
| Reagent | Final concentration | Quantity or Volume |
| Fine powder MgSO4 | 1 M | 246.47 ng |
| Deionized water | n/a | 1 mL |
| Total | 1 mL |
Laboratory supplies
1. Costar 96-well black polystyrene plate (Corning, catalog number: COS3915)
2. 1–10 μL pipette (Kirgen, catalog number: KG-Pro10)
3. 2–20 μL pipette (Kirgen, catalog number: KG-Pro20)
4. 20–200 μL pipette (Kirgen, catalog number: KG-Pro200)
5. 100–1,000 µL pipette (Kirgen, catalog number: KG-Pro1000)
Equipment
1. Wide mini-sub cell GT Cell (Bio-Rad, catalog number: 1704468EDU)
2. PowerPacTM basic power supply (Bio-Rad, catalog number: 1645050)
3. Spark multimode microplate reader (Tecan, model: Spark-10M)
4. Thermocycler (DNK-Technology, model: DTlite 4S1)
5. ChemiDocTM touch gel imaging system (Bio-Rad, catalog number: 1708370)
Software and datasets
1. Basic Local Alignment Search Tool (BLAST) available on the National Center for Biotechnology Information (NCBI) platform to analyze the sequence data
2. LAMP primer design tool provided by New England Biolabs (NEB)
3. GraphPad Prism v. 9.3.1.
Procedure
A. DAMP primer design
1. Identify specific genes that are unique to the targeted pathogens. Whenever possible, refer to previously established specificity genes for these pathogens, as documented in the existing literature.
Note:
a. For Klebsiella pneumoniae, the target gene is phoE (accession: EF197995.1)
b. For Haemophilus influenzae, the target gene is hpd (accession: MN488730)
c. For Neisseria meningitidis, the target gene is ctrA (accession: NZ_CP021520)
d. For Streptococcus pneumoniae, the target gene is lytA (accession: AE005672)
2. To confirm that the selected regions are highly conserved among various strains of the target species, perform a BLAST search for each gene to verify its presence and specificity across the targeted species.
a. Open the Nucleotide BLAST tool.
b. Paste the sequence of the target gene or upload the FASTA file into the provided input box.
c. In the "Organism" field, exclude the target pathogen by specifying it.
d. Click the BLAST button to initiate the search.
Note: In some cases, the BLAST search results may show no matches, indicating that the target gene is highly specific to the intended pathogen. However, there may be instances where the search reveals that the target gene or part of its sequence is also present in other species. In such cases, it is still possible to proceed with the primer design process, as further optimization and specificity testing during primer validation will help ensure the accuracy of the assay.
3. Open the NEB LAMP Primer design tool and follow these steps:
a. Paste the sequence of the target gene or upload the FASTA file into the provided input box.
b. Under the Specificity Parameter Selection, select the option Automatically assign based on %GC.
c. Navigate to the Preferences tab and set the following parameters for Distances (min/max):
(F2–B2): 76–224
Loop (F1c–F2): 18–40
F2–F3: 0–40
F1c–B1c: 0–100
Note: If the GC content of the target sequence is higher than 65%, adjust the Loop (F1c–F2) distance to 15–40.
d. Leave the fields for FL/FB in the sections Tm (min/max) and Lengths (min/max) empty.
e. Retain the default settings for all other parameters, as no further adjustments are needed.
f. Click the Save button to save the entered parameters, and then click the Generate Primers button to create the primer set.
g. A number of primer sets will be generated. Choose the first primer set, then extract the sequence that will be amplified by the F3 and B3 primers.
h. Perform a BLAST search as in step A2 using this sequence to ensure its specificity to the target species. If this primer set is not specific, proceed to the second primer set and repeat the BLAST search. Continue this process until a specific primer set is identified.
4. After identifying the specific primer set, the tool will display a list of eight primers. Out of these, six primers are needed: the four primers highlighted in orange along with the F1c and B1c primers. These primers should be selected for the amplification process:
a. Incorporate the F1c and B1c primers independently into the reaction as standalone primers, denoted as FC and RC, respectively.
b. The F3 and B3 primers are responsible for specificity and should be denoted as FO and RO, respectively.
c. The FI and RI primers consist of the F1c + F2 and B1c +B2 primer sections, respectively.
d. Add a four-thymine oligonucleotide spacer (TTTT spacer) between the F1c and F2 regions for the FI primer, and between the B1c and B2 regions for the RI primer. This spacer should be incorporated to enhance the efficiency of the primer binding.
B. Extraction of genomic DNA
1. Use the standard phenol-chloroform extraction method to extract genomic DNA from the cell culture of the targeted and non-targeted pathogens [12]. This method can be applied uniformly to both types of samples, cell cultures, and CSF clinical samples without requiring modifications. The bacterial strains used in this study are classified as biosafety level 2 (BSL-2) organisms due to their potential to cause disease in humans. Proper safety measures according to the rules of your country of presence should be strictly followed to minimize any risks to laboratory personnel and prevent exposure.
C. Characterization of DAMP
1. DAMP reaction mix preparation:
a. Prepare the stock solutions of the components as mentioned in Table 1.
Notes:
1. The stock solution of the primers can be prepared in advance and stored in a -20 °C freezer for up to seven days.
2. Completely thaw the primer solutions 30 min before using. Gently mix by tapping (do not intensively vortex). Spin down the solution using a microcentrifuge (quickly, for a few seconds).
b. Mix the DAMP reaction component while working on ice in the order specified in Table 1.
Table 1. DAMP reaction mix
| Component | Stock concentration | Final concentration | Volume |
|---|---|---|---|
| Bst buffer | 10× | 1× | 2.5 μL |
| dNTPs | 25 μM | 1.6 μM | 1.6 μL |
| MgSO4 | 100 μM | 4 μM | 1 μL |
| FO/RO | 10 μM | 0.2 μM | 0.5 μL |
| FI/RI | 40 μM | 1.6 μM | 1 μL |
| FC/RC | 40 μM | 1.6 μM | 1 μL |
| Bst polymerase | 8,000 U/mL | 320 U/mL | 1 μL |
| Genomic DNA | n/a | 1 ng | n/a |
| Nuclease-free water | n/a | n/a | 14.65 μL |
| Total | 25 μL |
Note: Bst polymerase should be stored at -20 °C and kept on ice while working.
c. Prepare the negative control of the DAMP reaction by repeating step C1b and substituting the genomic DNA with nuclease-free water.
d. Mix the tubes gently and spin down the solution.
e. Incubate at 60 °C for 40 min.
2. Limit of detection of the DAMP reaction:
a. Prepare 10-fold serial dilutions of genomic DNA ranging from 10,000 to 1 genome equivalent.
Note: Use nuclease-free water for making serial dilutions.
b. Repeat steps C1b–e and substitute the genomic DNA with the prepared serial dilutions in the reaction mix.
Note: Calculation of genome equivalent (GE):
GE of an organism (g/mol) = Weight of the reference genome (bp) * 615.96 g/mol/bp
3. Specificity of DAMP:
a. Prepare 1,000 GE of DNA of the non-target pathogens.
b. Repeat steps C1b–e and substitute the genomic DNA with 1,000 GE of DNA of the non-target pathogens.
Note: Each set of primers should be tested against its target pathogen and the DNA of the other non-target pathogens.
D. DAMP detection
1. DAMP fluorescent detection:
a. Prepare the DAMP detection mixture by mixing the components mentioned in Table 2 in the following order at room temperature in 0.5 mL microcentrifuge tubes.
Table 2. DAMP detection mix
| Reagent | Final concentration | Volume |
|---|---|---|
| Brilliant Green | 30 μM | 18 μL |
| DAMP reaction | n/a | 3 μL |
| Water | n/a | 39 μL |
| Total | 60 μL |
Note: Brilliant green can be stored in a -20 °C freezer and should be completely thawed 30 min before using.
b. Prepare the blank tube by repeating step D1a and substituting the DAMP reaction sample with nuclease-free water. This is used to assess the basic background of Brilliant Green stability.
c. Gently mix by tapping (do not intensively vortex). Spin down the solution using a microcentrifuge (quickly, for 30 s).
d. Incubate at room temperature for 5 min.
e. Transfer the mixes into a black 96-well microplate reader.
f. Measure fluorescence at an excitation wavelength of 610 nm and emission wavelength of 610 nm.
2. Agarose gel electrophoresis of DAMP:
a. Prepare the 2% agarose: mix 0.5 g of fine-powder agarose in 25 mL of 1× TAE buffer. Heat and mix carefully and slowly until it becomes transparent.
b. Move the mixture to the gel cassette and let it polymerize.
c. Mix 1 μL of the DAMP reaction sample with 2 μL of 1× TAE buffer and 1 μL of 4× loading dye and load the mixture into the gel.
d. Load 3 μL of 50 bp + DNA ladder into the gel.
e. Put the cassette into the chamber, fill it with a 1× TAE buffer, and let the gel run for 20 min at 80 V.
f. Dye the gel with ethidium bromide and visualize it using a gel-documentation system.
Note: Although ethidium bromide was used in this study for DNA visualization, safer DNA-binding alternatives such as SYBR Safe, GelRed, or similar dyes can also be used for this purpose, ensuring proper laboratory safety and environmental considerations.
Data analysis
1. Fluorescent detection analysis:
The sensitivity limit was reached at 100 GE (Figure 1). The sample-to-background ratio exceeded the established threshold, where the reaction with 0 GE of DNA was considered as the background. The DAMP specificity test demonstrated the absence of any discernible cross-reactivity among the targeted DNA fragments (Figure 2), as they were detected by measuring the relative fluorescence with the addition of dye.
2. Gel electrophoresis analysis
The amplified fragments in DAMP demonstrate a ladder-like pattern on an agarose gel, starting from 100 bp and above, due to the unique "dual-priming" mechanism that generates diverse intermediate products and basic structures during the amplification cycle. These structures differ in size due to the simultaneous "self-priming" and "pairing-priming" strand extensions, which produce the ladder-like pattern of amplicons (Figures 3 and 4). This phenomenon is similar to the ladder-like pattern seen in LAMP and reflects the diversity of amplified DNA fragments generated during the cycling amplification process.
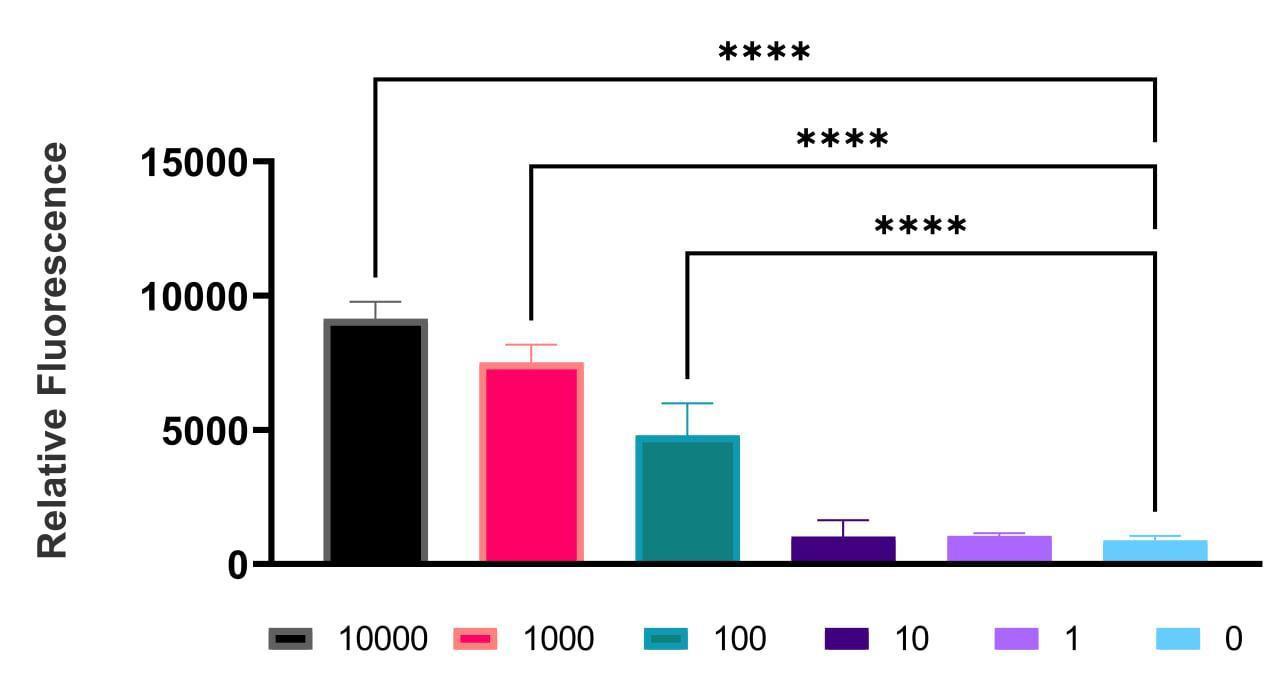
Figure 1. Example of data collected demonstrating the relative fluorescence of the DAMP product for Klebsiella pneumoniae with different genome equivalents (GEs) ranging from 10,000 to 1 GE using Brilliant Green.
Data are expressed as mean ± SD using GraphPad Prism 9.3.1 software. The experiment was carried out three times in three technical repetitions. **** p < 0.0001.
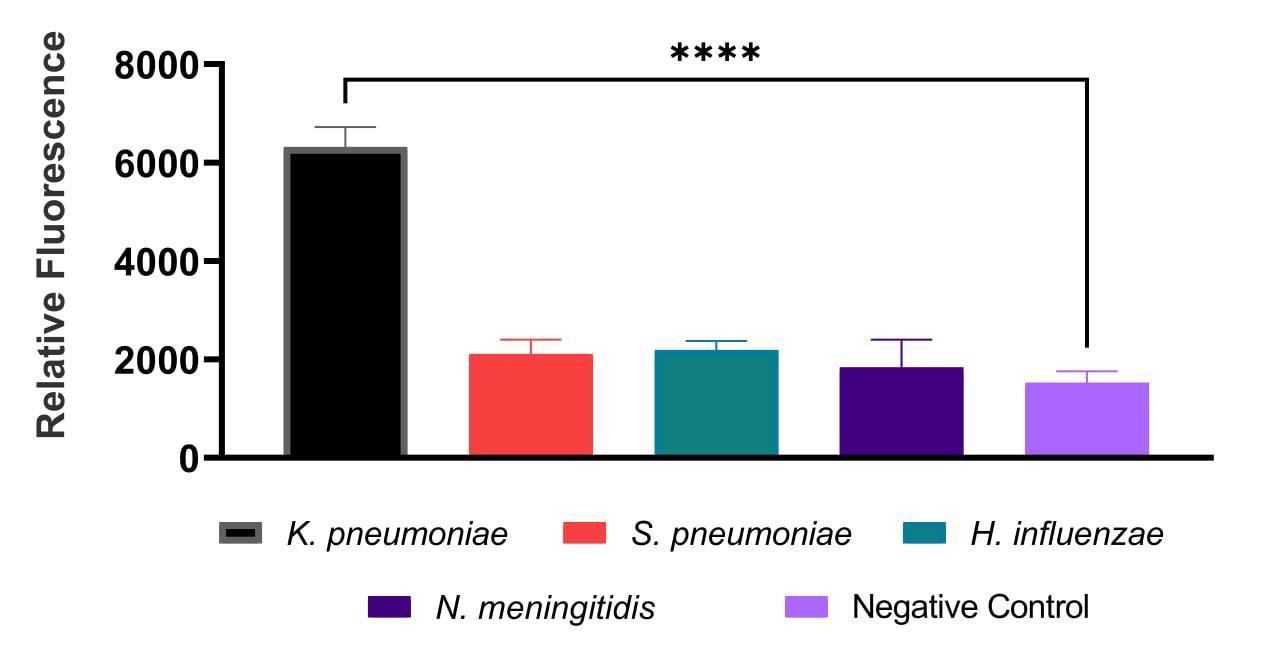
Figure 2. Example of data collected demonstrating the cross-reaction assay for the Klebsiella pneumoniae primer set performed on DNA isolated from Streptococcus pneumoniae, Haemophilus influenzae, and Neisseria meningitidis. Relative fluorescence of the DAMP amplification product for the primer set using Brilliant Green. Data are expressed as mean ± SD using GraphPad Prism 9.3.1 software. The experiment was carried out three times in three technical repetitions. **** p < 0.0001.
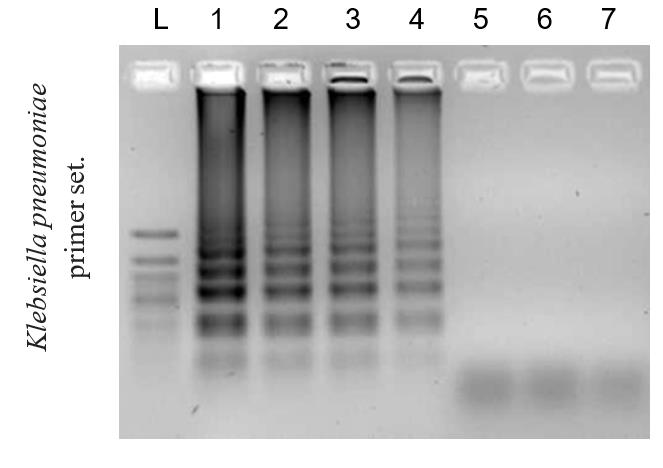
Figure 3. Example of the limit of detection of the DAMP assay. Agarose 2% gel electrophoresis of DAMP amplicons using Klebsiella pneumoniae primer set. Reaction corresponding to DNA in concentrations of (1) 100,000 genome equivalents (GE), (2) 10,000 GE, (3) 1,000 GE, (4) 100 GE, (5) 20 GE, (6) 1 GE, and (7) 0 GE. (L) 50 bp + DNA marker.
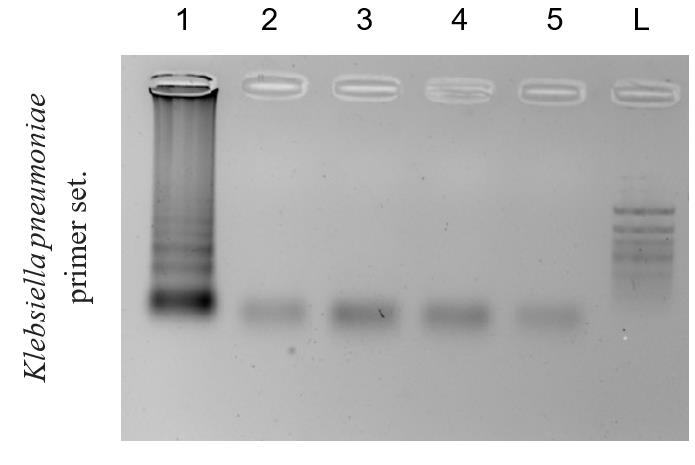
Figure 4. Example of the cross-reaction of the DAMP assay. Agarose 2% gel electrophoresis of DAMP amplicons using Klebsiella pneumoniae primer set. Reaction with DNA of (1) Klebsiella pneumoniae, (2) Streptococcus pneumoniae, (3) Haemophilus influenzae, (4) Neisseria meningitidis, and (5) negative control. (L) 50 bp+ DNA marker.
Validation of protocol
This protocol was initially validated in our previously published article titled “A DAMP-Based Assay for Rapid and Affordable Diagnosis of Bacterial Meningitis Agents: Haemophilus influenzae, Neisseria meningitidis, and Streptococcus pneumoniae” [1]. In that study, we successfully tested the protocol on clinical CSF samples, demonstrating excellent specificity and sensitivity. In this article, we extended the validation of the protocol to include the diagnosis of Klebsiella pneumoniae, further broadening its applicability to additional bacterial pathogens.
General notes and troubleshooting
General notes
Perform the reaction in a DNA-free environment. Decontaminate the area before the reaction assembly. Separate the reaction assembly and visualization in space. Use separate lab clothes, shoes, and tools for different lab areas. In case of contamination, expose the area to hydrogen peroxide or sodium hypochlorite.
Troubleshooting
Problem 1: False positive results (amplification products present in negative control samples).
Possible causes: Primers create dimeric structures that are recognized by the polymerase as a template.
Solution 1: Optimization of magnesium ion concentration in the reaction:
a. Perform the reaction using the standard protocol with 6 mM magnesium ions. If amplification products appear in the negative control, adjust the magnesium concentration to 4, 6, 8, 10, or 12 mM.
b. Run electrophoresis for each condition and select the magnesium concentration that eliminates amplification in the negative control while maintaining amplification in positive samples.
Solution 2: Optimization of primer concentrations: If the issue of false positive results persists after optimizing the magnesium concentration, it is essential to further optimize the concentrations of primers RI/FI and RC/FC. Reactions at each magnesium concentration should be performed with different concentrations of these primers: 0.8, 1, 1.2, 1.4, 1.6, 1.8, 2.0, 2.2, and 2.4 μM.
Solution 3: Designing a new primer set: If the issue of false positives remains unresolved after the above optimizations, selecting a new primer set is recommended.
Problem 2: Absence of amplification products in positive samples.
Possible cause: Degradation of the DNA sample or the polymerase activity in the reaction.
Solution 1: Use a control sample to verify reagent functionality. Perform the reaction with a positive control sample. If amplification occurs in the control but not in the experimental sample, check the integrity of the DNA in the experimental sample via electrophoresis.
Solution 2: Verify DNA integrity. If the DNA is degraded, re-extract or replace the sample with intact DNA.
Solution 3: If the DNA is not degraded, it is necessary to optimize the reaction according to the presented scheme to solve the problem of false positive results.
Acknowledgments
The authors would like to thank E.V. Nikitina, V.V. Gostev, and S.V. Sidorenko for the provision of clinical samples and bacterial DNA, and G.A. Bobkov and M.A. Kolesnikova for their contributions to the conceptualization of this article. The authors would also like to extend their gratitude to the Russian Science Foundation for supporting scientific research. This research was funded by the Russian Science Foundation, grant number 22-75-10073.
Competing interests
The authors declare no competing financial interests.
References
- Shkodenko, L. A., Mohamed, A. A., Ateiah, M., Rubel, M. S. and Koshel, E. I. (2024). A DAMP-Based Assay for Rapid and Affordable Diagnosis of Bacterial Meningitis Agents: Haemophilus influenzae, Neisseria meningitidis, and Streptococcus pneumoniae. Int J Mol Sci. 25(15): 8282. https://doi.org/10.3390/ijms25158282
- van de Beek, D., Cabellos, C., Dzupova, O., Esposito, S., Klein, M., Kloek, A., Leib, S., Mourvillier, B., Ostergaard, C., Pagliano, P., et al. (2016). ESCMID guideline: diagnosis and treatment of acute bacterial meningitis. Clin Microbiol Infect. 22: S37–S62. https://doi.org/10.1016/j.cmi.2016.01.007
- Wu, Q., Suo, C., Brown, T., Wang, T., Teichmann, S. A. and Bassett, A. R. (2021). INSIGHT: A population-scale COVID-19 testing strategy combining point-of-care diagnosis with centralized high-throughput sequencing. Sci Adv. 7(7): eabe5054. https://doi.org/10.1126/sciadv.abe5054
- Michael, B., Menezes, B. F., Cunniffe, J., Miller, A., Kneen, R., Francis, G., Beeching, N. J. and Solomon, T. (2010). Effect of delayed lumbar punctures on the diagnosis of acute bacterial meningitis in adults. Emerg Med J. 27(6): 433–438. https://doi.org/10.1136/emj.2009.075598
- Ghafari, S., Namakin, K., Khooban, A. R., Askari, P., Yousefi, M. and Ziaee, M. (2023). Comparison of Multiplex Quantitative Real-Time PCR and Culture Methods for the Diagnosis of Bacterial Meningitis in Patients with Suspected Meningitis. Infect Epidemiol Microbiol. 9(3): 219–228. https://doi.org/10.61186/iem.9.3.219
- Moehling, T. J., Choi, G., Dugan, L. C., Salit, M. and Meagher, R. J. (2021). LAMP Diagnostics at the Point-of-Care: Emerging Trends and Perspectives for the Developer Community. Expert Rev Mol Diagn. 21(1): 43–61. https://doi.org/10.1080/14737159.2021.1873769
- Kim, S. H., Lee, S. Y., Kim, U. and Oh, S. W. (2023). Diverse methods of reducing and confirming false-positive results of loop-mediated isothermal amplification assays: A review. Anal Chim Acta. 1280: 341693. https://doi.org/10.1016/j.aca.2023.341693
- Gill, P. and Ghaemi, A. (2008). Nucleic Acid Isothermal Amplification Technologies—A Review. Nucleosides, Nucleotides Nucleic Acids. 27(3): 224–243. https://doi.org/10.1080/15257770701845204
- Ding, X., Xu, Z., Yin, K., Sfeir, M. and Liu, C. (2019). Dual-Priming Isothermal Amplification (DAMP) for Highly Sensitive and Specific Molecular Detection with Ultralow Nonspecific Signals. Anal Chem. 91(20): 12852–12858. https://doi.org/10.1021/acs.analchem.9b02582
- Fujino, M., Yoshida, N., Yamaguchi, S., Hosaka, N., Ota, Y., Notomi, T. and Nakayama, T. (2005). A simple method for the detection of measles virus genome by loop-mediated isothermal amplification (LAMP). J Med Virol. 76(3): 406–413. https://doi.org/10.1002/jmv.20371
- Tomita, N., Mori, Y., Kanda, H. and Notomi, T. (2008). Loop-mediated isothermal amplification (LAMP) of gene sequences and simple visual detection of products. Nat Protoc. 3(5): 877–882. https://doi.org/10.1038/nprot.2008.57
- Gautam, A. (2022). Phenol-Chloroform DNA Isolation Method. In: DNA and RNA Isolation Techniques for Non-Experts. Techniques in Life Science and Biomedicine for the Non-Expert. Springer, Cham. 33–39. https://doi.org/10.1007/978-3-030-94230-4_3
Article Information
Publication history
Received: Nov 5, 2024
Accepted: Feb 4, 2025
Available online: Feb 27, 2025
Published: Mar 20, 2025
Copyright
© 2025 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Shkodenko, L. A., Ateiah, M., Mohamed, A., Rubel, M. S. and Koshel, E. I. (2025). A Highly Sensitive and Selective DAMP Assay for the Detection of Bacterial Pathogens Associated With Brain Inflammation. Bio-protocol 15(6): e5240. DOI: 10.21769/BioProtoc.5240.
Category
Microbiology > Pathogen detection > Biosensor
Molecular Biology > DNA > DNA quantification
Neuroscience > Nervous system disorders
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.