- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Rapid and Efficient Isolation of Total RNA-Bound Proteomes by Liquid Emulsion–Assisted Purification of RNA-Bound Protein (LEAP-RBP)
Published: Vol 14, Iss 14, Jul 20, 2024 DOI: 10.21769/BioProtoc.5236 Views: 2828
Reviewed by: Dipak Kumar PoriaThirupugal GovindarajanAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Sep 2023

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
The critical roles of RNA-binding proteins (RBPs) in all aspects of RNA biology fostered the development of methods utilizing ultraviolet (UV) crosslinking and method-specific RNA enrichment steps for proteome-wide identification and assessment of RBP function. Despite the substantial contributions of these UV-based RNA-centric methods to our understanding of RNA–protein interaction networks, their utility is constrained by biases in RBP recovery and significant noise contributions, which can confound meaningful interpretation. To overcome these issues, we recently developed a method termed Liquid Emulsion–Assisted Purification of RNA-Bound Protein (LEAP-RBP) and introduced quantitative signal-to-noise (S:N)-based metrics for the proteome-wide identification of RNA interactomes and accurate assessment of global RBP occupancy dynamics. Compared to existing methodologies, LEAP-RBP provides significant advantages in speed, cost, efficiency, and selectivity for RNA-bound proteins. In this work, we provide a step-by-step guide for the successful application of the LEAP-RBP method for both small- and large-scale investigations of RNA-bound proteomes.
Key features
• Unbiased and efficient isolation of total RNA-bound protein, RNA, and protein from biological samples.
• Cost-effective identification of proteome-wide RNA interactomes and validation of direct RNA-binding protein functionality.
• Robust and accurate assessment of context- and/or condition-dependent RBP occupancy state dynamics.
Keywords: LEAP-RBPGraphical overview
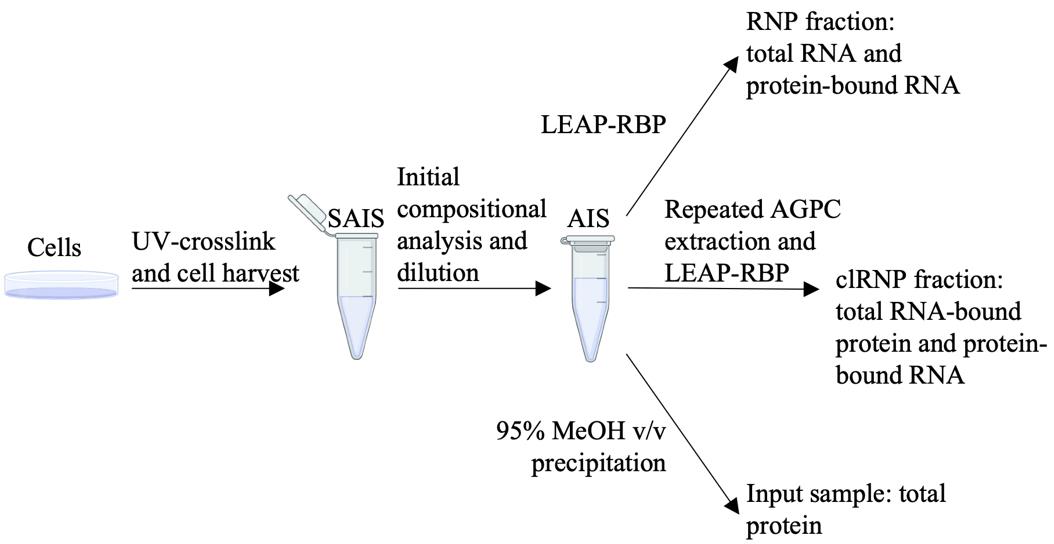
Background
With the growing interest in the identification and study of RNA interactomes, an expanding toolkit of methods to capture RNA-protein complexes has appeared [1–9]. The most widely utilized methods employ UV crosslinking and different RNA-enrichment or organic phase–separation conditions for selective isolation of RNA–protein complexes and can be categorized as UV-based RNA-centric methods, or RNA-centric methods for short [10–17]. When paired with downstream quantitative mass spectrometry, these methods address the need for proteome-wide identification of candidate RNA-binding proteins (RBPs). We recently reported an RNA-centric method termed Liquid Emulsion–Assisted Purification of RNA-Bound Protein (LEAP-RBP), which can be distinguished from existing methods by its high selectivity for protein–RNA complexes via the use of a lithium-supplemented heterogeneous solvent system [18]. Although the precise biochemical/biophysical mechanism for the high selectivity of LEAP-RBP for protein–RNA adducts remains conjecture, we postulate that lithium complexation with the RNA component of protein–RNA adducts promotes selective precipitation, consistent with the established, but not well understood, capacity for lithium to precipitate RNA [19]. Importantly, the high selectivity of the LEAP-RBP method for protein–RNA complexes enables signal-to-noise (S:N)-based metrics for quantitative evaluation of protein–RNA interactions. Combined, LEAP-RBP addresses limitations in existing methodologies, which include biases in RBP recovery due to the inclusion of stringent denaturing washes, confounding levels of experimental noise (i.e., free protein recovery in the RBP fraction), and a paucity of quantitative metrics for assessing RNA binding function [18]. The utility of the LEAP-RBP method has been further elaborated in the identification of RBPs undergoing a condition-dependent change in RNA occupancy following short-term inhibition of translation initiation [18]. These included both RBPs with well-established roles in translation-related processes such as UPF1 and RPS3 and proteins with previously unknown roles in RNA regulation such as ABCF3 [18]. In summary, the high protein–RNA complex specificity and efficiency of the LEAP-RBP method supports rapid and inexpensive orthogonal validation of RBP occupancy state changes and RNA-binding function of candidate RBPs, using basic laboratory techniques such as SDS-PAGE RNase-dependent Assay (SRA). The simplicity and broad applicability of the LEAP-RBP method position it as a valuable tool for low- and high-throughput identification of RNA-binding proteins and rigorous assessment of RBP occupancy state change dynamics.
Materials and reagents
2-mercaptoethanol (Sigma-Aldrich, catalog number: M7522)
Acidic phenol pH 4.5 (VWR, catalog number: 0981)
Chloroform, ethanol stabilizer (Thermo Fisher Scientific, catalog number: MCX10601)
Isopropanol (VWR, catalog number: BDH1133)
Methanol (MeOH) (VWR, catalog number: BDH1135)
Sodium citrate (VWR, catalog number: BDH9288)
Citric acid (Sigma-Aldrich, catalog number: C0759)
EDTA disodium salt dihydrate (VWR, catalog number: 0105)
N-lauroylsarcosine sodium salt (Sigma-Aldrich, catalog number: L9150)
Guanidine thiocyanate (Chem-Impex, catalog number: 00522)
Diethyl pyrocarbonate (DEPC) (RPI, catalog number: D43060)
Hydrochloric acid (Ricca Chemical, catalog number: RABH0010)
TRIS base (VWR, catalog number: 97062)
LiDS (Thermo Fisher Scientific, catalog number: J32816)
LiCl (VWR, catalog number: ALFA10515; Sigma-Aldrich, catalog number: 213233)
Sodium hydroxide (VWR, catalog number: BDH9292)
Turbo DNase kit (Thermo Fisher Scientific, catalog number: AM2238)
Pierce BCA kit (Thermo Fisher Scientific, catalog number: 23225)
Dulbecco’s phosphate-buffered saline (PBS) (Thermo Fisher Scientific, catalog number: 14190144)
Solutions
DEPC-treated ultrapure H2O (see Recipes)
5 M GT (see Recipes)
750 mM sodium citrate pH 7.0 (see Recipes)
10% N-lauroylsarcosine (m/v) (see Recipes)
0.5 M EDTA pH 8.0 (see Recipes)
GT buffer (see Recipes)
AGP (see Recipes)
1.0 M Tris-HCl pH 8.0 (see Recipes)
10% LiDS (m/v) (see Recipes)
1% LiDS TE (see Recipes)
2% LiDS TE (see Recipes)
10 M LiCl (see Recipes)
Precipitation solution (see Recipes)
95% MeOH (see Recipes)
Recipes
DEPC-treated ultrapure H2O (1 L)
Reagent Final concentration Amount or volume Ultrapure H2O n/a 999.0 mL Diethyl pyrocarbonate (DEPC) 0.1% v/v 1 mL Total n/a 1 L Prepare in an airtight glass container. Shake vigorously for 5 s to dissolve DEPC. Incubate overnight at 37 °C and autoclave to decompose unreacted DEPC. Shelf life is undetermined, > 1 year.
5 M GT (0.5 L)
Reagent Final concentration Amount or volume Guanidine thiocyanate 5 M 295.4 g DEPC-treated ultrapure H2O n/a 271 mL Total n/a 0.5 L Add in the order listed. Weigh guanidine thiocyanate and adjust the volume of DEPC-treated ultrapure H2O accordingly. Prepare in a beaker using a magnetic stir bar and hot plate stirrer set to 37 °C. Clarify by filtering twice using Whatman paper or by standing incubation overnight and transferring the clarified portion. Store in an airtight glass container at room temperature (RT). Shelf life is undetermined, > 1 year. Protect from light during all stages of the preparation procedure.
750 mM sodium citrate pH 7.0 (50 mL)
Reagent Final concentration Amount or volume Citric acid anhydrous n/a 0.13 g Sodium citrate dihydrate n/a 10.83 g DEPC-treated ultrapure H2O n/a Adjust volume to 50 mL Total n/a 50 mL Add in the order listed. Prepare in a 50 mL conical tube. Weigh citric acid anhydrous and adjust the amount or volume of other components accordingly. Dissolve by incubating on a rotator at RT. Filter using a syringe equipped with a 0.2 µm cellulose acetate syringe filter. Store at RT. Shelf life is undetermined, > 1 year.
10% N-lauroylsarcosine m/v (10 mL)
Reagent Final concentration Amount or volume N-lauroylsarcosine sodium salt 10% m/v 1 g DEPC-treated ultrapure H2O n/a 9 mL Total n/a 10 mL Add in the order listed. Prepare in a 15 mL conical tube. Weigh N-lauroylsarcosine sodium salt and adjust the volume of DEPC-treated ultrapure H2O accordingly. Dissolve by incubating on a rotator at RT. Store at 4 °C. Shelf life is undetermined, > 6 months.
0.5 M EDTA pH 8.0 (50 mL)
Reagent Final concentration Amount or volume Sodium hydroxide pellets n/a 1.28 g DEPC-treated ultrapure H2O n/a 44.27 mL EDTA disodium salt dihydrate 0.5 M 9.31 g Total n/a 50 mL Add in the order listed. Prepare in a 50 mL conical tube. Weigh sodium hydroxide pellets and adjust the amount or volume of other components accordingly. Dissolve by incubating on a rotator at RT. Filter using a syringe equipped with a 0.2 µm cellulose acetate syringe filter. Store at RT. Shelf life is undetermined, > 1 year.
GT buffer (50 mL)
Reagent Final concentration Amount or volume DEPC-treated ultrapure H2O n/a 5 mL 5 M GT 4 M 40 mL 750 mM sodium citrate pH 7.0 25 mM 1.67 mL 0.5 M EDTA pH 8.0 5 mM 500 µL 10% N-lauroylsarcosine m/v 0.5% m/v 2.5 mL 2-mercaptoethanol 0.7% v/v 350 µL Total n/a 50 mL Add in the order listed. Prepare in a 50 mL conical tube. Can be prepared without N-lauroylsarcosine and 2-mercaptoethanol and stored at RT. Protect from light. Shelf life of GT buffer (−N-lauroylsarcosine m/v and 2-mercaptoethanol) is undetermined, > 1 year. Use within a day after adding of N-lauroylsarcosine and 2-mercaptoethanol or store at -80 °C (shelf life is undetermined, > 1 year). Mix stock solutions before adding.
AGP (10 mL)
Reagent Final concentration Amount GT buffer 66% v/v 6.67 mL Acidic phenol pH 4.5 n/a 3.33 ml Total n/a 10 mL Add in the order listed. Combine reagents in a 15 mL conical tube and store at RT. Protect from light. Use within a day or store at -80 °C (shelf life is undetermined, > 1 year).
1.0 M Tris-HCl pH 8.0 (50 mL)
Reagent Final concentration Amount or volume TRIS base 1.0 M 6.06 g DEPC-treated ultrapure H2O n/a 43.1 mL Hydrochloric acid n/a 2.35 mL Total n/a 50 mL Add in the order listed. Prepare in a 50 mL conical tube. Dissolve by incubating on a rotator at RT. Filter using a syringe equipped with a 0.2 µm cellulose acetate syringe filter. Store at RT. Shelf life is undetermined, > 1 year.
10% LiDS m/v (50 mL)
Reagent Final concentration Amount or volume LiDS 10% m/v 5 g DEPC-treated ultrapure H2O n/a Adjust volume to 50 mL Total n/a 50 mL Add in the order listed. Prepare in a 50 mL conical tube. Weigh LiDS and adjust the volume of DEPC-treated ultrapure H2O accordingly. Dissolve by incubating on a rotator at RT. Store at 4 °C. Shelf life is undetermined, > 6 months.
1% LiDS TE (50 mL)
Reagent Final concentration Amount or volume DEPC-treated ultrapure H2O n/a 44.4 mL 1 M Tris-HCl pH 8.0 10 mM 500 µL 0.5 M EDTA pH 8.0 1 mM 100 µL 10% LiDS m/v 1% m/v 5 mL Total n/a 50 mL Add in the order listed. Prepare in a 50 mL conical tube and store at RT. Shelf life is undetermined, > 1 year.
2% LiDS TE (2 mL)
Reagent Final concentration Amount or volume DEPC-treated ultrapure H2O n/a 1,552 µL 1 M Tris-HCl pH 8.0 20 mM 40 µL 0.5 M EDTA pH 8.0 2 mM 8 µL 10% LiDS m/v 2% m/v 400 µL Total n/a 2 mL Add in the order listed. Prepare in a 2 mL microcentrifuge tube and store at RT. Shelf life is undetermined, > 1 year.
10 M LiCl (50 mL)
Reagent Final concentration Amount or volume LiCl 10 M 21.20 g DEPC-treated ultrapure H2O n/a 39.76 mL Total n/a 50 mL Add in the order listed. Prepare in a 50 mL conical tube. Add LiCl quickly and close the cap. Adjust the volume of DEPC-treated ultrapure H2O accordingly to limit moisture absorption. Add water to LiCl salt while on ice. Close tube and incubate on ice with occasional inversion. Finish dissolving LiCl by incubating on a rotator at RT. Avoid exposing the tube seal to LiCl salt to prevent leakage. Clarify by centrifugation at 3,000–3,200× g for 30 min at RT, transfer 90% v/v of the clarified portion to a new 50 mL conical tube, and store at RT. Shelf life is undetermined, > 1 year.
Precipitation solution (40 mL)
Reagent Final concentration Amount or volume DEPC-treated ultrapure H2O n/a 5 mL 10 M LiCl 3.75 M 15 mL 100% isopropanol v/v 50% v/v 20 mL Total n/a 40 mL Add reagent components to DEPC-treated ultrapure H2O. Prepare in a 50 mL conical tube and store at RT. Shelf life is undetermined, > 1 year. A small decrease in volume to 36–38 mL after mixing components is expected.
95% MeOH (1 L)
Reagent Final concentration Amount or volume 100% MeOH v/v 95% v/v 950 mL DEPC-treated ultrapure H2O n/a 50 mL Total n/a 1 L Add in the order listed. Store in a glass airtight bottle and/or aliquot into 50 mL conical tubes. Store in flammable storage cabinet when not in use. Shelf life is undetermined, > 1 year. Alternatively, add 210.5 mL of DEPC-treated ultrapure H2O to an unopened 4 L bottle of MeOH.
Laboratory supplies
15 cm culture plates (e.g., Corning, catalog number: 430599)
Cell lifter (e.g., BIOLOGIX, catalog number: 70-2180)
30 mL syringe (e.g., VWR, catalog number: 76124-668)
18–22 G hypodermic needles (e.g., BD, catalog number: 305187)
Sterile syringe filter w/ 0.2 µm cellulose acetate membrane (e.g., VWR, catalog number: 28145-477)
1.5 and 2.0 mL microcentrifuge tubes (1.5 mL tubes must have round bottoms) (e.g., VWR, catalog numbers: 490004-444, 525-1136)
0.2 mL thermocycler tubes (e.g., VWR, catalog number: 20170-012)
15 and 50 mL conical tubes (e.g., Corning, catalog numbers: 430790, 430828)
0.5 and 1 L airtight glass bottles (e.g., VWR, catalog numbers: 89000-238, 89000-240)
Whatman paper (e.g., Sigma-Aldrich, catalog number: WHA1001150)
10, 200, and 1 mL pipette tips (e.g., Genesee Scientific, catalog numbers: 24-121RL, 24-150RS, 23-165RL)
10, 100, 200, and 1,000 µL pipettes (e.g., Gilson or Eppendorf)
10 and 25 mL serological pipettes (e.g., Genesee Scientific, catalog numbers: 12-104, 12-106)
Pipette controller (e.g., Thermo Fisher Scientific, catalog number: 14-387-165)
Kimwipes (e.g., VWR, catalog number: 82003-820)
96-well microplate (e.g., VWR, catalog number: 82050-678)
Thermal adhesive sealing film (e.g., Thermo Fisher Scientific, catalog number: 08-408-240)
Nitrile gloves (e.g., Microflex, catalog number: 92-134)
Repeater pipette (e.g., Eppendorf, catalog number: 022260201)
5 mL Combitips (e.g., Sigma-Aldrich, catalog number: EP0030089456)
Equipment
UV crosslinker (e.g., Stratagene, model: Stratalinker UV 2400 Crosslinker)
Analytical balance (e.g., Mettler Toledo, model: ME54E)
Sample tube rotator (e.g., Fisher Scientific Hematology Chemistry Mixer, model: 346)
Tube rack (e.g., VWR, catalog number: 82010-750)
Refrigerated microcentrifuge (e.g., Eppendorf, model: 5417R)
Centrifuge with 15 mL and 50 mL conical tube holders (e.g., Beckman, model: CS-6R)
Thermocycler (e.g., Bio-Rad, model: T100 PCR Thermocycler)
Hot plate stirrer (e.g., Accuplate, model: D0310)
UV spectrophotometer (e.g., NanoDrop, model: ND-100)
Microplate reader (e.g., VersaMax, model: M5e)
Dry bath/heating block (e.g., Thermolyne, model: 17600)
Mini centrifuge (e.g., ISC BioExpress, model: C1301P-ISC)
37 °C incubator (e.g., Baxter Scientific Products, model: J1450-3)
4 °C refrigerator (e.g., Kenmore, model: 253.6880201E)
-20 °C freezer (e.g., Kelvinator, model: KFU21M7LW3)
-80 °C freezer (e.g., VWR, model: 5706)
Software and datasets
Microsoft Excel for Mac version 16.66.1
SoftMax Pro version 7.2
NanoDrop 1000 version 3.8.1
Procedure
Cell culturing, UV crosslinking, and cell harvest
Grow cells to the desired confluency in a 15 cm plate.
Note: Adjust volumes used during subsequent steps accordingly when employing plates of varying sizes.
Decant media and place the cell culture dish on an ice-filled culture dish lid.
Wash cells twice with 12 mL of ice-cold 1× PBS.
Tilt the plate to an 80° angle while on the ice-filled culture dish lid, incubate for 1 min, and aspirate residual PBS.
Irradiate cells while on the ice-filled culture dish lid with the desired amount of UV energy and at the desired wavelength (e.g., 0.4 J/cm2, 254 nm) using a Stratalinker or other suitable UV crosslinker (General note 1).
Lyse, scrape, and transfer cells into a 2.0 mL microcentrifuge tube on ice using two 0.4 mL aliquots of GT buffer and a cell scraper.
Critical: Pre-wet pipette tips for pipetting accuracy (General note 2).
Add 0.4 mL of acidic phenol for a total of 1.2 mL of AGP (i.e., 0.8 mL of GT and 0.4 mL of acidic phenol) and sheer the lysate by passaging through a 19–20 G needle 15 times. Add additional AGP to a final concentration of 80% v/v.
Note: Keep samples at RT after sheering unless indicated otherwise. Sheered lysates represent starting AGP input suspensions (Table S1).
Caution: Perform this step in a fume hood.
Caution: Avoid exposing tube seals to AGP to prevent sample leakage (General note 3).
Critical: Pre-wet pipette tips for pipetting accuracy (General note 2).
Critical: Mix stock solutions and sample suspensions containing AGP < 10 s prior to sampling (General note 4).
Pause point: Samples can be stored at RT in AGP for 16 h or at -80 °C for long-term storage (General note 5).
For initial compositional analysis of starting AGP input suspensions, transfer two 10–50 µL aliquots to separate 1.5 mL microcentrifuge tubes containing an appropriate volume of AGP for a final volume of 200 µL. Isolate RNP fractions according to the LEAP-RBP method (steps C1–C9), suspend in 10–50 µL of 1% LiDS TE as described in step F1, and perform RNA (UV spectrophotometry) quantitation as described in step F3. Estimate RNA and protein yields of the starting AGP input suspension using the tools included as part of Table S1.
Caution: Perform dilution of starting AGP input suspensions in fume hood.
Caution: Avoid exposing tube seals to AGP to prevent sample leakage (General note 3).
Critical: Pre-wet pipette tips for pipetting accuracy (General note 2).
Critical: Mix stock solutions and sample suspensions containing AGP < 10 s prior to sampling (General note 4).
Pause point: Samples can be stored at RT for 16 h or at -80 °C for long-term storage (General Note 5).
Add AGP to the undiluted starting AGP input suspension for a final concentration of 10–250 ng of RNA and DNA/µL and > 86% AGP v/v for isolation of RNP fractions by LEAP-RBP or 25–2,000 ng of protein/µL for isolation of total input samples by 95% MeOH v/v precipitation (Sections C, E).
Note: A dilution calculator is included as part of Table S1.
Caution: Perform this step in a fume hood.
Caution: Avoid exposing tube seals to AGP to prevent sample leakage (General note 3).
Critical: Pre-wet pipette tips for pipetting accuracy (General note 2).
Critical: Mix stock solutions and sample suspensions containing AGP < 10 s prior to sampling (General note 4).
Pause point: Samples can be stored at RT for 16 h or at -80 °C for long-term storage (General note 5).
Repeated AGPC extraction
Transfer 1 mL aliquots of an AGP input suspension containing > 80% AGP v/v to separate 2.0 mL microcentrifuge tubes (General note 8). Add 200 µL of chloroform per milliliter of AGP (v/v), vortex (10 s, maximum setting), and centrifuge at 10,000× g for 10 min at 4 °C with slow brake setting. Carefully remove 80%–90% of the aqueous and organic phases without removing the AGPC interphase (General note 6).
Note: Slow or “soft” brake settings can be found on many tabletop centrifuges (e.g., Eppendorf, model: 5417R). By slowing the rate of deceleration, the likelihood of disturbing the pellet fraction is reduced. Not including this setting when listed may impact yield and/or introduce biases, although this has not yet been established.
Caution: Perform this step in a fume hood.
Caution: Avoid exposing tube seals to AGP or AGPC to prevent sample leakage (General note 3).
Critical: Pre-wet pipette tips for pipetting accuracy (General note 2).
Critical: Mix stock solutions and sample suspensions containing AGP < 10 s prior to sampling (General note 4).
Perform additional AGPC extractions as described in the previous step using 800 µL of AGP and 160 µL of chloroform per 2.0 mL microcentrifuge tube per extraction.
Note: AGPC interphase samples can be pooled 2:1 before each additional AGPC extraction (General note 7). The required number of repeated AGPC extractions depends on protein UV-crosslinking efficiency and sample composition (General note 9).
Caution: Perform this step in a fume hood.
Caution: Avoid exposing tube seals to AGP or AGPC to prevent sample leakage (General note 3).
Critical: Remove most of the organic phase before the next step.
Critical: Pre-wet pipette tips for pipetting accuracy (General note 2).
Critical: Mix stock solutions and sample suspensions containing AGP < 10 s prior to sampling (General note 4).
Add 1 mL of AGP to each final AGPC interphase sample and pool if applicable. Determine RNA yield according to step A8. Dilute final AGPC interphase suspensions to 10–250 ng of RNA/µL for isolation of RNP fractions by LEAP-RBP (Section C).
Caution: Perform this step in a fume hood.
Caution: Avoid exposing tube seals to AGP or AGPC to prevent sample leakage (General note 3).
Critical: Pre-wet pipette tips for pipetting accuracy (General note 2).
Critical: Mix stock solutions and sample suspensions containing AGP < 10 s prior to sampling (General note 4).
Pause point: Samples can be stored at RT for 16 h or at -80 °C for long-term storage (General note 5).
Isolation of RNP fractions by LEAP-RBP
Transfer 200 µL aliquots of an AGP input suspension or final AGPC interphase suspension to separate 1.5 mL microcentrifuge tubes (General note 8). Mix by continuous vortex within 1 h of the next step (10 s, setting 4–5). Keep samples off the lid.
Caution: Perform this step in a fume hood.
Caution: Avoid exposing tube seals to AGP or AGPC to prevent sample leakage (General note 3).
Critical: Pre-wet pipette tips for pipetting accuracy (General note 2).
Critical: Mix stock solutions and sample suspensions containing AGP < 10 s prior to sampling (General note 4).
Pause point: Samples can be stored at RT for 16 h or at -80 °C for long-term storage (General note 5).
To each 1.5 mL microcentrifuge tube: Add 14 µL of chloroform and mix by pulse vortex until an emulsion forms (1 s intervals, setting 4–5). Then, mix by continuous vortex (10 s, setting 4–5).
Note: Final AGPC interphase suspensions contain residual chloroform. Use a single 200 µL aliquot to determine the optimal amount of chloroform for the remaining aliquots. Briefly, add 12 µL of chloroform and mix by pulse vortex (1 s intervals, setting 4–5). If an emulsion does not form and/or if the emulsion takes on a more greyish appearance (see Kristofich and Nicchitta [18], Supplementary Figure 11c), add an additional 2 µL of chloroform and use 14 µL of chloroform for each of the remaining 200 µL aliquots. Otherwise, use 12 µL of chloroform.
Caution: Perform this step in a fume hood.
Caution: Avoid exposing tube seals to AGP or AGPC to prevent sample leakage (General note 3).
Critical: Pre-wet pipette tips for pipetting accuracy (General note 2).
Critical: Close sample tubes within 10 min of adding chloroform and vortex within 1 h to avoid a decrease in yield (General note 10).
If samples were stored at -80 °C and/or if starting from Turbo DNase digests, mix samples by vortex before adding chloroform (10 s, setting 4–5).
Gently add/layer 856 µL of RT precipitation solution (3.75 M LiCl, 50% isopropanol v/v) on top of each AGPC mixture and close the tubes.
Note: Add the first 100–200 µL slowly to the inner side of the sample tubes below the seal line.
Caution: Perform this step in a fume hood.
Caution: Avoid exposing tube seals to AGP, AGPC, or precipitation solution to prevent sample leakage (General note 3). Note that water condensation near the seal of the tube, which forms above the precipitation solution, will not cause sample leakage during subsequent steps.
Critical: Pre-wet pipette tips for pipetting accuracy (General note 2).
Critical: Perform this step within 1 h of the previous step to avoid a decrease in yield (General note 10).
Using a sample tube rack, gently invert tubes to a 90° angle (> 1 s) and hold until the AGPC mixtures become displaced from the bottom of the tubes (0.5–1.0 s). Revert the tubes back to an upright position at a slightly faster speed (0.5–1.0 s) and let stand for 1 min at RT. Repeat this process twice more in alternating directions for three rounds of gentle inversions/reversions followed by additional rounds with increasing angle and speed. Once most of the AGPC mixture becomes displaced from the bottom of vertically standing sample tubes, perform a final round inversion with a forceful reversion, let stand for 30 s, and vortex (5 s, maximum setting).
Note: Sample appearance and behavior will differ depending on clRNP and protein content of AGP suspensions. While this process can be completed after only five inversions, trying to complete the process in ten or more inversions will reduce the chance of overmixing without compromising results (Video S1).
Critical: Perform the first inversion/reversion step within 1 h of adding the precipitation solution and/or complete all inversion/reversion steps within 1 h and 10 min to avoid an increase in free protein recovery.
Pause point: Samples can be stored at RT for 16 h after mixing by vortex (General note 5).
Using a sample tube rack, perform two inversions with forceful inversions in opposite directions. Then, centrifuge sample tubes at 14,000× g for 5 min at 20 °C.
Critical: Not performing inversions with sufficient force results in scattered or non-uniform RNP pellets, which can negatively impact yields.
Pause point: Samples can be stored at RT for 16 h (General note 5).
Remove supernatants and partially close lids. Add 1 mL of RT 95% MeOH v/v to each sample tube and close lids. Complete the following process for one sample tube at a time: Invert three times, pausing briefly for < 1 s before each reversion, and quickly remove/discard the MeOH washes using a syringe equipped with an 18–22 G needle. Keep tube lids partially closed between MeOH washes. Repeat the process twice more using two instead of three inversions.
Note: Allow samples to incubate in 95% MeOH v/v for at least 5 min across all three washes. The white precipitate, which forms after adding 95% MeOH v/v, will be removed during the washes. It is not necessary to remove residual 95% MeOH v/v adhering to the cap or sides of tubes.
Caution: Perform this step in a fume hood.
Critical: Not removing the washes quickly after inverting tubes allows precipitates containing free protein to settle and contaminate RNP fractions.
Critical: If an RNP pellet is not dislodged from the bottom of the tube before removing the final MeOH wash, gently nudge the top and/or side of the pellet with a syringe or pipette tip and resume at the inversion step.
Critical: If RNP pellets are small and thus fragile, add and remove 95% MeOH v/v washes using a P1000 to avoid sample loss.
Pause point: Samples can be stored at RT in any of the three 95% MeOH v/v washes for 16 h (General note 5).
Transfer and/or pool RNP pellets into a new 1.5 mL microcentrifuge tube. Briefly, pour 1 mL of RT 95% MeOH v/v from a new tube into the tube containing the RNP pellet at a moderate rate for 0.5–1.0 s until the RNP pellet becomes displaced from the bottom of the tube and then quickly pour it back into the new tube.
Note: If pooling multiple RNP pellets, wait until RNP pellets have settled in the new tube before repeating the process.
Caution: Perform this step in a fume hood or limit exposure to MeOH.
Pause point: RNP pellets can be stored in 95% MeOH v/v at RT for 16 h or at -80 °C for long-term storage after being transferred to a new tube (General note 5).
Remove most of the 95% MeOH v/v leaving 50–200 µL. Centrifuge briefly and use a P1000 pipette equipped with a P10 pipette tip attached to a P1000 pipette tip to remove residual 95% MeOH v/v. Air dry pellets by incubating the tube with the lid open for 5–10 min at RT.
Note: RNP pellets, which have been pooled into the same tube, should be spread out while removing residual 95% MeOH v/v so they dry more uniformly.
Caution: Perform this step in a fume hood or limit exposure to MeOH.
Pause point: Opened sample tubes can be incubated overnight at RT if necessary. Dried RNP pellets can be stored at -80 °C for long-term storage (General note 5).
For RNP fractions isolated from diluted starting AGP input suspension (step A8), resuspend as described in step F1, perform UV-spectrophotometric analysis of unclarified sample suspensions as described in step F3, and analyze the data using the tools provided in Table S1. Otherwise, proceed to DNA depletion (Section D).
DNA depletion
Add 15 µL of TE buffer to dry RNP pellet(s) containing 10–50 µg of RNA and DNA and incubate for > 2 min at RT. Using the same pipette tip, pipette 5 µL of the sample suspension 8 times without submerging the pipette tip in the sample suspension. Incubate for > 2 min and repeat the pipetting step using the same pipette tip. During the second pipetting step, scrap the bottom of the tube while ejecting.
Note: Pooling RNP pellets to maximize the amount of RNA and DNA per tube reduces the time-sensitivity of the second LEAP step following Turbo DNase digestion (General note 10). An empty pipette tip box is a convenient way to store pipette tips between pipetting steps.
Critical: Not maintaining sample suspensions at the bottom of the tube during the resuspension procedure results in scattered precipitates during the second LEAP step and lower yields.
Critical: If dried RNP pellets have become dislodged from the bottom of the tube prior to adding TE buffer, centrifuge sample tubes at 3,000× g for 5 s.
Critical: Submerging the pipette tip and/or scraping the bottom of the tube during the first pipetting step can result in hydrated RNP pellets becoming stuck to the side of the pipette tip. In these scenarios, gently remove the pipette tip and allow it to incubate while submerged in the sample suspension between pipetting steps. Do not attempt to rub the pipette tip against the sides of the tube.
Pause point: Sample suspensions, which were kept at the bottom of the tubes, can be stored at RT for 1 h.
Using a new pipette tip for each sample, add 5.0 µL of a pre-mixed master mix containing 2.0 µL of 10× Turbo DNase Buffer and 1.0 µL of Turbo DNase enzyme per 10.0 µg of DNA (0.2 µL minimum and 1.5 µL maximum) supplemented with TE buffer to 5 µL.
Note: The pre-mixed master mix should be prepared after the previous step. DNA quantity based on the change in RNA quantity (UV spectrophotometry) is observed after the second LEAP step when using the maximum amount of Turbo DNase.
Critical: Not maintaining sample suspensions at the bottom of the tube results in scattered precipitates during the second LEAP step and lower yields.
Critical: UV-crosslinked samples begin to adhere to pipette tips after adding Turbo DNase buffer. Add the entire volume of the pre-mixed master mix directly to the sample suspension and swirl the pipette tip for < 3 s to mix without pipetting.
Incubate samples for 15 min at 37 °C.
Add 180 µL of AGP and mix by vortex (10 s, setting 5).
Note: Aliquots of AGP can be prepared ahead of time and stored at -80 °C (General note 5).
Caution: Perform this step in a fume hood.
Caution: Avoid exposing tube seals to AGP or AGPC to prevent sample leakage (General note 3).
Critical: Pre-wet pipette tips for pipetting accuracy (General note 2).
Critical: Perform this step within 15 min of the previous step to avoid a decrease in yield (General note 10).
Pause point: Sample tubes can be centrifuged briefly at 3,000× g (quick spin) and stored at RT for 16 h or -80 °C for long-term storage (General note 5).
Isolate RNP fractions by LEAP-RBP (start at step C2).
Isolation of input (total protein) samples
Transfer 50 µL aliquots of an AGP input suspension containing > 80% AGP v/v and 25–2,000 ng of protein/µL to separate 2 mL microcentrifuge tubes.
Caution: Perform this step in a fume hood.
Caution: Avoid exposing tube seals to AGP or AGPC to prevent sample leakage (General note 3).
Critical: Pre-wet pipette tips for pipetting accuracy (General note 2).
Critical: Mix stock solutions and sample suspensions containing AGP < 10 s prior to sampling (General note 4).
Pause point: Samples can be stored at RT for 16 h or at -80 °C for long-term storage (General note 5).
Mix samples by vortex (5 s, maximum setting), add 950 µL of RT 100% MeOH v/v, and vortex (10 s, maximum setting).
Caution: Perform this step in a fume hood.
Caution: Avoid exposing tube seals to AGP or AGPC to prevent sample leakage (General note 3).
Critical: Pre-wet pipette tips for pipetting accuracy (General note 2).
Pause point: Samples can be stored at RT for 16 h or at -80 °C for long-term storage (General note 5).
Incubate sample tubes on a rotator for 1 h at RT and centrifuge at 20,000× g for 10 min at 20 °C with slow-brake setting. Remove supernatants using a syringe equipped with an 18–22 G needle and partially close tube lids.
Caution: Perform this step in a fume hood.
Caution: Avoid exposing tube seals to AGP or AGPC to prevent sample leakage (General note 3).
Add 1 mL of RT 95% MeOH v/v to each sample tube, close lids, and vortex (5 s, maximum setting). Incubate sample tubes on a rotator for 10 min at RT, centrifuge at 20,000× g for 10 min at 20 °C with slow-brake setting, and remove supernatants using a syringe equipped with an 18–22 G needle. Repeat this process once or twice if pooling > 100 µg of protein during the next step. Keep tube lids partially closed between MeOH washes.
Note: Slow or “soft” brake settings can be found on many tabletop centrifuges (e.g., Eppendorf 5417R). By slowing the rate of deceleration, the likelihood of disturbing the pellet fraction is reduced. Not including this setting when listed may impact yield and/or introduce biases although this has not yet been established.
Caution: Perform this step in a fume hood.
Critical: Not removing the washes quickly and gently after centrifugation can result in sample loss.
Critical: Keep precipitates off the lid during centrifugation by holding sample tubes vertically and moving them in a single up-down motion. Repeat the process until white precipitates are no longer visible on the bottom of tube lids.
Pause point: Samples can be stored at RT in 95% MeOH v/v washes for 16 h (General note 5).
Use three 400 µL aliquots of RT 95% MeOH v/v to recover precipitates and transfer them into a new 1.5 mL microcentrifuge tube (General note 11). Keep tube lids partially closed after, between removing and adding 95% MeOH v/v.
Note: This process can be used to pool precipitates from multiple 2 mL microcentrifuge tubes. Additional aliquots can be used if necessary.
Caution: Perform this step in a fume hood or limit exposure to MeOH.
Pause point: Precipitates can be stored in 95% MeOH v/v at RT for 16 h or at -80 °C for long-term storage after being transferred to a new tube (General note 5).
Incubate sample tubes vertically for 30 min at RT to allow precipitates to settle at the bottom of the tube. Centrifuge at 20,000× g for 10 min at 20 °C with a slow-brake setting and remove most of the supernatant, leaving 50–200 µL. Repeat the centrifugation step and remove residual 95% MeOH v/v using a P1000 pipette equipped with a P10 pipette tip attached to a P1000 pipette tip.
Caution: Perform this step in a fume hood or limit exposure to MeOH.
Critical: Not removing the washes quickly and gently after centrifugation can result in sample loss.
Use a P20 pipette and 2 µL of RT 95% MeOH v/v to break up and spread out precipitates so they dry more uniformly. Incubate opened sample tubes for 10–30 min at RT or until precipitates are dry.
Note: Precipitates should be spread out before they start to dry.
Caution: Perform this step in a fume hood or limit exposure to MeOH.
Critical: Not spreading out precipitates will make resuspension more difficult (Section F).
Pause point: Opened sample tubes can be incubated overnight at RT if necessary. Dried precipitates can be stored at -80 °C for long-term storage (General note 5).
Sample resuspension, clarification, and quantitation
Add 1% LiDS TE to dried RNP pellets or precipitates and incubate for 30 min at RT with occasional pipetting at the 2, 15, and 30 min mark. Briefly, pipette 90% of the sample volume eight times, scraping the bottom of the tube while ejecting the sample. During the initial pipetting step (2 min mark), pipette samples until most of the visible precipitates are suspended and not adhering to the sides of the tube.
Note: The volume of 1% LiDS TE added to samples and the desired concentration depends on the intended downstream application(s). In particular, steps F2–4 are intended for samples suspended above their working concentration (i.e., 4.0 µg of RNA and DNA/µL for RNP fractions and 5.0 µg of protein/µL for input samples). If samples are suspended below their working concentrations, proceed to step F5, and adjust sample dilutions during step F6 accordingly (if applicable) so they fall within the linear quantitation range of UV spectrophotometer and/or BCA (Table S2). Efforts should be made to not introduce bubbles during the pipetting steps. Nonetheless, they will be removed during the clarification procedure. The same pipette tip can be used for all three pipetting steps by storing them in a pipette tip box between pipetting steps.
Critical: Do not store samples on ice. Samples do not contain active proteases and/or nucleases, so storage on ice is not necessary.
Critical: Samples should not be stored after starting the resuspension procedure and prior to the clarification step.
Mix samples by vortex (5 s, setting 5) and quickly (< 10 s) transfer 2 µL to a 0.2 mL thermocycler tube containing 8 µL of 1% LiDS TE for a 1:5 dilution. Rinse pipette tips after transferring by pipetting the added volume three times.
Critical: Samples suspended well above their working concentrations are difficult to pipette accurately. If this is the case, measure the pipetted sample volume using the 2 µL graduation mark that can be found on most standard P10 pipette tips.
Analyze 1–2 µL of diluted (1:5) sample suspensions by UV spectrophotometry (Nanodrop, RNA setting) and calculate the concentration of undiluted sample suspensions (Table S2).
Note: DNA and protein contributions confound RNA quantitation. Nonetheless, it provides a reliable and rapid means for diluting samples to their working concentration.
Critical: Pipette 90% of the sample volume five times < 10 s before analyzing samples.
Critical: The recommended range for quantitation of 1% LiDS TE sample suspensions is 0.1–1.8 µg of RNA&DNA for RNP fractions and 0.1–1.0 µg of RNA&DNA for input sample as determined by UV spectrophotometry (Nanodrop, RNA setting).
Critical: Read the same 1–2 µL aliquot twice. If the values vary by more than 10%, rigorously clean the pedestal of the Nanodrop UV spectrophotometer and repeat step F3. This often occurs with RNP fractions containing a high amount of crosslinked RNA-protein when they are suspended above 1.8 µg of RNA/µL and/or when not adequately mixing samples prior to UV-spectrophotometric analysis.
Add 1% LiDS TE to undiluted sample suspensions to a final concentration of 1 µg of RNA/µL for input samples and 4 µg of RNA/µL for RNP fractions as determined by UV-spectrophotometric analysis. Mix samples by brief vortex (5 s, setting 5) and incubate for 5 min at RT.
Pause point: Diluted sample suspensions can be stored at RT for 2 h.
Incubate samples in a dry bath/heating block or thermocycler at 65 °C for 3 min, vortex (5 s, maximum setting), and centrifuge at 270–310× g for 5 min at 20 °C with soft-brake setting. Carefully transfer 90% of the clarified fraction to a separate 1.5 mL microcentrifuge tube.
Note: If working with sample volumes < 100 µL, this step should be performed in 0.2 mL thermocycler tubes.
Clarified input (total protein) and RNP (total RNA-bound protein) fractions should be diluted as described in step F2 for RNA and protein quantitation.
Note: Sample dilutions can be calculated using the tools included as part of Table S2.
Pause point: Diluted and/or undiluted clarified sample suspensions can be used immediately or stored at -80 °C (General note 5).
RNA quantitation of clarified RNP fractions should be performed as described in step F3 and data should be analyzed using the tools included in Table S3 for calculation of total RNA yields.
Perform protein quantitation using the BCA microplate assay. Briefly, dilute 1 mL of a BSA standard (2 mg/mL) with 1 mL of 2% LiDS TE (D1) and use 500 µL to prepare seven 1:2 serial dilutions using 1% LiDS TE (D2–D8). Prepare the BCA working reagent and place a 96-well microplate on ice. Mix diluted clarified sample suspensions prepared in step F6 by vortex (5 s, setting 5) and quickly (< 10 s) transfer two 2–4 µL aliquots to separate wells (Table S2). Add two 2 µL aliquots of 1% LiDS TE and each of the BSA standards to separate well in descending order of their dilution number (D#). Then, add an additional two 4 µL aliquots of D1 to separate wells. Promptly (< 3 min) add 200 µL of the BSA working reagent to each well containing BSA standards in ascending order of their dilution number (D#). Lastly, add 200 µL of the BSA working reagent to each of the remaining wells containing either 1% LiDS TE or aliquoted sample suspensions. Incubate the plate on ice for 5 min, attach thermal adhesive sealing film, and incubate at 37 °C for 2.5–3 h. Remove the sealing film and measure absorbance at 562 nm using a microplate reader and appropriate software (e.g., SoftMax Pro version 7.2, standard curve setting: bi-exponential).
Note: Protein quantitation data can be exported and analyzed using the tools included in Table S3 for the determination of total protein and RNA-bound protein yields. BSA standards can be prepared ahead of time, aliquoted, and stored at -80 °C. Once thawed, BSA standards can be kept at 4 °C for 6 months. The appearance of precipitates in BSA standard D1 when stored at 4 °C is expected and will be resolved after equilibrating it to RT. Load > 0.25 µg of protein per well; non-crosslinked RNP fractions are an exception (General note 12).
Critical: Keep samples and BSA standards at room temperature when setting up the plate and use a new pipette tip for each aliquot. Add aliquots directly to the bottom side of the wells without touching the bottom, as this can scratch the plate and confound results. Add the BCA working reagent to the same side of the well.
Critical: Set up the microplate on ice to avoid the impact that differences in incubation times have on protein quantitation data.
Data analysis
Data analysis methods are detailed in the original LEAP-RBP study as part of the Supplementary Methods [18]. Briefly, LEAP-RBP fractions can be analyzed by SRA to evaluate UV-dependent enrichment and S/N of proteins. Input samples and LEAP-RBP fractions can be normalized to micrograms of protein, RNase-digested, and compared by SDS-PAGE to evaluate total and RNA-bound abundance, respectively. Total protein and total RNA crosslinking efficiencies can be estimated using RNA and protein yields of input (total protein) samples, LEAP-RBP fractions isolated from AGP input suspensions containing total RNA and total RNA-bound protein, and LEAP-RBP fractions isolated and final AGPC interphase suspensions containing total RNA-bound protein and total protein-bound RNA. SILAC LC-MS/MS analysis of LEAP-RBP fractions isolated from pooled cellular samples containing equivalent amounts of UV-crosslinked and non-crosslinked cells allows comprehensive identification of UV-enriched candidate RBPs. Comparison of input samples and corresponding LEAP-RBP fractions by LC-MS/MS informs condition- and/or context-dependent differences in total and RNA-bound abundances, respectively. Lastly, LC-MS/MS analysis of input samples and LEAP-RBP fractions allows proteome-wide determination of UV-crosslinking efficiencies (%CL) for functional validation of candidate RBPs and %CL-based ranking of protein features and classifications [20].
Validation of protocol
Validation of LEAP-RBP and the methods herein can be found in the original publication [18]. Briefly, the ability of LEAP-RBP to rapidly recover total RNA and/or bound proteins from AGP sample suspensions with near 100% efficiency was validated by SRA analysis of precipitated and unprecipitated fractions. The importance of components added during the LEAP step and the liquid–liquid interphase was evidenced by a change in RNA and/or protein yields. RNA-centricity of the LEAP-RBP method was validated by performing LEAP-RBP on RNase-treated clRNP fractions resulting in a loss of detectable protein by SDS-PAGE and Coomassie Blue staining. Efficiency of DNA depletion steps was validated by qPCR and SRA analysis. RNA isolated by LEAP-RBP method was found to be of high integrity by Bioanalyzer analysis (RIN > 9), and an RNA-seq analysis showed broad representation of diverse sRNA species displaying broad genome distributions. The efficiency of DNA and/or RNA depletion steps used for the preparation of total protein and total RNA-bound protein samples for MS proteomic was validated by compositional analyses (i.e., RNA and protein quantitation) and by comparison using both TBE gel analysis and SDS-PAGE with SYBR Safe (RNA and DNA), Coomassie Blue (protein), and Silver Stain (RNA, DNA, and protein) staining.
General notes and troubleshooting
General notes
Differences in UV-crosslinking conditions will affect total protein and protein-specific crosslinking efficiencies (Supplementary Figure 13 [18]). Importantly, this includes factors such as plate height, plate diameter, and position in the UV crosslinker. While most crosslinkers have a sensor meant to increase exposure time as the bulb energy output declines from age, it is not meant to measure the UV dose from individual bulbs and make the necessary adjustments. Because not all RNA–protein interactions exhibit similar crosslinking dose-responsiveness (Supplementary Figure 6d [18]), we highly recommend experiments to account for position-dependent differences in UV dose across experimental sample groups.
Pre-wet pipette tips by pipetting to and from a sample and/or stock solution a few times before transferring a set volume (Figure S1, Video S2).
Exposing the seal of standard laboratory tubes to AGP, AGPC, or lithium-supplemented precipitation solution (3.75 M LiCl, 50% isopropanol v/v) prior to closing the lid results in sample leakage. If this occurs, clean the seal using a Kimwipe prior to closing the tube. A snapping sound when closing the tube is indicative of a tight seal. Importantly, opening microcentrifuge tubes while AGP is on the bottom of the cap will expose gloves as evidenced by a white residue. Performing the procedures as recommended should prevent exposure and/or sample leakage. Acidic phenol poses serious health risks and can penetrate most standard laboratory gloves within minutes. Use appropriate gear and/or replace contaminated gloves under their reported penetration time. Always maintain open containers and/or sample tubes containing AGP in a fume hood.
AGP will slowly separate into two phases over time. Therefore, mix samples and/or stock solutions that contain AGP by vortex (if < 50% of tube volume: 5 s, setting 5; otherwise, 5 s, maximum setting), inversion (four times), or by pipetting (> 50% sample volume four times) < 10 s prior to sampling.
When indicated, samples can be stored at RT for a short time or at -80 °C for long-term storage (i.e., undetermined, > 1 year). We have observed negligible impact on RNA and/or protein integrity for samples even after six freeze/thaw cycles in AGP and/or 1% LiDS TE. Therefore, we recommend storing samples at -80 °C regardless of the anticipated duration. If desired, RNA integrity can be evaluated by diluting RNP fractions with TE buffer to 0.16% LiDS and analyzing 0.5 µg of RNA by TBE gel electrophoresis, while protein integrity can be evaluated by SRA analysis and total protein staining as described in the original publication [18]. Samples and/or stock solutions (e.g., aliquots of AGP) stored at -80 °C should be thawed by incubating on the bench for 30 min at RT before being used. Notably, volumes larger than 1.5 mL may require longer incubation times to thaw and equilibrate to RT.
A detailed description of the repeated AGPC extraction procedure has been included as part of the Methods and Supplementary Methods in the original publication [18]. Briefly, use a P1000 pipette tip to remove most of the aqueous phase leaving 100–150 µL. Carefully tilt the sample tube and, using a gel loading pipette tip equipped with a new P1000 pipette tip, slide down against the bottom side of the tube through a now exposed region of the organic phase until it touches the bottom of the tube. Carefully begin to aspirate the organic phase while observing the AGPC interphase and stop once it starts to break apart and/or gets close to the pipette tip. Note that this often occurs just as the AGPC interphase falls below the 250 µL graduation mark on the side of the tube. After pausing, slowly return the tube to a vertical position and attempt to remove more of the organic phase. Lastly, resume the tilted position as before and quickly remove the pipette tip at the previously used entry point.
Pooling AGPC interphase samples is an effective strategy for concentrating crosslinked RNA–protein complexes from very dilute samples. Briefly, use a new P1000 pipette to aspirate 800 µL of the AGP stock solution and eject 200–300 µL into one of two AGPC interphase samples that are going to be pooled. Continuing, aspirate the second AGPC interphase sample and transfer it to the tube containing the first AGPC interphase sample by ejecting 200–300 µL. Lastly, use the 500–600 µL of AGP remaining in the pipette tip to rinse the tube that contained the second AGPC interphase sample before transferring the entire volume back to the tube containing both AGPC interphase samples. A similar process should be used to transfer one or two AGPC interphase sample(s) to a clean 2 mL microcentrifuge tube if the seals become compromised (General note 3).
We find that accurate and consistent sampling of AGP input and final AGPC interphase suspensions requires mixing samples and pre-wetting pipette tips after every four to five aliquots using standard pipettes (General notes 2, 4). However, using a repeater pipette greatly simplifies the process and reduces the likelihood of sample leakage when sample volumes are large. For example, consider a scenario where 200 µL aliquots of a 10 mL AGP input suspension need to be transferred to separate 1.5 mL microcentrifuge tubes. If this is approached using a repeater pipette equipped with a 5 mL Combitip, the sample can be mixed by pipetting 5 mL four times before and/or after every eight 200 µL aliquots without the need for pre-wetting the pipette tip. The alternative approach using a P200 or P1000 pipette would require pre-wetting the pipette tip thereby reducing the number of aliquots that can be transferred before having to mix the sample down to four. If taking the second approach, we recommend using an appropriately sized tube to allow mixing by vortex without exposing the seal of the tube. For example, a 10 mL AGP input suspension can be mixed by vortex when it is contained in a 50 mL conical tube without it touching the tube seal (5 s, setting 5). Aliquoted samples should be centrifuged briefly (stop before getting to 3,000× g) before storing them at -80 °C (Video S1).
We recommend performing repeated AGPC extractions until the size of the interphase stops reducing in size. However, additional AGPC extractions can be employed to pool AGPC interphase samples (General note 7). The number of repeated AGPC extractions required is often between 2 and 8. Notably, additional AGPC extractions can be performed after resuspending AGPC interphase samples in AGP if desired, including those stored at -80 °C while performing an initial yield assessment (General note 5).
The time sensitivity of all steps of the LEAP-RBP has been thoroughly investigated. Briefly, we note that the time after adding chloroform and before mixing to form an AGPC mixture or after mixing to form an AGPC mixture and before adding/layering the precipitation solution can affect RNA-bound protein recovery, while the time after adding/layering the precipitation solution and before inversions/reversions can affect free protein recovery. Importantly, we did not observe an effect when limiting the duration of these steps to 1 h, and extending the duration of any step by incubating overnight at room temperature had no discernible effect on RNA integrity. More generally, we note that the time-sensitivity of steps that impact yield can be reduced by increasing the concentration of RNA and/or RNA-bound protein in AGP suspensions and the time-sensitivity of steps that impact free protein recovery can be reduced by decreasing the concentration of free protein in AGP suspensions. The most likely situation where time sensitivity becomes an issue is when performing LEAP-RBP on Turbo DNase digests containing a low amount of RNA and/or RNA-bound protein. Here, the time after adding chloroform and before mixing to form an AGPC mixture or after mixing to form an AGPC mixture and before adding/layering the precipitation solution can negatively affect yield. Therefore, we highly recommend pooling RNP pellets isolated from more diluted samples prior to the DNA depletion step and/or reducing the number of samples being processed in parallel during the time-sensitivity steps noted above.
To facilitate pooling and/or resuspension of input samples in small volumes, use three 400 µL aliquots to recover precipitates adhering to the sides of tubes and transfer them to a new 1.5 mL microcentrifuge tube. Note that this process is aided by using two P1000 pipettes and setting one to 425 µL and the other to 1 mL. Setting the pipette to 425 µL removes the need to pre-wet the pipette tip during the procedure. Briefly, following the removal of the final methanol wash, add 400 µL of RT 95% MeOH v/v to one of the sample tubes for each sample group and partially close the lids. Then, for one sample group at a time, use the second pipette to aspirate 200–400 µL of the methanol wash using the second pipette and carefully scrape down the sides of the tube while ejecting to dislodge precipitates. Typically, this step requires two complete passes around the inside of the tube. If pooling precipitates from multiple sample tubes, aspirate the entire volume, partially close the cap, and add it to the next tube to be pooled. After completing this process for all sample tubes of a given sample group, transfer the methanol wash and precipitates to a 1.5 mL microcentrifuge tube. For the remaining two aliquots of RT 95% MeOH v/v, closing the tube and mixing by vortex (3 s, maximum setting) is sufficient for collecting any remaining precipitates.
Non-crosslinked RNP fractions lack quantifiable amounts of protein and should not be used as a background control for UV-crosslinked RNP fractions due to the observation that recovery of non-crosslinked proteins by LEAP-RBP is dependent on RNA-bound proteins [18]. Nonetheless, for instances where they are included for comparison to UV-crosslinked samples, we recommend using a similar amount of RNA. For example, if UV-crosslinked samples containing > 0.25 µg of protein contain > 2.86 µg of RNA, use > 2.86 µg of RNA for non-crosslinked samples. Alternatively, use an equivalent percent fraction given a comparable amount of starting material.
For experiments where precision is key, we suggest increasing the number of aliquots used when preparing sample fractions and pooling prior to resuspension and/or after RNA and protein quantitation. For example, an RNP fraction prepared from one 200 µL aliquot of an AGP input suspension is less representative of the entire starting sample than an RNP fraction prepared from three 200 µL aliquots. Furthermore, processing each of the three 200 µL aliquots separately would help to reduce the variability introduced during specific steps of the LEAP-RBP procedure. This could include, for example, performing inversions/reversions for each aliquot independently by placing them on three separate sample racks. These types of measures do not replace the need for biologically independent replicates when studying biology-related phenomena, but they do help to reduce the likelihood that technical sources of variation (i.e., batch effects) impact biologically relevant observations.
Troubleshooting
| Problem or issue | Likely cause(s) | Possible solution(s) |
| Tugging of the AGPC interphase when removing the aqueous phase during the repeated AGPC extraction procedure. | Insufficient shearing of cell lysates during cell harvest. | Centrifuge sample tubes prior to shearing, increase the number of passages when shearing lysates, increase the speed of passaging. |
| RNP pellets do not resuspend during the Turbo DNase digestion step. | Attempting to add Turbo DNase Buffer to dry pellets. | Complete resuspension of RNP pellets in TE buffer prior to adding Turbo DNase Buffer. |
| RNP pellets are brittle and break apart during 95% MeOH v/v washes. | Centrifugation at 4 °C following inversions/reversions. | Centrifuge samples at 20 °C. |
| Adding an excessive amount of Turbo DNase buffer during the DNA depletion step. | Do not pre-wet pipette tips before adding pre-mixed master mix containing Turbo DNase buffer, Turbo DNase, and TE buffer to TE suspensions. | |
| Forceful inversions during the washing procedure. | Reduce speed and force when performing inversions. | |
| Insufficient forceful reversion(s) prior to the centrifugation during the LEAP-RBP procedure. | Using a sample tube rack, invert sample tubes until mixtures become displaced from the bottom of tubes and then perform a forceful reversion. Repeat the process in the opposite direction. | |
| High free-protein recovery in LEAP-RBP fractions. | Incubating for longer than recommended after adding/layering precipitation solution to AGPC mixtures. | Decrease the time to complete inversions/reversions after adding/layering the precipitation solution. |
| Not removing 95% MeOH v/v washes quickly after inverting tubes. | Begin to remove the 95% MeOH v/v wash before the pellet is allowed to settle. | |
| Attempting to invert and quickly remove 95% MeOH v/v washes from multiple sample tubes at the same time. | Invert and quickly remove the 95% MeOH v/v wash from one tube at a time. | |
| Insufficient denaturing of non-crosslinked RNA–protein and protein–protein interactors due to insufficient mixing of AGPC mixtures. | Mix samples after adding chloroform by vortex for at least 10 s at the highest speed setting. | |
| Input samples and/or RNP pellets are not resuspended in 30 min using the recommended procedure. | Attempting to dry a large number of precipitates or RNP pellets in the same sample tube. | Spread out precipitates or RNP pellets so they dry more uniformly. |
| Colored flakes in samples. | Degradation of graduation marks on serological pipettes after contacting phenol. | Work quickly when transferring phenol using serological pipettes, < 2 min. |
| Plastic in samples. | Melting of serological pipettes made of polystyrene after contacting chloroform or degradation of polypropylene tubes used to store chloroform for >1 h. | Use pipette tips to transfer chloroform and decrease the time that chloroform is stored in polypropylene tubes to less than 1 h. |
| Unexpected decrease in LEAP-RBP yield. | Chloroform stock has degraded. | Replace the chloroform stock solution or increase the volume of chloroform and precipitation solution used to 15 µL and 860 µL, respectively. |
| RNP pellets extend up along the side of the tube after the first LEAP step. | Not performing a sufficient number of inversions/reversions and/or not performing the final reversion with force. | Increase the number of inversions/reversions to ten or more and increase the force and speed of the final reversion. |
| AGP input or final AGPC interphase suspensions contain more than 300 ng of RNA and DNA/µL. | Dilute AGP input or final AGPC interphase suspensions with AGP. | |
| RNP pellets appear scattered after the second LEAP step. | Not maintaining TE-suspended RNPs at the bottom of tubes. | Follow the recommended pipetting technique. |
| Insufficient mixing before and after adding chloroform. | Ensure that Turbo DNase digests are mixed by vortex after adding 9 parts AGP (10 s, setting 5) and again after adding chloroform (10 s, maximum setting). | |
| Inaccurate or widely variable RNA and protein quantitation data. | Reagents have not been clarified, allowing particulates from the manufacturing processes to contaminate samples and react with the BCA reagent. | Ensure that reagents have been clarified and/or that reagents being used are of sufficient quality. |
| Large sample volumes have made the recommended clarification procedure inadequate. | Aliquot the sample prior to clarification. | |
| Trying to process too many samples in parallel during the clarification step and/or not transferring the clarified portion quickly after centrifugation. | Reduce the number of sample suspensions being clarified and transfer the clarified portion quickly (< 5 min) after centrifugation. | |
| Samples suspended in 1% LiDS TE have been stored at 4 °C or RT for extended periods of time before RNA and protein quantitation. | Quantitate or store samples at -80 °C within 6 h of them being resuspended in 1% LiDS TE. | |
| Phenol and/or GT have not been efficiently depleted from sample fractions during 95% MeOH v/v washes; for representative UV-spectrophotometric profiles of uncontaminated sample fractions, see Table S2. | Remove additional supernatant following centrifugation during 95% MeOH v/v washes and/or increase the volume or number of 95% MeOH v/v washes used. |
Acknowledgments
We thank members of the Nicchitta lab and in particular Rose Homoelle for helpful comments and critical feedback. Funding was provided by a grant from the NIH to CVN (GM139480).
Competing interests
The authors certify that they have no affiliations with or involvement in any organization or entity with any financial or non-financial interest in the subject matter and/or materials presented in this manuscript.
References
- Ascano, M., Gerstberger, S. and Tuschl, T. (2013). Multi-disciplinary methods to define RNA–protein interactions and regulatory networks. Curr Opin Genet Dev. 23(1): 20–28.
- Marchese, D., de Groot, N. S., Lorenzo Gotor, N., Livi, C. M. and Tartaglia, G. G. (2016). Advances in the characterization of RNA‐binding proteins. Wiley Interdiscip Rev RNA. 7(6): 793–810.
- McHugh, C. A., Russell, P. and Guttman, M. (2014). Methods for comprehensive experimental identification of RNA-protein interactions. Genome Biol. 15(1): 203.
- Ramanathan, M., Porter, D. F. and Khavari, P. A. (2019). Methods to study RNA–protein interactions. Nat Methods. 16(3): 225–234.
- Smith, J. M., Sandow, J. J. and Webb, A. I. (2021). The search for RNA-binding proteins: a technical and interdisciplinary challenge. Biochem Soc Trans. 49(1): 393–403.
- Vieira-Vieira, C. H. and Selbach, M. (2021). Opportunities and Challenges in Global Quantification of RNA-Protein Interaction via UV Cross-Linking. Front Mol Biosci. 8: e669939.
- Wheeler, E. C., Van Nostrand, E. L. and Yeo, G. W. (2017). Advances and challenges in the detection of transcriptome‐wide protein–RNA interactions. Wiley Interdiscip Rev RNA. 9(1): e1436.
- Nechay, M. and Kleiner, R. E. (2020). High-throughput approaches to profile RNA-protein interactions. Curr Opin Chem Biol. 54: 37–44.
- Van Nostrand, E. L., Freese, P., Pratt, G. A., Wang, X., Wei, X., Xiao, R., Blue, S. M., Chen, J. Y., Cody, N. A. L., Dominguez, D., et al. (2020). A large-scale binding and functional map of human RNA-binding proteins. Nature. 583(7818): 711–719.
- Van Ende, R., Balzarini, S. and Geuten, K. (2020). Single and Combined Methods to Specifically or Bulk-Purify RNA–Protein Complexes. Biomolecules. 10(8): 1160.
- Esteban‐Serna, S., McCaughan, H. and Granneman, S. (2023). Advantages and limitations of UV cross‐linking analysis of protein–RNA interactomes in microbes. Mol Microbiol. 120(4): 477–489.
- Trendel, J., Schwarzl, T., Horos, R., Prakash, A., Bateman, A., Hentze, M. W. and Krijgsveld, J. (2019). The Human RNA-Binding Proteome and Its Dynamics during Translational Arrest. Cell. 176: 391–403.e19.
- Queiroz, R. M. L., Smith, T., Villanueva, E., Marti-Solano, M., Monti, M., Pizzinga, M., Mirea, D. M., Ramakrishna, M., Harvey, R. F., Dezi, V., et al. (2019). Comprehensive identification of RNA–protein interactions in any organism using orthogonal organic phase separation (OOPS). Nat Biotechnol. 37(2): 169–178.
- Urdaneta, E. C., Vieira-Vieira, C. H., Hick, T., Wessels, H. H., Figini, D., Moschall, R., Medenbach, J., Ohler, U., Granneman, S., Selbach, M., et al. (2019). Purification of cross-linked RNA-protein complexes by phenol-toluol extraction. Nat Commun. 10(1): 990.
- Perez-Perri, J. I., Rogell, B., Schwarzl, T., Stein, F., Zhou, Y., Rettel, M., Brosig, A. and Hentze, M. W. (2018). Discovery of RNA-binding proteins and characterization of their dynamic responses by enhanced RNA interactome capture. Nat Commun. 9(1): 4408.
- Castello, A., Fischer, B., Eichelbaum, K., Horos, R., Beckmann, B. M., Strein, C., Davey, N. E., Humphreys, D. T., Preiss, T., Steinmetz, L. M., et al. (2012). Insights into RNA Biology from an Atlas of Mammalian mRNA-Binding Proteins. Cell. 149(6): 1393–1406.
- Baltz, A. G., Munschauer, M., Schwanhäusser, B., Vasile, A., Murakawa, Y., Schueler, M., Youngs, N., Penfold-Brown, D., Drew, K., Milek, M., et al. (2012). The mRNA-Bound Proteome and Its Global Occupancy Profile on Protein-Coding Transcripts. Mol Cell. 46(5): 674–690.
- Kristofich, J. and Nicchitta, C. V. (2023). Signal-noise metrics for RNA binding protein identification reveal broad spectrum protein-RNA interaction frequencies and dynamics. Nat Commun. 14(1): 5868.
- Barlow, J., Mathias, A., Williamson, R. and Gammack, D. (1963). A simple method for the quantitative isolation of undegraded high molecular weight ribonucleic acid. Biochem Biophys Res Commun. 13(1): 61–66.
- Kristofich, J. and Nicchitta, C. (2024). High-throughput quantitation of protein-RNA UV-crosslinking efficiencies as a predictive tool for high confidence identification of RNA binding proteins. RNA. 30(6): 644–661.
Supplementary information
The following supporting information can be downloaded here:
- Figure S1. Graphical representation of pipette tip wetting procedure.
- Table S1. Compositional analysis of starting AGP input suspensions and dilutions calculator for downstream isolation of sample fractions.
- Table S2. Dilution calculator for dilution of sample fractions prior to the clarification step.
- Table S3. Compositional analysis of clarified sample fractions.
- Video S1. Example of the LEAP-RBP inversion step being performed on eight 200 µL aliquots of an AGP suspension containing UV-crosslinked(0.4 J/cm2, 254 nm) HCT116 cells. Notably, samples were incubated for at least 1 min after the first nine inversions and 30 s after the final inversion.
- Video S2. Example of the pre-wetting procedures used for aliquoting AGP input suspensions and chloroform precipitation solution.
Article Information
Publication history
Received: Mar 30, 2024
Accepted: Jun 19, 2024
Available online: Jul 5, 2024
Published: Jul 20, 2024
Copyright
© 2024 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Kristofich, J. and Nicchitta, C. V. (2024). Rapid and Efficient Isolation of Total RNA-Bound Proteomes by Liquid Emulsion–Assisted Purification of RNA-Bound Protein (LEAP-RBP). Bio-protocol 14(14): e5236. DOI: 10.21769/BioProtoc.5236.
Category
Biochemistry > RNA > RNA-protein interaction
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.