- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Protocol to Mine Unknown Flanking DNA Using PER-PCR for Genome Walking
(*contributed equally to this work) Published: Vol 15, Iss 4, Feb 20, 2025 DOI: 10.21769/BioProtoc.5188 Views: 1670
Reviewed by: Alba BlesaMichael KotikAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Mar 2024
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Genome walking, a molecular technique for mining unknown flanking DNAs, has a wide range of uses in life sciences and related areas. Herein, a simple but reliable genome walking protocol named primer extension refractory PCR (PER-PCR) is detailed. This PER-PCR-based protocol uses a set of three walking primers (WPs): primary WP (PWP), secondary WP (SWP), and tertiary WP (TWP). The 15 nt middle region of PWP overlaps the 3' region of SWP/TWP. The 5' regions of the three WPs are completely different from each other. In the low annealing temperature cycle of secondary or tertiary PER-PCR, the short overlap mediates the annealing of the WP to the previous WP site, thus producing a series of single-stranded DNAs (ssDNA). However, the 5' mismatch between the two WPs prevents the template DNA from synthesizing the WP complement at its 3' end. In the next high annealing temperature cycles, the target ssDNA is exponentially amplified because it is defined by both the WP and sequence-specific primer, while non-target ssDNA cannot be amplified as it lacks a binding site for at least one of the primers. Finally, the target DNA becomes the main PER-PCR product. This protocol has been validated by walking two selected genes.
Key features
• The current protocol builds upon the technique developed by Li et al. [1], which is universal to any species.
• The developed protocol relies on the partial overlap among a set of three WPs.
• Two rounds of nested PER-PCRs can generally result in a positive walking result.
Keywords: Genome walkingGraphical overview
Background
In life science–related research, genome walking has achieved various applications, such as identifying transgenes, discovering new genetic resources, and cloning full-length genes, by mining unknown genomic regions flanking known DNA [2–4]. Therefore, genome walking has played an essential role in advancing life sciences–related disciplines like genetics, genomics, and microbiology [5–8].
At present, there are many genome walking methods available. The earliest genome walking method depended on constructing and then screening a genome library, which is laborious and time-consuming. Therefore, this method is seldom adopted now [9,10]. In contrast, PCR-based genome walking has received special attention due to its rapidity, low cost, and ease of operation [11–13]. To date, dozens of PCR-based genome walking methods have been established [14–17]. The existing PCR-based methods can be divided into two groups according to whether genome preprocessing is required preceding PCR: genome preprocessing-dependent PCR (such as panhandle PCR, inverse PCR, and restriction site extension PCR) and random PCR (such as wristwatch PCR, SiteFinding-PCR, and thermal asymmetric interlaced PCR). The latter has gradually become the mainstream walking method since it is free of the time-consuming genome preprocessing step. Although there are many random PCR walking methods available now, developing a practical analogous method is still popular [1,18].
Herein, a novel protocol based on primer extension refractory PCR (PER-PCR) is detailed for rapid and reliable genome walking. The PER-PCR relies on a set of three walking primers (WP): primary WP (PWP), secondary WP (SWP), and tertiary WP (TWP). The middle 15 nt of PWP overlaps the 3' part of SWP/TWP. The 5' parts of the three WPs are different from each other. The 15 nt overlap allows the WP to anneal to its predecessor site in the low annealing temperature (50 °C) cycle. However, the WP complement is unable to be produced on the DNA template due to the mismatch of the two WPs 5' regions. Clearly, only target DNA is amplified in the next 65 °C cycles. The disadvantage of PER-PCR is the complexity of designing a practical WP set. However, the three WP sets provided in this study are universal to any species. Users need only to design the required sequence-specific primer (SSP) set. This PER-PCR protocol can be used in many aspects, such as identifying new genes, unveiling regulatory DNAs, or clarifying structures of genetic elements [1].
Materials and reagents
Biological materials
1. Genomic DNA of rice was obtained from the lab of Dr. Xiaojue Peng at Nanchang University
2. Genomic DNA of Levilactobacillus brevis CD0817 [19–22] was extracted by our lab
Reagents
1. 10× LA PCR buffer (Mg2+ plus) (Takara, catalog number: RR042A)
2. 6× Loading buffer (Takara, catalog number: 9156)
3. LA Taq polymerase (hot-start version) (Takara, catalog number: RR042A)
4. dNTP mixture (Takara, catalog number: RR042A)
5. DL 5,000 DNA marker (Takara, catalog number: 3428Q)
6. 1× TE buffer (Sangon, catalog number: B548106)
7. Agarose (Sangon, catalog number: A620014)
8. 1 M NaOH (Yuanye, catalog number: B28412)
9. 0.5 M EDTA (Solarbio, catalog number: B540625)
10. Green fluorescent nucleic acid dye (10,000 ×) (Solarbio, catalog number: G8140)
11. Tris (Solarbio, catalog number: T8060)
12. Boric acid (Solarbio, catalog number: B8110)
13. DiaSpin DNA Gel Extraction kit (Sangon, catalog number: B110092)
14. Primers (Sangon)
PWP1: AAAGTAGTCATGTATCTGCGTCCTAGTC
PWP2: AAAGTAGTCATGTATCTGGCAGTCATAG
PWP3: AAAGTAGTCATGTATCTGTGCTGTCTGA
SWP: GTCGGTCTGGGTAGTCATGTATCTG
TWP: ACCCTGTGCCGTAGTCATGTATCTG
SSP1-gadR: TCCTTCGTTCTTGATTCCATACCCT
SSP2-gadR: CCATTTCCATAGGTTGCTCCAAGGTCA
SSP3-gadR: TAGGATACTGGCTAAAATGAATTAACTCGGAT
SSP1-hyg: GGAAGTGCTTGACATTGGGGAGT
SSP2-hyg: AAGACCTGCCTGAAACCGAACTGC
SSP3-hyg: CAAGGAATCGGTCAATACACTACATGGC
Solutions
1. 2.5× TBE buffer (see Recipes)
2. 0.5× TBE buffer (see Recipes)
3. 100 μM primer (see Recipes)
4. 10 μM primer (see Recipes)
5. 1.5% agarose gel (see Recipes)
Recipes
1. 2.5 × TBE buffer
| Reagent | Final concentration | Amount |
| 0.5 M EDTA solution | 5 mM | 10 mL |
| Tris | 225 mM | 27 g |
| Boric acid | 225 mM | 13.75 g |
| ddH2O | n/a | 980 mL |
| Total | n/a | 1,000 mL |
| Adjust pH to 8.3 with 1 M NaOH and then replenish the solution to 1,000 mL with ddH2O. | ||
2. 0.5× TBE buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| 2.5× TBE buffer | 0.5× | 200 mL |
| ddH2O | n/a | 800 mL |
| Total | n/a | 1,000 mL |
3. 100 μM primer
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| Powdery primer | 100 μM | n/a |
| 1× TE buffer | 1× | Volume specified in the sheet of primer synthesis |
| Total | n/a | Volume specified in the sheet of primer synthesis |
Note: Dilute a portion of the 100 μM primer to prepare 10 μM primer and store the remaining portion at -80 °C.
4. 10 μM primer
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| 100 μM primer | 10 μM | 1 μL |
| 1× TE buffer | 1× | 9 μL |
| Total | n/a | 10 μL |
Note: Prepare extra volume of a 10 μM primer and divide it into multiple 1.5 mL microcentrifuge tubes; then, store the microcentrifuge tubes at -80 °C. Take one tube at a time and store it at -20 °C after use.
5. 1.5% agarose gel
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| Agarose | 1.5% | 1.5 g |
| 0.5× TBE buffer | 0.5× | 100 mL |
| Green fluorescent nucleic acid dye (10,000×) | 1× | 10 μL |
| Total | n/a | 100 mL |
Laboratory supplies
1. 0.2 mL thin-wall PCR tubes (Kirgen, catalog number: KG2311)
2. 10 μL pipette tips (Sangon, catalog number: F600215)
3. 200 μL pipette tips (Sangon, catalog number: F600227)
4. 1,000 μL pipette tips (Sangon, catalog number: F630101)
5. 1.5 mL microcentrifuge tubes (Labselect, catalog number: MCT-001-150)
Equipment
1. PCR apparatus (Analtytikjena, model: Biometra TAdvanced)
2. Electrophoresis apparatus (Beijing Liuyi, model: DYY-6C)
3. Gel imaging system (Bio-Rad, model: ChemiDoc XRS+)
4. Microcentrifuge (Tiangen, model: TGear)
Software and datasets
1. Oligo 7 software (Molecular Biology Insights, Inc., USA)
2. DNASTAR Lasergene software (DNASTAR, Inc., USA)
Procedure
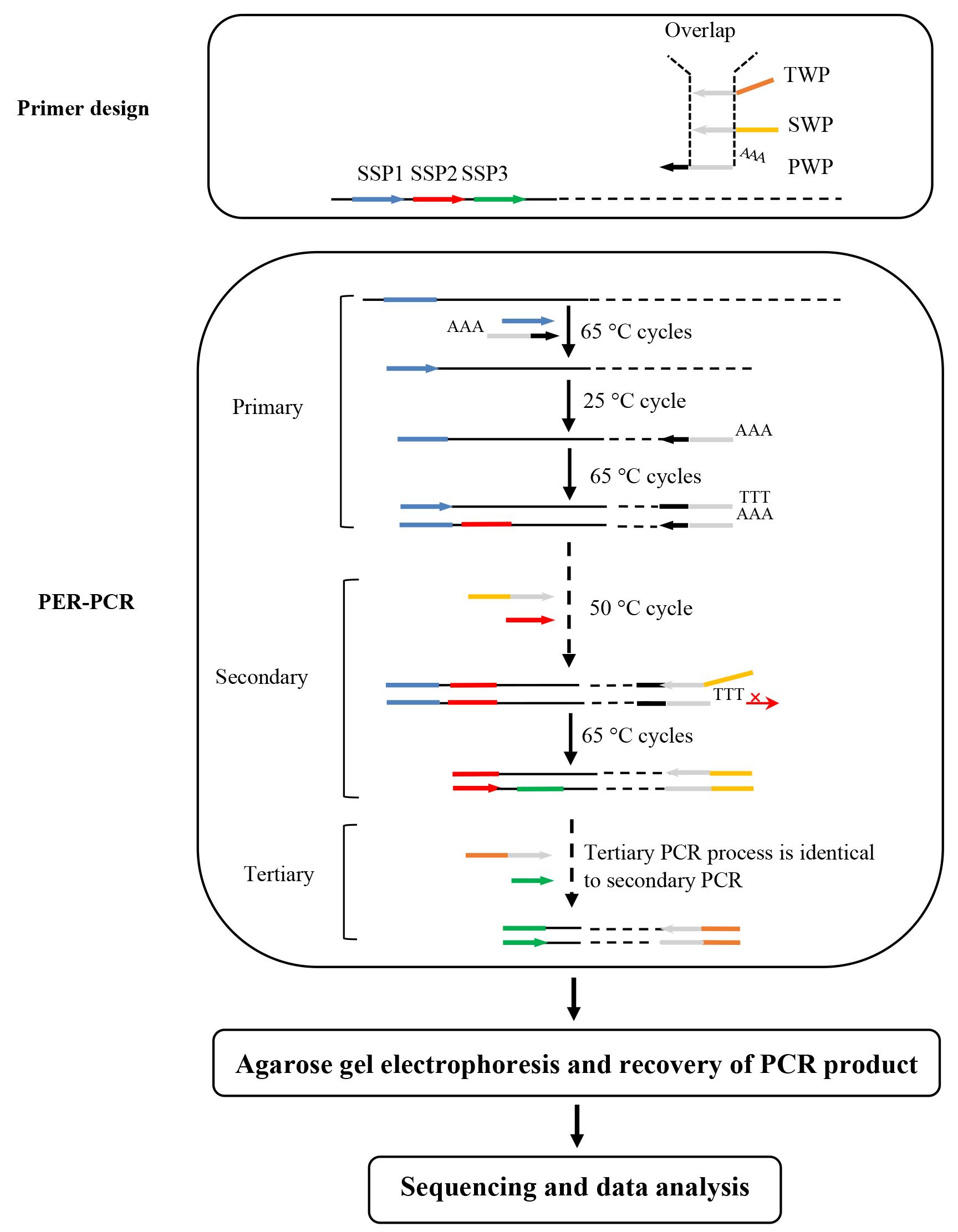
The overview of PER-PCR is shown in Figure 1.
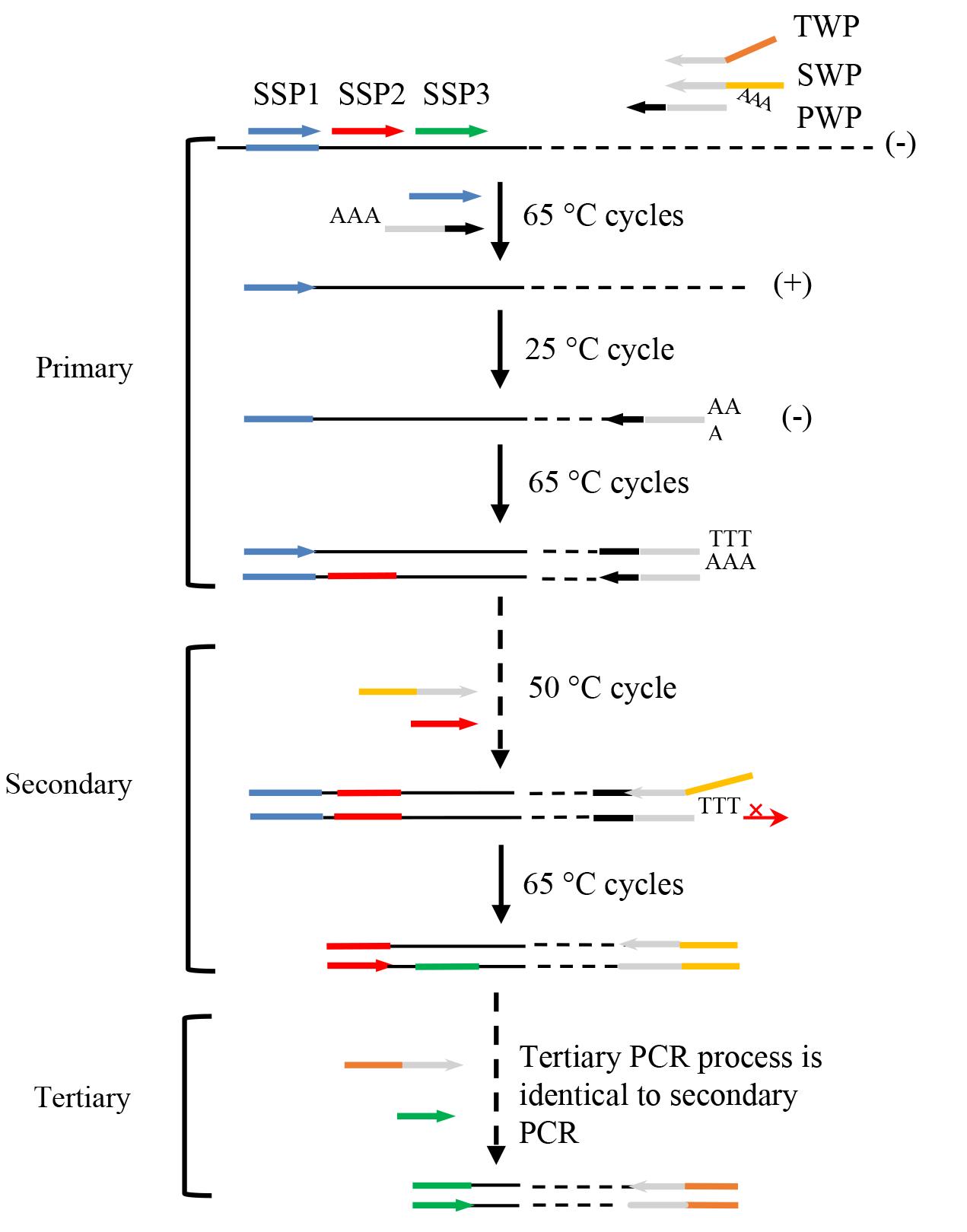
Figure 1. Schematic overview of PER-PCR. PWP: primary walking primer; SWP: secondary walking primer; TWP: tertiary walking primer; SSP: sequence-specific primer. Thin solid line: known sequence; thin dotted line: unknown sequence; arrows: primers; thick lines: primer complements; symbol +: plus strand; and symbol -: minus strand.
Note: In primary PER-PCR, the initial 65 °C cycles only permit SSP1 to hybridize with its complement on known region, thus producing several target plus strands (+). In the subsequent 25 °C cycle, PWP randomly anneals somewhere on the unknown region of this strand and synthesizes the target minus strand (-). In the following 65 °C cycles, this target minus strand is exponentially amplified. Any non-target single strand formed in the 25 °C cycle cannot be amplified because it lacks an authentic binding site for any primer. In secondary PCR, the three types (type I, primed by SSP1; type II, primed by SSP1 and PWP; and type III, primed by PWP) of primary PCR non-target products are easily removed, as they lack an exact binding site for at least one of the primers. Nevertheless, the target product is exponentially amplified. Similarly, only the target product is further amplified in tertiary PCR.
A. Design of primers
1. Design WP sets (Figure 2).

Figure 2. The three WP sets designed in this study. The three PWPs (PWP1, PWP2, and PWP3) have a length of 28 nt, comprising three regions. Region 1 is the 5' head of three adenosines (AAA); region 2 is the 15 nt overlap in the middle; and region 3 is the 3' tails (10 nt) different from each other. SWP or TWP has a length of 25 nt, with the 15 nt overlap at the 3' part. The 5' parts of SWP and TWP are different from each other and also different from region 1 of PWP. PWP: primary walking primer, SWP: secondary walking primer, and TWP: tertiary walking primer.
Note: The DNA sequences of three WPs (PWP, SWP, and TWP) in a WP set are arbitrary, but any WP should a) have a Tm value between 60 and 65 °C; b) have an even distribution of G, C, T, and A, with a GC content of about 50%; and c) avoid a hairpin or dimer structure with a Tm value exceeding 40 °C. The Tm value of the 15 nt overlap is about 35 °C. Simultaneously designing more than one WP set can perform parallel PER-PCRs in a single walking cycle. n WP sets comprise n PWPs, one SWP, and one TWP. The SWP and WTP are universal to the n PWPs. In general, 2–3 parallel PER-PCR sets are suggested for a single walking cycle.
a. Open the Oligo 7 software, then click File and New Sequence (Figure 3a); type in a 28 nt arbitrary sequence as the initial PWP1 in the Edit Sequence dialog box (Figure 3b). Next, click Accept/Discard and Accept (Figure 3c).
Note: Here, the design of WP set 1 (including PWP1, SWP, and TWP) is provided as an example.
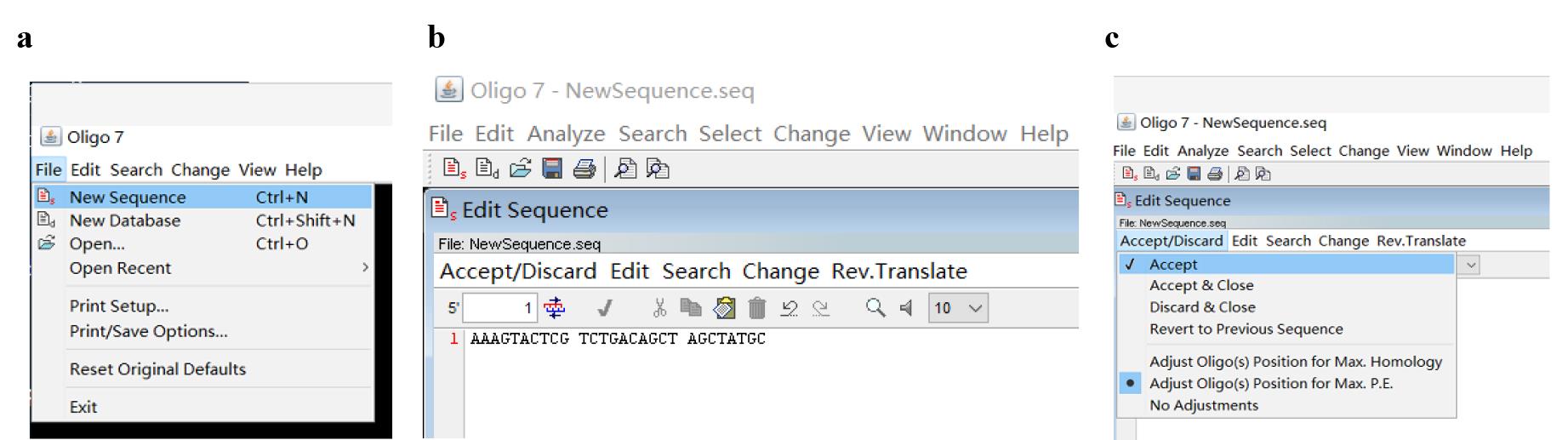
Figure 3. Screenshots displaying how to enter the initial primary walking primer in the Oligo 7 software. Locations of New Sequence (a), Edit Sequence dialog box (b), and Accept (c).
b. Click Analyze, Duplex Formation, and Current Oligo (Figure 4a) to analyze primer dimer (Figure 4b).
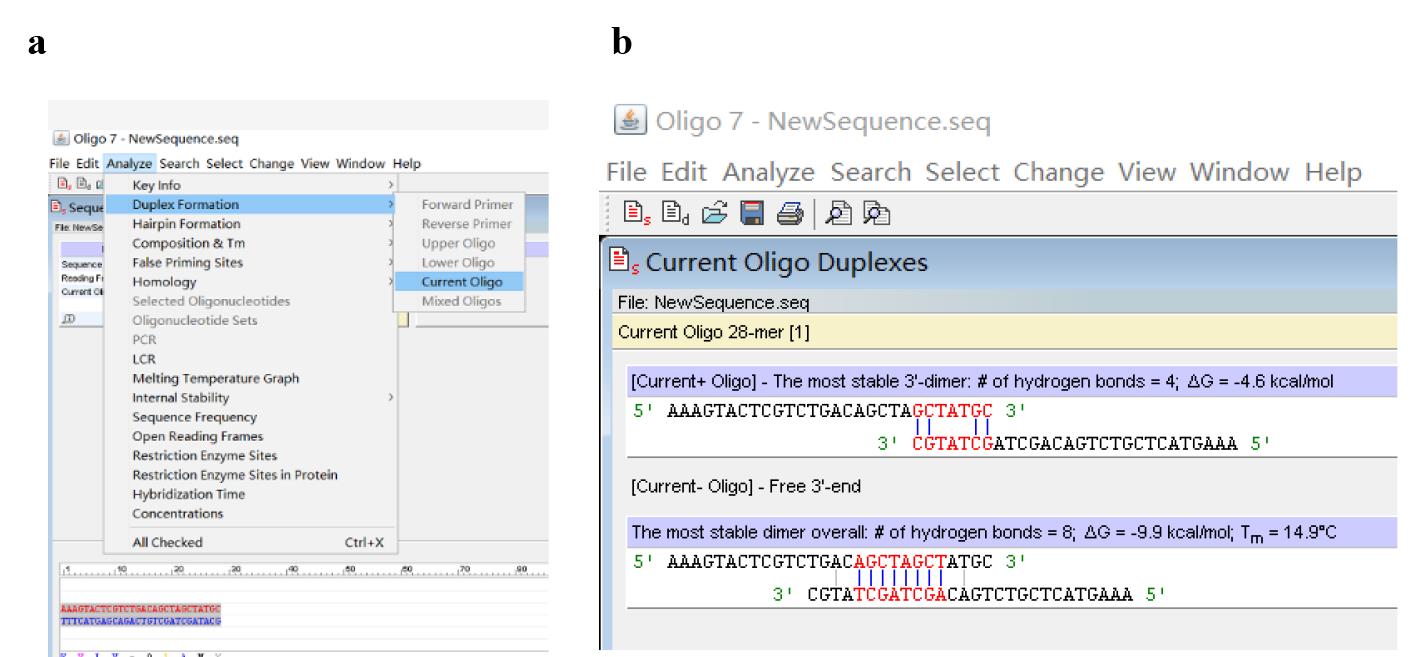
Figure 4. Screenshots displaying how to analyze primer dimer. (a) Locations of Duplex Formation and Current Oligo. (b) Predicted primer dimers.
c. Click Analyze, Hairpin Formation, and Current Oligo (Figure 5a) to analyze primer hairpin (Figure 5b).
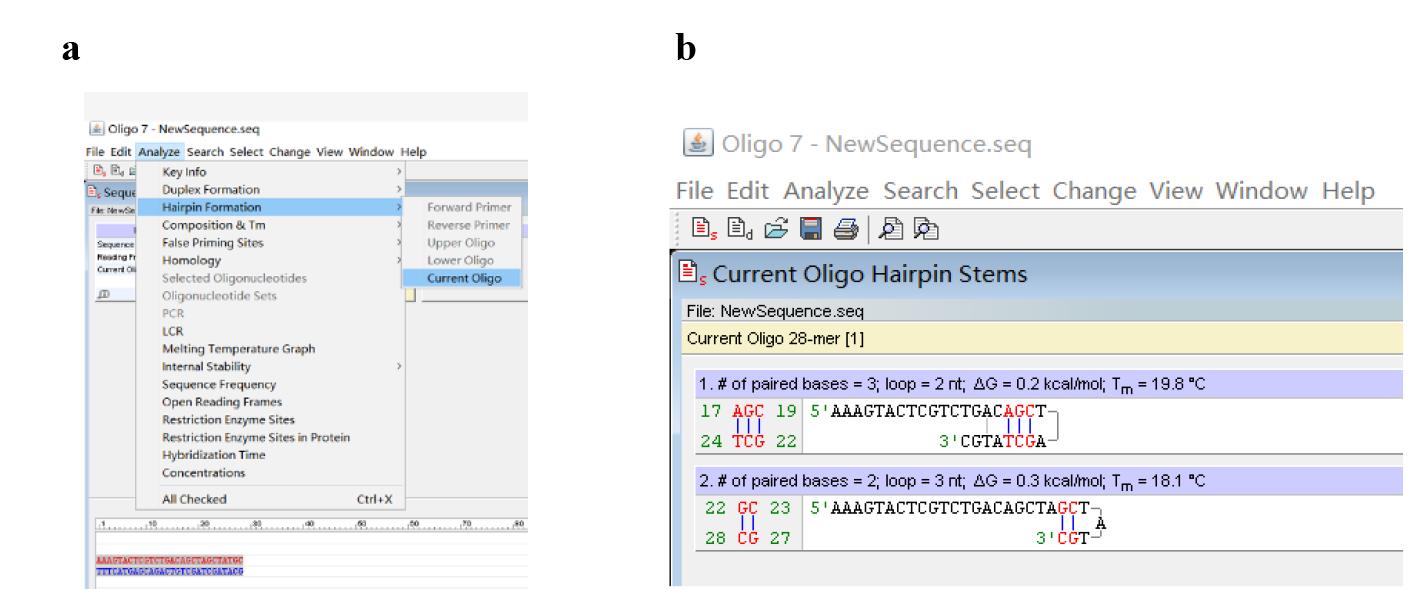
Figure 5. Screenshots displaying how to analyze primer hairpin. (a) Locations of Hairpin Formation and Current Oligo. (b) Predicted primer hairpins.
Note: Optimize the sequence of the initial PWP1 (see below) if it is unsatisfactory because it forms an obvious primer dimer(s) or hairpin(s).
d. Click Edit and Entire Sequence (Figure 6a) to return to the Edit Sequence dialog box (Figure 3b); edit the initial sequence according to the above analysis results, then click Accept/Discard and Accept (Figure 3c). Next, minimize this dialog box to display the dialog box shown in Figure 4a.
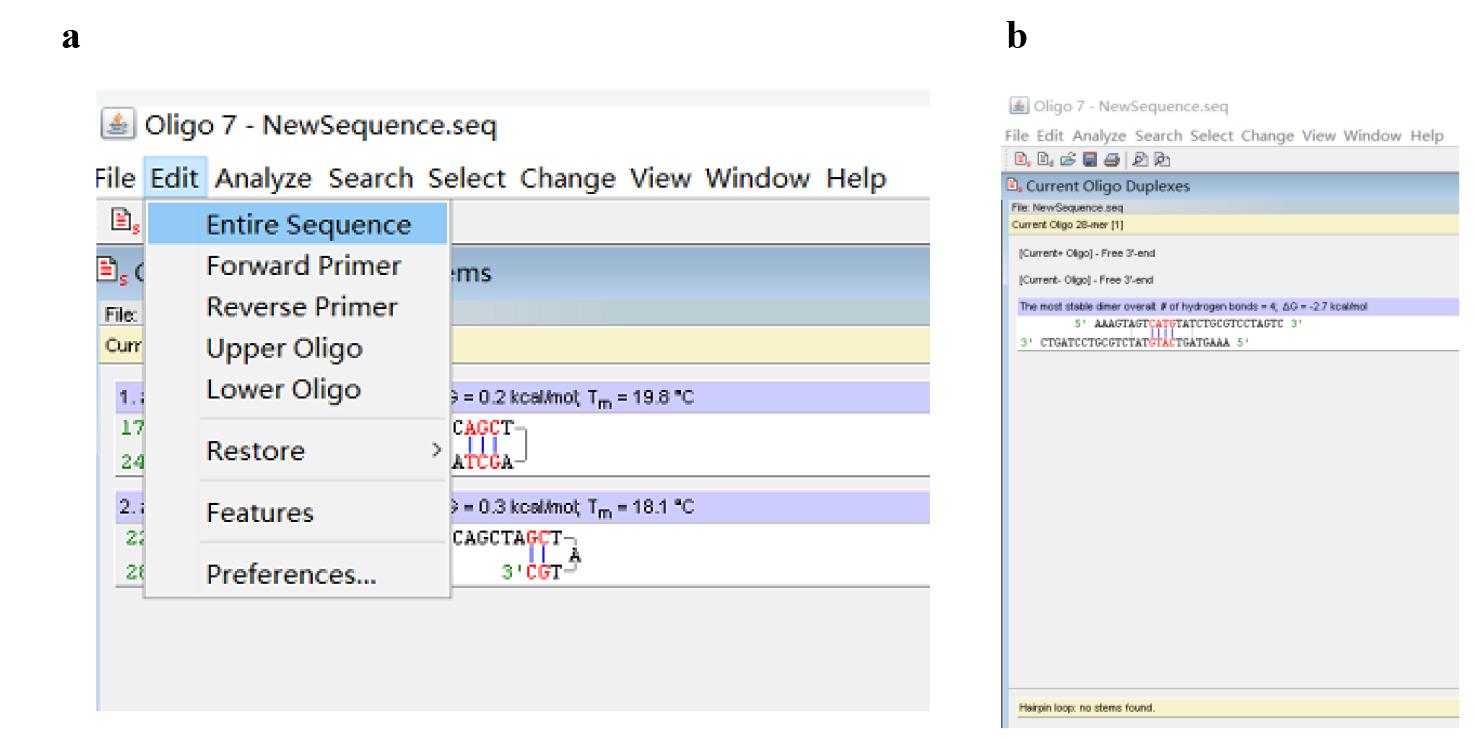
Figure 6. Screenshots displaying how to optimize the sequence of the initial PWP1. (a) Screenshot displaying how to return to the Edit Sequence dialog box. (b) Predicted primer hairpins.
e. Repeat steps A1b, c to evaluate the formation of primer dimer and hairpin in the edited PWP1 until a satisfactory PWP1 (Figure 6b) is obtained.
Note: The PWP1 shown in Figure 6b is satisfactory because this oligo only forms an acceptable primer dimer and does not form any hairpin.
f. Select region 2 (the middle 15 nt overlap) of the satisfactory PWP1.
g. Attach a 10 nt arbitrary oligo to the 5’ end of this overlap, creating the initial SWP.
h. Assess this SWP using the above method (corresponding to the steps A1a–e) until a satisfactory SWP is obtained.
i. Design a satisfactory TWP like designing the SWP.
Critical: An obvious primer dimer should be evaded between any two WPs in a WP set.
2. Select an SSP set of three SSPs (outmost SSP1, middle SSP2, and innermost SSP3) from a known DNA (Figure 7).
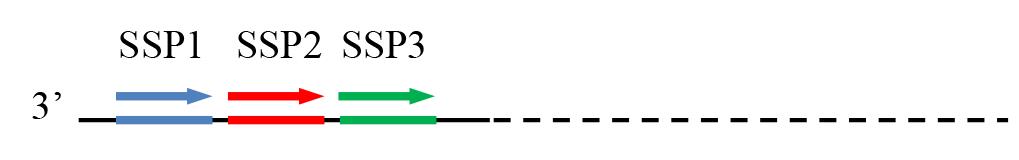
Figure 7. Mutual positional relationship of SSP1, SSP2, and SSP3. SSP: sequence-specific primer; thin solid line: known sequence; thin dotted line: unknown sequence; arrows: primers; thick lines: primer complements.
Note: An SSP set of three SSPs is within the same known DNA, with an extension direction toward the unknown flanking region. An SSP should (i) have a length of approximately 25 nt, with a melting temperature (Tm) between 60 and 65 °C; (ii) have an even distribution of guanine (G), cytosine (C), thymine (T), and adenine (A), with a GC content of about 50%; (iii) avoid repetition sequences; and (iv) avoid a hairpin or dimer structure with a Tm exceeding 40 °C. In general, there is no limit of distance between the two adjacent SSPs, but a distance of approximately 20–50 nt is suggested if possible.
j. Open the Oligo 7 software, then click File and Open to enter the known DNA sequence of the gadR gene (Figure 8a).
Note: Here, the design of SSP1-gadR is provided as an example.
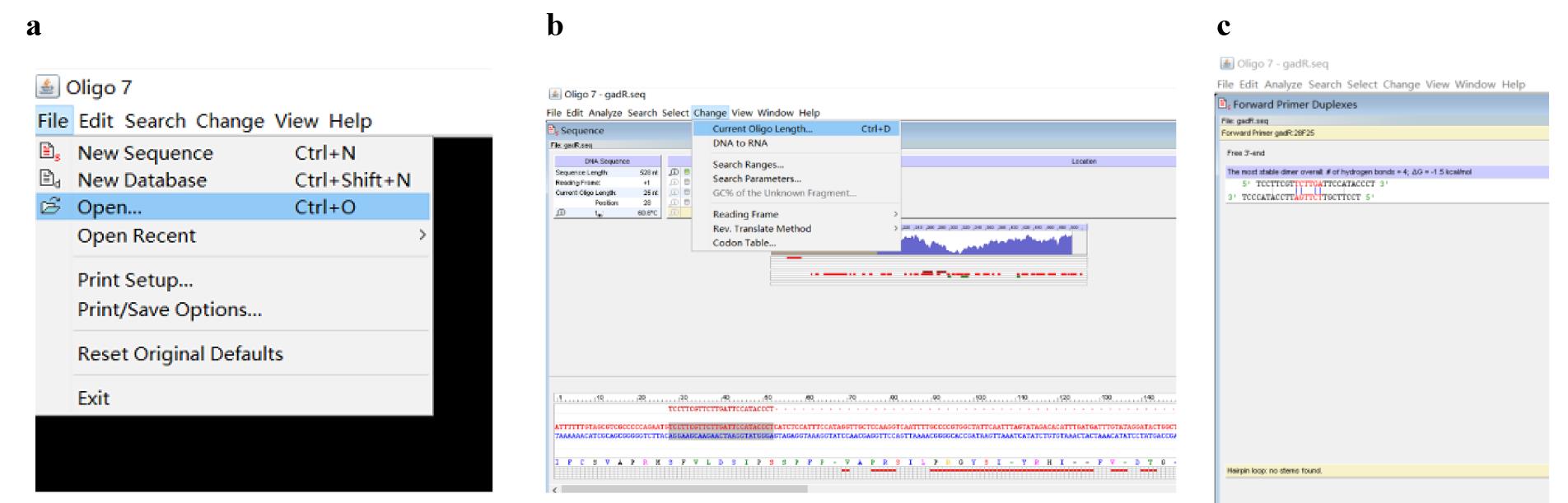
Figure 8. Screenshots displaying how to design SSP1-gadR. (a) Screenshot displaying how to enter the known DNA sequence. (b) Screenshot displaying how to choose an oligo with a defined length. (c) The predicted primer dimers and hairpin.
k. Click Change and Current Oligo Length to define the length (for instance 25 nt) of oligo to be analyzed; then, move the mouse to a position of the entered sequence (the 25 nt following the mouse is automatically shaded) and click to select this shaded 25 nt (Figure 8b).
l. Assess the selected 25 nt oligo according to the method (corresponding to the steps of Figures 4 and 5) for assessing PWP1.
Note: In case the current SSP1-gadR is unsatisfactory due to forming an obvious primer dimer(s) or hairpin(s), re-select and assess a new oligo by repeating steps A1a–c until a satisfactory SSP1-gadR (Figure 8c) is obtained.
m. Design the nested SSP2-gadR and SSP2-gadR, just like designing the SSP1-gadR.
Critical: According to Mazars et al. [23], each SSP shows a melting temperature of 65–70 °C and should avoid forming severe hairpin or dimer structure.
B. PER-PCR amplification
The current protocol comprises three rounds of nested PCRs. Primary PCR is driven by PWP and SSP1; secondary PCR is driven by SWP and SSP2; and tertiary PCR is driven by TWP and SSP3.
1. Primary PER-PCR
a. Pipette primary PER-PCR components (Table 1) into a 0.2 mL PCR tube.
Table 1. Primary PER-PCR mix
| Reagent | Final concentration | Amount (μL) |
|---|---|---|
| Genomic DNA | Microbe, 0.2–2 ng/μL; plant or animal, 2–20 ng/μL | 1 |
| LA Taq polymerase (5 U/μL) | 0.05 U/μL | 0.5 |
| PWP (10 μM) | 0.2 μM | 1 |
| SSP1 (10 μM) | 0.2 μM | 1 |
| 10× LA PCR buffer II (Mg2+ plus) | 1× | 5 |
| dNTP mixture (2.5 mM each) | 0.4 mM each | 8 |
| ddH2O | n/a | 33.5 |
| Total | n/a | 50 |
b. Completely mix the components with a pipette.
c. Centrifuge for 10–20 s with a microcentrifuge to gather the mixture.
d. Run the amplification in the PCR apparatus (Table 2).
Table 2. Primary PER-PCR cycling conditions
| Step | Temp. (°C) | Duration | Cycle |
| Initial denaturation | 95 | 1 min | 1 |
| Denaturation | 95 | 20 s | 5 |
| Annealing | 65 | 30 s | |
| Extension | 72 | 2 min | |
| Denaturation | 95 | 20 s | 1 |
| Annealing | 25 | 30 s | |
| Extension | 72 | 2 min | |
| Denaturation | 95 | 20 s | 30 |
| Annealing | 65 | 30 s | |
| Extension | 72 | 2 min | |
| Final extension | 72 | 5 min | 1 |
| Hold | 4 | forever | |
e. Put the PCR product onto ice.
f. Take 1 μL of the product as the template of secondary PER-PCR.
g. Store the remaining product at -20 °C for future assays.
2. Secondary PER-PCR
a. Pipette secondary PER-PCR components (Table 3) into a 0.2 mL PCR tube.
Table 3. Secondary PER-PCR mix
| Reagent | Final concentration | Amount (μL) |
|---|---|---|
| Primary PCR product | n/a | 1 |
| LA Taq polymerase (5 U/μL) | 0.05 U/μL | 0.5 |
| SWP (10 μM) | 0.2 μM | 1 |
| SSP2 (10 μM) | 0.2 μM | 1 |
| 10× LA PCR buffer II (Mg2+ plus) | 1× | 5 |
| dNTP mixture (2.5 mM each) | 0.4 mM each | 8 |
| ddH2O | n/a | 33.5 |
| Total | n/a | 50 |
Critical: Dilute primary PER-PCR product 10–10,000 folds if necessary.
b. Completely mix the components with a pipette.
c. Centrifuge for 10–20 s with a microcentrifuge to gather the mixture.
d. Run the amplification in the PCR apparatus (Table 4).
Table 4. Secondary PER-PCR cycling conditions
| Step | Temp. (°C) | Duration | Cycle |
| Initial denaturation | 95 | 1 min | 1 |
| Denaturation | 95 | 20 s | 1 |
| Annealing | 50 | 30 s | |
| Extension | 72 | 2 min | |
| Denaturation | 95 | 20 s | 25 |
| Annealing | 65 | 30 s | |
| Extension | 72 | 2 min | |
| Final extension | 72 | 5 min | 1 |
| Hold | 4 | forever | |
e. Put the PCR product onto ice.
f. Take 1 μL of the product as the template of tertiary PER-PCR.
g. Store the remaining product at -20 °C for future assays.
3. Tertiary PER-PCR
a. Pipette tertiary PER-PCR components (Table 5) into a 0.2 mL PCR tube.
Table 5. Tertiary PER-PCR mix
| Reagent | Final concentration | Amount (μL) |
|---|---|---|
| Secondary PCR product | n/a | 1 |
| LA Taq polymerase (5 U/μL) | 0.05 U/μL | 0.5 |
| TWP (10 μM) | 0.2 μM | 1 |
| SSP3 (10 μM) | 0.2 μM | 1 |
| 10× LA PCR buffer II (Mg2+ plus) | 1× | 5 |
| dNTP mixture (2.5 mM each) | 0.4 mM each | 8 |
| ddH2O | n/a | 33.5 |
| Total | n/a | 50 |
Critical: Dilute secondary PER-PCR product 10–10,000 folds if necessary.
b. Completely mix the components with a pipette.
c. Centrifuge for 10–20 s with a microcentrifuge to gather the mixture.
d. Run the amplification in the PCR apparatus (Table 6).
Table 6. Tertiary fork PCR cycling conditions
| Step | Temp. (°C) | Duration | Cycle |
| Initial denaturation | 95 | 1 min | 1 |
| Denaturation | 95 | 20 s | 1 |
| Annealing | 50 | 30 s | |
| Extension | 72 | 2 min | |
| Denaturation | 95 | 20 s | 25 |
| Annealing | 65 | 30 s | |
| Extension | 72 | 2 min | |
| Final extension | 72 | 5 min | 1 |
| Hold | 4 | forever | |
e. Store the PCR product at -20 °C for future assays.
C. Gel electrophoresis
1. Completely mix 5 μL of PER-PCR product and 1 μL of 6× loading buffer.
2. Load the mixture into a 1.5% agarose gel supplemented with 1× green fluorescent nucleic acid dye.
3. Set the electrophoresis apparatus to a voltage of 150 V (the distance between the two electrodes is 30 cm).
4. Check the PCR product using the ChemiDoc XRS+ imaging system after approximately 25 min of electrophoresis (Figure 9).
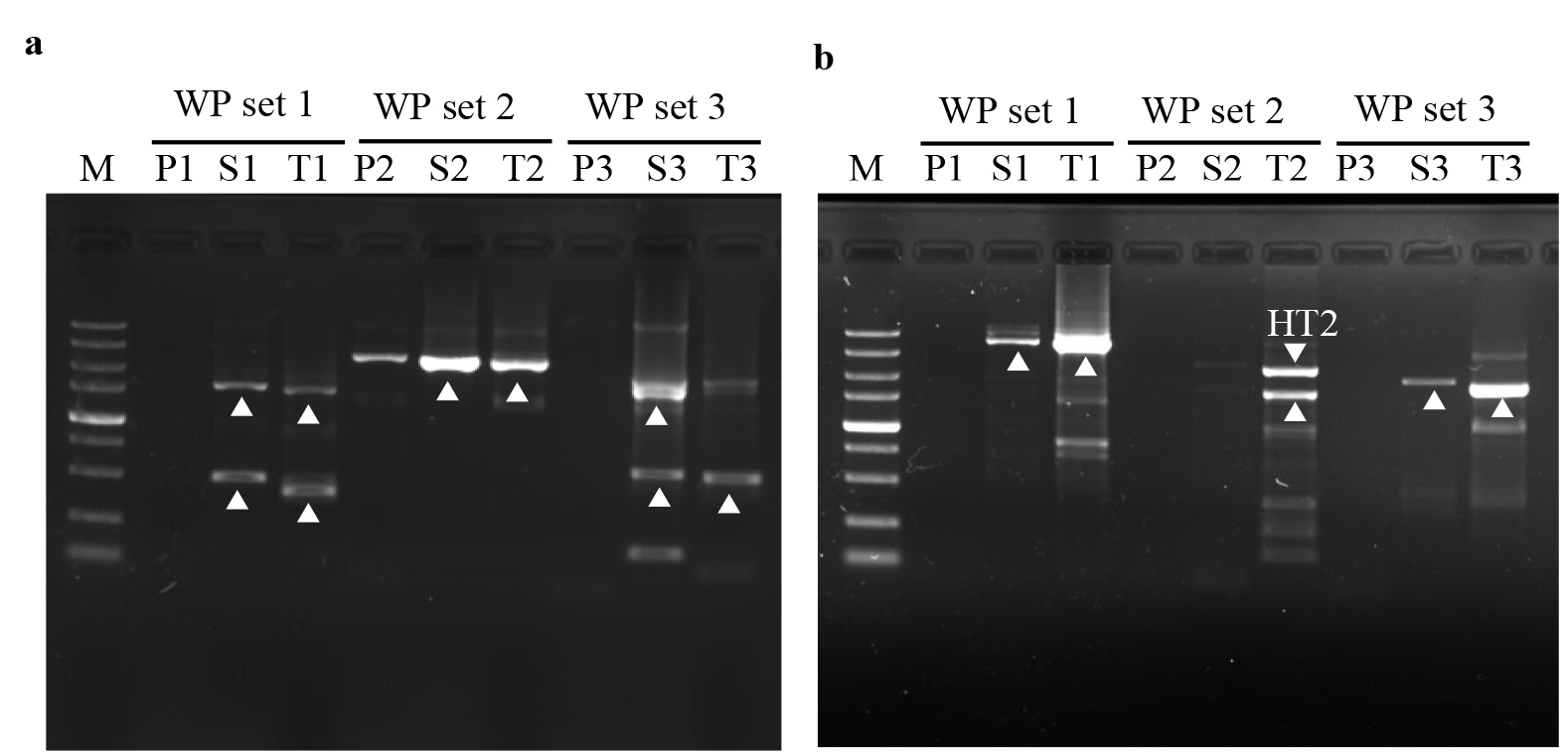
Figure 9. Mining unknown flanks of gadR (A) and hyg (B). The three parallel sets of PER-PCRs in each walking are shown. Lanes P1, P2, and P3: primary products; Lanes S1, S2, and S3: secondary products; Lanes T1, T2, and T3: tertiary products; and Lanes Ms: DNA5000 Marker (5,000, 3,000, 2,000, 1,500, 1,000, 750, 500, 250, and 100 bp). The clear PCR amplicons are shown with white arrows.
D. Recovery of PCR product
1. Completely mix 40 μL of secondary/tertiary PER-PCR product and 8 μL of 6× loading buffer.
2. Load the mixture into a 1.5% agarose gel supplemented with 1× green fluorescent nucleic acid dye.
3. Set the electrophoresis apparatus to a voltage of 150 V (the distance between the two electrodes is 30 cm).
4. Visualize the PCR product using the ChemiDoc XRS+ imaging system. Subsequently, cut out clear DNA band(s) using a knife.
5. Extract the DNA band(s) from the cut gel using the DiaSpin DNA Gel Extraction kit.
6. Confirm the extracted DNA band(s) with 1.5% agarose gel electrophoresis.
E. DNA Sequencing
Sequence the extracted DNA band(s) at Sangon Biotech Co., Ltd.
Data analysis
Perform sequence analysis using the By Clustal W Method function in MegAlign software. In general, walking is considered correct if one end of the obtained DNA overlaps with one end of the known DNA [24–26].
1. Open the MegAlign software, then click File and Enter Sequences (Figure 10a) to input a PER-PCR product and the corresponding known DNA sequence (Figure 10b).
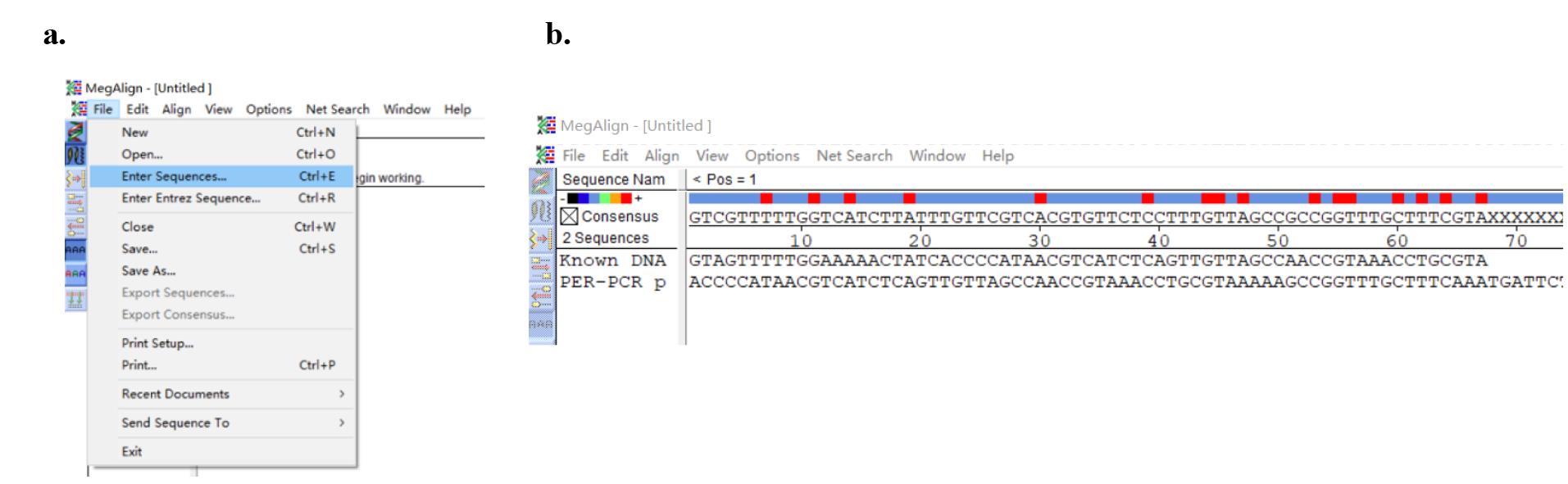
Figure 10. Screenshots displaying how to enter DNA sequences. (a) Screenshot displaying the location of Enter Sequences. (b) Input DNA sequences.
2. Click Align and By Clustal W Method (Figure 11a) to output the alignment outcome (Figure 11b).
Note: Genome walking is regarded as successful if the SSP3-sided region of the PER-PCR product overlaps the known DNA (an example is shown in Figure 11b).
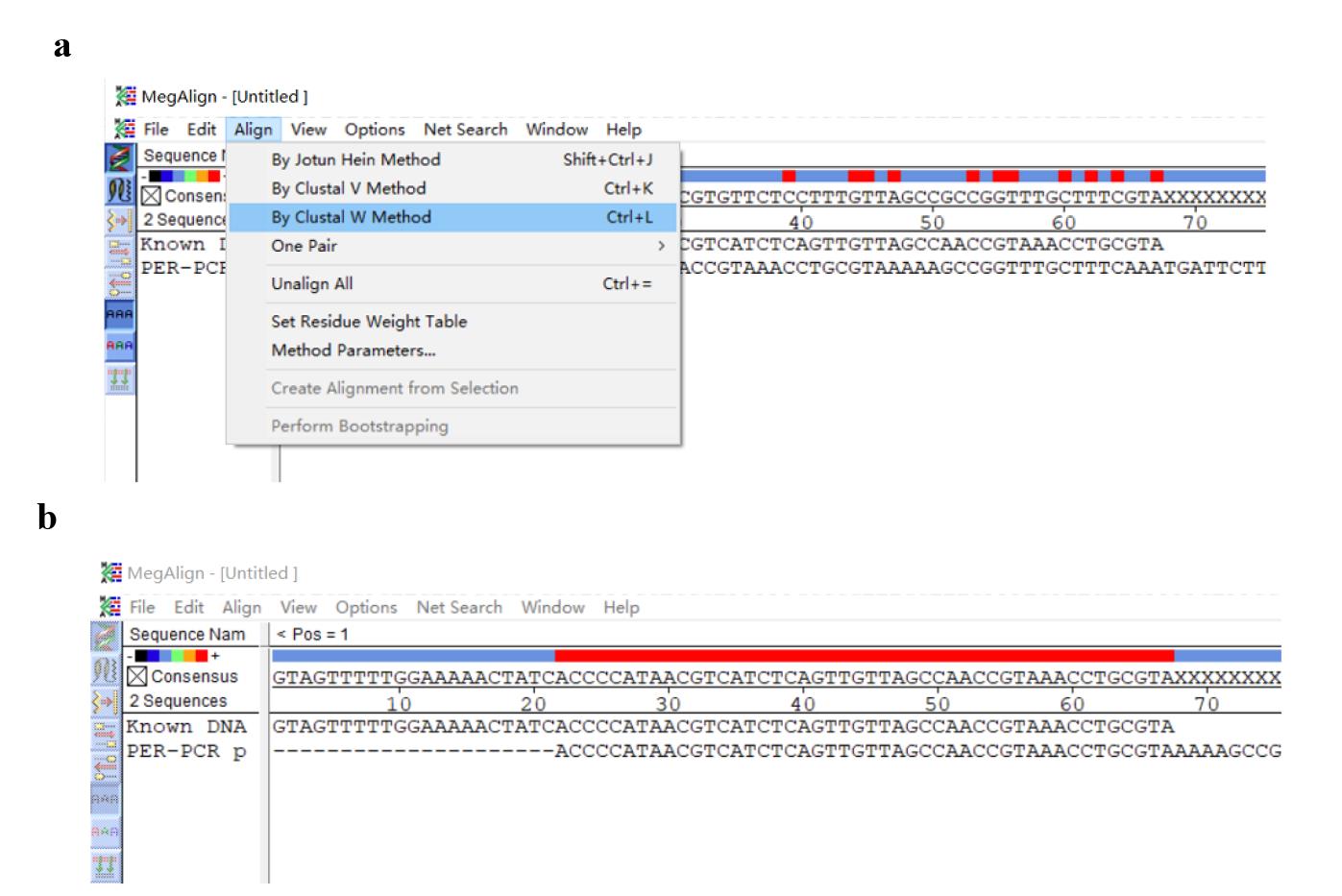
Figure 11. Screenshots displaying how to align the entered sequences. (a) Screenshot showing the location of By Clustal W. (b) Alignment outcome.
Validation of protocol
This protocol or parts of it has been used and validated in the following research article:
Li et al. [1] Primer extension refractory PCR: an efficient and reliable genome walking method. Mol Genet Genomics (Figure 9).
General notes and troubleshooting
General notes
1. PER-PCR comprises three rounds of nested PCRs. However, secondary PCR generally suffices to release a positive outcome.
2. In general, it is not necessary to check the primary PER-PCR product.
3. Before tertiary PER-PCR, secondary PER-PCR product is analyzed using agarose gel electrophoresis so as to see if a clear DNA band(s) appears. If a clear DNA band(s) appears, it is recovered and sequenced. It is not necessary to perform tertiary PCR if this DNA band(s) is the target product.
4. Like the other existing PCR-based genome walking protocols, the current PER-PCR protocol also suffers from the multiple-band issue. However, if multiple DNA bands appear, only the largest one needs to be assayed.
5. Simultaneously design n WP sets (2–3 sets are suggested) so as to do n parallel PER-PCRs in a single walking cycle. The parallel PER-PCR design of this protocol is conducive to improving the success and efficiency of genome walking. n sets of WPs comprise n PWPs, one SWP, and one TWP. The SWP and WTP are universal to the n PWPs.
6. The current PER-PCR protocol is applicable to any species.
Troubleshooting
Problem 1: There is no clear DNA band(s) in secondary or even tertiary PER-PCR.
Possible cause: (i) In primary PER-PCR, target amplification is weak while non-target amplification is strong. (ii) the GC content of the genomic DNA template may be high.
Solution: (i) Dilute primary PER-PCR product 10–10,000 times and then use 1 μL of each dilution as the template in secondary PER-PCR. Next, tertiary PER-PCR is performed using each secondary PER-PCR product as the template. If there is still no clear DNA(s) in any secondary/tertiary PER-PCR, redesign an SSP set. (ii) Add a PCR enhancer (for example betaine) to PCR reaction solution, increase the working concentration of primers, or lengthen the 5’ head of walking primer to enhance its Tm value; meanwhile, use an SSP with a higher Tm value.
Problem 2: A PER-PCR product cannot be directly sequenced.
Possible cause: There is interference from non-target products.
Solution: Clone the obtained DNA band and then sequence.
Problem 3: A clear DNA band(s) is the non-target product.
Possible cause: The genome has sites homologous to the SSP complements in other region(s).
Solution: Redesign an SSP set at different sites of known DNA.
Problem 4: The PCR product cannot be separated by agarose gel electrophoresis, showing as a large block in the gel.
Possible cause: The walking primer has multiple major partial annealing sites on unknown flanks that are very close to each other.
Solution: Recover the upper part of this block from the gel and then directly use SSP3 or an SSP inner to SSP3 to sequence it or clone it and then sequence.
Acknowledgments
This work was funded by the Jiangxi Provincial Department of Science and Technology (grant No. 20225BCJ22023), China, and the National Natural Science Foundation of China (grant No 32160014). This PER-PCR-based genome walking protocol has been originally described and validated in Li et al. [1].
Competing interests
The authors declare no competing interests.
References
- Li, H., Lin, Z., Guo, X., Pan, Z., Pan, H. and Wang, D. (2024). Primer extension refractory PCR: an efficient and reliable genome walking method. Mol Genet Genomics. 299(1): 27. https://doi.org/10.1007/s00438-024-02126-5
- Thirulogachandar, V., Pandey, P., Vaishnavi, C. and Reddy, M. K. (2011). An affinity-based genome walking method to find transgene integration loci in transgenic genome. Anal Biochem. 416(2): 196–201. https://doi.org/10.1016/j.ab.2011.05.021
- Myrick, K. V. and Gelbart, W. M. (2002). Universal Fast Walking for direct and versatile determination of flanking sequence. Gene. 284: 125–131. https://doi.org/10.1016/s0378-1119(02)00384-0
- Lin, Z., Wei, C., Pei, J. and Li, H. (2023). Bridging PCR: An efficient and reliable scheme implemented for genome-walking. Curr Issues Mol Biol. 45(1): 501–511. https://doi.org/10.3390/cimb45010033
- Kotik, M. (2009). Novel genes retrieved from environmental DNA by polymerase chain reaction: Current genome-walking techniques for future metagenome applications. J Biotechnol. 144(2): 75–82. https://doi.org/10.1016/j.jbiotec.2009.08.013
- Leoni, C., Volpicella, M., De Leo, F., Gallerani, R. and Ceci, L. R. (2011). Genome walking in eukaryotes. FEBS J. 278(21): 3953–3977. https://doi.org/10.1111/j.1742-4658.2011.08307.x
- Pei, J., Sun, T., Wang, L., Pan, Z., Guo, X. and Li, H. (2022). Fusion primer driven racket PCR: A novel tool for genome walking. Front Genet. 13: 969840. https://doi.org/10.3389/fgene.2022.969840
- Fraiture, M. A., Papazova, N. and Roosens, N. H. (2021). DNA walking strategy to identify unauthorized genetically modified bacteria in microbial fermentation products. Int J Food Microbiol. 337: 108913. https://doi.org/10.1016/j.ijfoodmicro.2020.108913
- Li, H., Ding, D., Cao, Y., Yu, B., Guo, L. and Liu, X. (2015). Partially overlapping primer-based PCR for genome walking. PLoS One. 10(3): e0120139. https://doi.org/10.1371/journal.pone.0120139
- Chang, K., Wang, Q., Shi, X., Wang, S., Wu, H., Nie, L. and Li, H. (2018). Stepwise partially overlapping primer-based PCR for genome walking. AMB Express. 8(1): 77. https://doi.org/10.1186/s13568-018-0610-7
- Wei, C., Lin, Z., Pei, J., Pan, H. and Li, H. (2023). Semi-site-specific primer PCR: A simple but reliable genome-walking tool. Curr Issues Mol Biol. 45(1): 512–523. https://doi.org/10.3390/cimb45010034
- Chen, H., Wei, C., Lin, Z., Pei, J., Pan, H. and Li, H. (2024). Protocol to retrieve unknown flanking DNA sequences using semi-site-specific PCR-based genome walking. STAR Protoc. 5(1): 102864. https://doi.org/10.1016/j.xpro.2024.102864
- Guo, X., Zhu, Y., Pan, Z., Pan, H. and Li, H. (2024). Single primer site-specific nested PCR for accurate and rapid genome-walking. J Microbiol Methods. 220: 106926. https://doi.org/10.1016/j.mimet.2024.106926
- Wang, L., Jia, M., Li, Z., Liu, X., Sun, T., Pei, J., Wei, C., Lin, Z. and Li, H. (2023). Protocol to access unknown flanking DNA sequences using Wristwatch-PCR for genome-walking. STAR Protoc. 4(1): 102037. https://doi.org/10.1016/j.xpro.2022.102037
- Wang, L., Jia, M., Li, Z., Liu, X., Sun, T., Pei, J., Wei, C., Lin, Z. and Li, H. (2022). Wristwatch PCR: A versatile and efficient genome walking strategy. Front Bioeng Biotechnol. 10: e792848. https://doi.org/10.3389/fbioe.2022.792848
- Alquezar‐Planas, D. E., Löber, U., Cui, P., Quedenau, C., Chen, W. and Greenwood, A. D. (2020). DNA sonication inverse PCR for genome scale analysis of uncharacterized flanking sequences. Methods Ecol Evol. 12(1): 182–195. https://doi.org/10.1111/2041-210x.13497
- Volpicella, M., Leoni, C., Fanizza, I., Rius, S., Gallerani, R. and Ceci, L. R. (2012). Genome walking by Klenow polymerase. Anal Biochem. 430(2): 200–202. https://doi.org/10.1016/j.ab.2012.08.008
- Pan, H., Guo, X., Pan, Z., Wang, R., Tian, B. and Li, H. (2023). Fork PCR: a universal and efficient genome-walking tool. Front Microbiol. 14: e1265580. https://doi.org/10.3389/fmicb.2023.1265580
- Gao, D., Chang, K., Ding, G., Wu, H., Chen, Y., Jia, M., Liu, X., Wang, S., Jin, Y., Pan, H., et al. (2019). Genomic insights into a robust gamma-aminobutyric acid-producer Lactobacillus brevis CD0817. AMB Express. 9(1): e1186/s13568–019–0799–0. https://doi.org/10.1186/s13568-019-0799-0
- Wang, L., Jia, M., Gao, D. and Li, H. (2024). Hybrid substrate-based pH autobuffering GABA fermentation by Levilactobacillus brevis CD0817. Bioprocess Biosyst Eng. 47(12): 2101–2110. https://doi.org/10.1007/s00449-024-03088-z
- Li, H., Sun, T., Jia, M., Wang, L., Wei, C., Pei, J., Lin, Z. and Wang, S. (2022). Production of gamma-aminobutyric acid by Levilactobacillus brevis CD0817 by coupling fermentation with self-Buffered whole-cell catalysis. Fermentation 8(7): 321. https://doi.org/10.3390/fermentation8070321
- Jia, M., Zhu, Y., Wang, L., Sun, T., Pan, H. and Li, H. (2022). pH auto-sustain-based fermentation supports efficient gamma-aminobutyric acid production by Lactobacillus brevis CD0817. Fermentation. 8(5): 208. https://doi.org/10.3390/fermentation8050208
- Mazars, G. R., Moyret, C., Jeanteur, P. and Theillet, C. G. (1991). Direct sequencing by thermal asymmetric PCR. Nucleic Acids Res. 19(17): 4783–4783. https://doi.org/10.1093/nar/19.17.4783
- Tian, B., Wu, H., Wang, R., Chen, H. and Li, H. (2024). N7-ended walker PCR: An efficient genome-walking tool. Biochem Genet. https://doi.org/10.1007/s10528-024-10896-1
- Sun, T., Jia, M., Wang, L., Li, Z., Lin, Z., Wei, C., Pei, J. and Li, H. (2022). DAR-PCR: a new tool for efficient retrieval of unknown flanking genomic DNA. AMB Express. 12(1): 131. https://doi.org/10.1186/s13568-022-01471-1
- Wu, H., Pan, H. and Li, H. (2025). Protocol to retrieve unknown flanking DNA using fork PCR for genome walking. Bio Protoc. 15(2): e5161. https://doi.org/10.21769/BioProtoc.5161
Article Information
Publication history
Received: Sep 14, 2024
Accepted: Dec 12, 2024
Available online: Dec 26, 2024
Published: Feb 20, 2025
Copyright
© 2025 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Yu, Z., Wang, D., Lin, Z. and Li, H. (2025). Protocol to Mine Unknown Flanking DNA Using PER-PCR for Genome Walking. Bio-protocol 15(4): e5188. DOI: 10.21769/BioProtoc.5188.
Category
Microbiology > Microbial genetics > DNA > PCR
Molecular Biology > DNA > PCR
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.