- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

CRISPR/Cas9-Based Protocol for Precise Genome Editing in Induced Pluripotent Stem Cells
Published: Vol 14, Iss 24, Dec 20, 2024 DOI: 10.21769/BioProtoc.5141 Views: 3324
Reviewed by: Samantha HallerPhilipp WörsdörferRakesh Bam
Original research article
The authors used this protocol in:

Apr 2024

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
The advent of clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9-based genome editing has marked a significant advancement in genetic engineering technology. However, the editing of induced pluripotent stem cells (iPSCs) with CRISPR presents notable challenges in ensuring cell survival and achieving high editing efficiency. These challenges become even more complex when considering the specific target site. P53 activation as a result of traditional CRISPR editing can lead to apoptosis, potentially worsening cell health or even resulting in cell death. Mitigating this apoptotic response can enhance cell survival post-CRISPR editing, which will ultimately increase editing efficiency. In our study, we observed that combining p53 inhibition with pro-survival small molecules yields a homologous recombination rate of over 90% when using CRISPR in human iPSCs. This protocol significantly streamlines the editing process and reduces the time and resources necessary for creating isogenic lines.
Key features
• The combination of p53 inhibition and pro-survival small molecules promotes cell survival and increases the efficiency of genome editing.
• Genome editing can be completed in as little as 8 weeks for iPSCs, significantly reducing the total time required.
• Achieves a homologous recombination rate of over 90% in human iPSCs.
Keywords: iPSCsGraphical overview
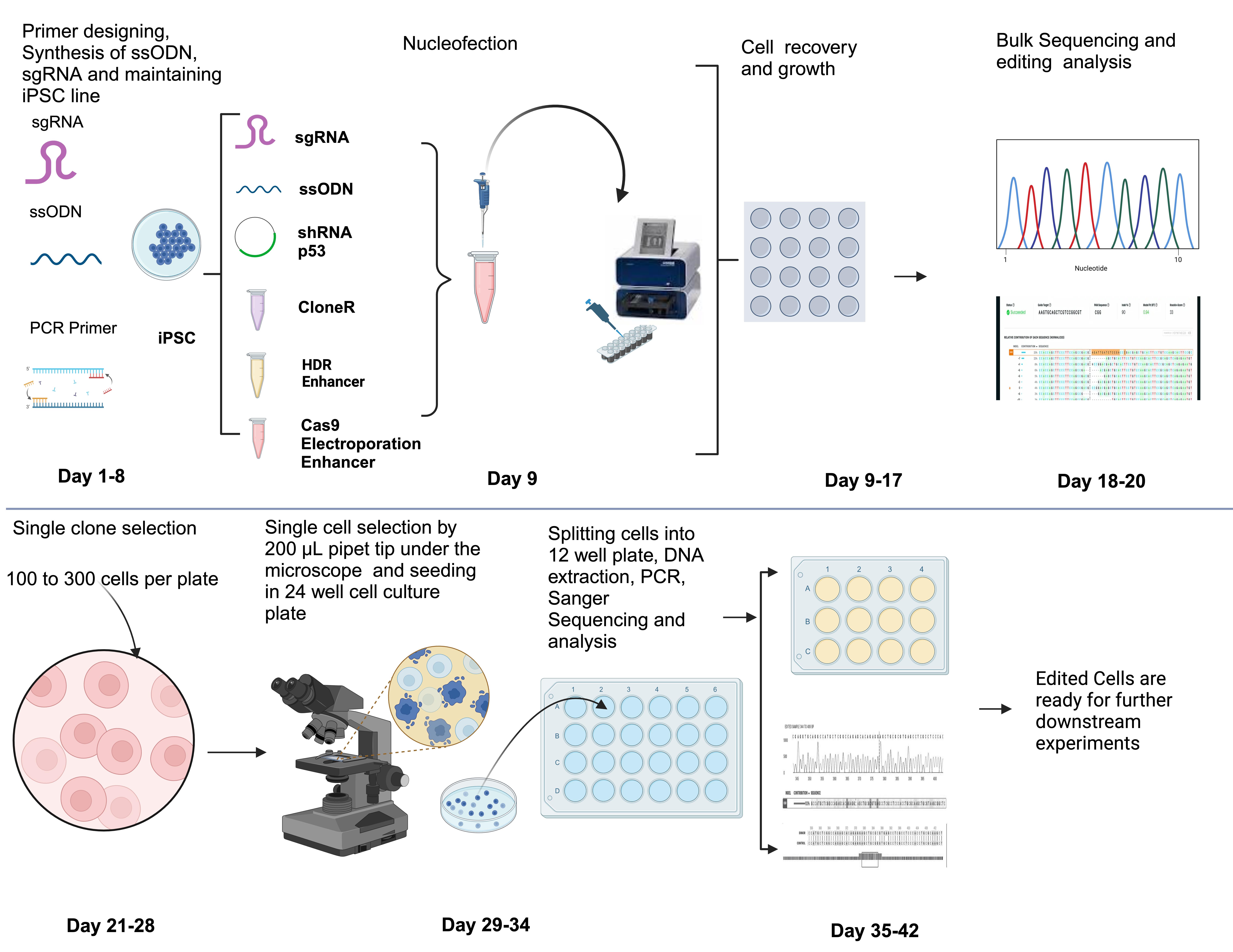
Outline of Protocol
Background
Genome editing has revolutionized biotechnology by allowing scientists to make precise changes to an organism's DNA. The ability to modify an organism's genome allows experts to study difficult evolutionary and medical problems in more complex systems [1]. The application of gene editing in induced pluripotent stem cells (iPSCs) to create genetically modified isogenic lines can enhance our understanding of how genetic mutations cause disease. Gene editing ranges from conceptually straightforward gene knockouts to more intricate modifications such as point mutations or whole gene insertions. However, currently, gene editing is still confronted with challenges [2–4]. In plant and animal models, clustered interspaced short palindromic repeats (CRISPR) have emerged as the tool of choice for genome editing [5]. Nowadays, the nuclease versions Cas9 and Cas12a are commonly used [1]. The CRISPR-Cas9 and Cas12a systems work with a guide RNA (gRNA) that is divided into two parts: a trans-activating CRISPR RNA (tracrRNA) that binds directly to the Cas nuclease to create ribonucleoprotein (RNP) and a CRISPR RNA (crRNA) that indicates the genomic target location [6]. When compared with previous editing techniques, the CRISPR-Cas innovation offers more precise targeting and editing proficiency as well as being easier and less expensive to configure, allowing it to be used more extensively [7–9]. However, the CRISPR-Cas editing systems still require improvement, particularly for point mutations. In certain cases, there may be several risk variants associated with a target gene, necessitating the creation of multiple lines with distinct single nucleotide polymorphisms (SNPs) or the sequential insertion of genetic modifications. Therefore, to enable more laboratories to produce the needed genetically modified lines, it is essential to develop a gene editing technique that is both highly efficient and easily adaptable. Currently, our attention is on overcoming the barriers to introducing point mutation in cell lines and improving the method to be as effective as possible. Editing efficiency may be increased by preventing cell death with the use of a Rho-related protein kinase (ROCK) inhibitor and blocking the p53 pathway [7]. In this protocol, we used pro-survival small molecules in conjunction with p53 inhibition to attain a homologous recombination rate exceeding 90%.
Materials and reagents
Biological materials
iPSC line (human NDC1) [10]
Reagents
mTeSR Plus basal (Stem Cell Technologies, catalog number: 100-0274)
mTeSR Plus 5× supplement (Stem Cell Technologies, catalog number: 100-0275)
CloneR (Stem Cell Technologies, catalog number: 05888 10 mL)
Revitacell supplement (Gibco, catalog number: A2644501 100×)
1× D-PBS (Gibco, catalog number: 10010049 500 mL)
DMEM/F12 (Gibco, catalog number: A4192001 500 mL)
Matrigel (Corning, catalog number: 47743-706 5 mL)
Accutase (VWR, catalog number: AT104 100 mL)
ReLeSR (Stem Cell Technologies, catalog number: 100-0484)
Alt-R Cas9 HDR enhancer (stock solution 3 mM) (IDT, catalog number: 1081062)
pmaxGFP (LONZA, catalog number: V4XP3032 50 μL at 1 μg/μL)
P3 primary cell nucleofector solution (LONZA, catalog number: V4XP3032 675 μL)
P3 primary cell nucleofector supplement (LONZA, catalog number: V4XP3032 150 μL)
Single-guide RNA (sgRNA) (IDT custom-made, 100 μM)
Alt-R S.p. HiFi Cas9 Nuclease V3 (IDT, catalog number: 10810559 500 μg at 10 μg/μL)
Single-strand oligo donor (ssODN) (IDT, 10 nmol)
shp53-f2 plasmid (Addgene)
Alt-R Cas9 electroporation enhancer (IDT, catalog number: 1075915 100 μm)
Molecular-grade H2O (Cytiva, catalog number: 82007-334 500 mL)
Zymo quick DNATM MicroPrep kit (Zymo Research, catalog number: D3021)
Agarose (VWR Life Sciences, catalog number: N605-500G)
Zymo CleanTM Gel DNA Recovery kit (Zymo Research, catalog number: D4002)
Solutions
Ribonucleoprotein (RNP) complex (see Recipes)
CloneR media (see Recipes)
Nucleofection reaction solution (see Recipes)
Nucleofection media (see Recipes)
P3 nucleofection solution mix (see Recipes)
mTeSR complete media (see Recipes)
Matrigel (see Recipes)
Recipes
RNP complex
This should be freshly prepared.
Reagent Final concentration Volume Cas9 nuclease V3 64 mM 0.8 μL sgRNA 100 µM 1.2 μL D-PBS (1×) 3.0 μL Total 5.0 μL CloneR media
This should be freshly prepared. Can be stored at 4 °C for one week.
Media component Volume mTeSR complete 8.9 mL Clone R 1 mL Revitacell 100 µL Total 10 mL Nucleofection reaction solution
This should be freshly prepared.
Component Test reaction GFP control No pulse control P3 primary cell nucleofector solution 1 μL 7 μL 1 μL shp53-f2 plasmid 1 μL 1 μL 1 μL Alt-R electroporation enhancer (100 μM) 1 μL 1 μL 1 μL ssODN (single-stranded oligodeoxynucleotide) (100 μM) 1 μL 0 μL 1 μL RNP complex 5 μL 0 μL 5 μL GFP (0.5 μg/μL) 0 μL 1 μL 0 μL Cells (1 million/reaction) (μL can be altered) 11 μL 11 μL 11 μL Total 20 μL 20 μL 20 μL Nucleofection media
This should be freshly prepared.
Component Volume CloneR media 1 mL Alt-R Cas9 HDR enhancer 10 µL P3 nucleofection solution mix
This should be freshly prepared.
Component Volume P3 primary cell nucleofector solution 16.4 μL P3 primary cell nucleofector supplement 3.6 μL Total 20 μL mTeSR Plus complete media
This should be freshly prepared and can be stored at 4 °C for two weeks.
Component Volume mTeSR Plus basal 40 mL mTeSR Plus 5× supplement 10 mL Total 50 mL Matrigel
Dilute Matrigel right before the experiment with DMEM/F12. The diluted Matrigel can be stored at 4 °C for 2 weeks.
Component Volume DMEM F12 11 mL Matrigel (1 mg/mL) 1 mL Total 12 mL
Laboratory supplies
6-well culture plate (Thermo Scientific, catalog number: 12556004)
24-well plate (Thermo Scientific, catalog number: 12556006)
12-well plate (Thermo Scientific, catalog number: 12556005)
1.7 mL microtube (Genesee Scientific, catalog number: 24-282C)
1,250 μL tips (GeneMate, catalog number: P-1237-1250)
200 μL tips (Neptune, catalog number: 89140-902)
20 μL tips (GeneMate, catalog number: P-1237-20)
10 μL tips (GeneMate, catalog number: P-1234-10)
15 mL Falcon tubes (Sarstedt, catalog number: 62.554.205)
16-well Nucleocuvette Strip (Lonza, catalog number: V4XP3032)
Equipment
Nucleofector (Lonza, model: 4D)
Centrifuge (Sorvall, model: ST16R)
Gel imager (Bio-Rad, model: ChemiDocTM MP Imaging System)
Gel electrophoresis machine (Labnet, model: Enduro GelXL)
NanoDrop (Thermo Scientific, model: Nanodrop One)
PCR thermocycler (Bio-Rad, model: T100TM thermal cycler)
Microscope (EVOS, Advance Microscopy group)
Incubator (SANYO CO2 incubator)
Biosafety cabinet (Nuaire, model: LabGard, Class II, Type A2, Biological Safety Cabinet)
Hemocytometer (Hausser Scientific Horsham, model: Bright-Line)
Software and datasets
Synthego.com (https://design.synthego.com/#/)
IDT (https://www.idtdna.com/site/order/designtool/index/CRISPR_CUSTOM)
ImageJ (https://ij.imjoy.io/)
Procedure
In silico design sgRNA and ssODN
Design sgRNA and ssODN by using IDT software: https://www.idtdna.com/site/order/designtool/index/HDRDESIGN.
The exact amount to resuspend sgRNA (Table 1) and ssODN (Table 2) to a 100 μM concentration can be determined using IDT Resuspension Calculator: https://www.idtdna.com/calc/resuspension/.
Table 1. Information of guide RNA used in this protocol
dsSNP ID Sequences PAM Site rs121918393 GAGGCGCACCCGCAGCTCCT CGG Table 2. Information on HDR templates used in this study
dsSNP ID HDR Template Sequences rs121918393 GGAGCCGCTTACGCAGCTTGCGCAGGTGGGAGGCGAGGCTCACGCGCAGCTCCTCTGTGCTCTGGCCGAGCATGGCCTGCACCTCGCCGCGGTACTGCAC
Cell culture and gene editing
Maintain the iPSC line in mTeSR Plus complete media (see Recipe 6).
Note: Maintain the iPSC line in mTeSR Plus complete media at 37 °C in a humidified 5% CO2 incubator. Change media every other day. Passage cells when 75%–80% confluence is achieved.
Change cell culture media to mTeSR Plus complete media with additional 1% Revitacell supplement 1 h before the start of nucleofection.
Prepare a Matrigel-coated 24-well plate (see Recipe 7) for the nucleofected cells >1 h prior.
After aspirating Matrigel, add 500 μL of nucleofection media (see Recipe 4). Use half these amounts if using a 48-well plate.
Aspirate media from the iPSC cell culture.
Add 2 mL of 1× D-PBS to clean/rinse the cell monolayer and aspirate.
Add 0.5 mL of Accutase, incubate at 37 °C for <10 min (8 min is optimal), and check that cells have been released using a microscope.
Add 2 mL of DMEM/F12 to neutralize the Accutase.
Gently wash cells off the plate using a p1000 pipette and transfer to a 15 mL conical tube.
Add additional DMEM/F12 to make up to 10 mL.
Take 10 μL of cell suspension and count using a hemocytometer or other cell counting methods. A cell concentration of 1 million cells per reaction is recommended (see cell counting protocol in File S1).
Note: We recommend the user to optimize the cell density when a new line is used.
Take the total number of cells (per milliliter of cell suspension) needed for the experiment depending on how many separate reactions are being done.
Note: For example, three reactions would need 3 million cells in total. If the cell concentration was 0.5 million/mL, then 6 mL would need to be taken from the counted suspension to have 3 million cells in total.
Centrifuge the cell suspension at 200× g for 7 min at 4 °C with acceleration at 9 and deceleration at 5.
Aspirate and resuspend cells in 11 μL per reaction of P3 primary cell nucleofector solution mix (see Recipe 5).
In each Nucleofection reaction mixture, add the cell suspension and 5 μL of the pre-prepared RNP complex and pmaxGFP, ssODN, shp53-f2 plasmid, and Alt-R Cas9 electroporation enhancer and add additional P3 primary cell nucleofector solution if required (see Recipe 3).
Pipette suspension up and down 2–3 times to mix and transfer 20 μL of the nucleofection reaction mixture to the corresponding 16-well nucleocuvette strip wells.
Gently tap the 16-well nucleocuvette strip on the counter inside the biosafety cabinet to ensure that there are no air bubbles.
Turn on the Lonza 4D nucleofector, select X, and select the 16-strip Nucleocuvette with the P3 program CA137.
Note: We recommend optimizing by testing 4–8 programs initially when using a new cell line.
Place the nucleocuvette strip into the Lonza 4D system and nucleofect.
Check corresponding wells to program. Ensure that the no-pulse control does not have an assigned program, then select START. The nucleofection will take only ~30 s (Figure 1). Any errors will show on the screen.
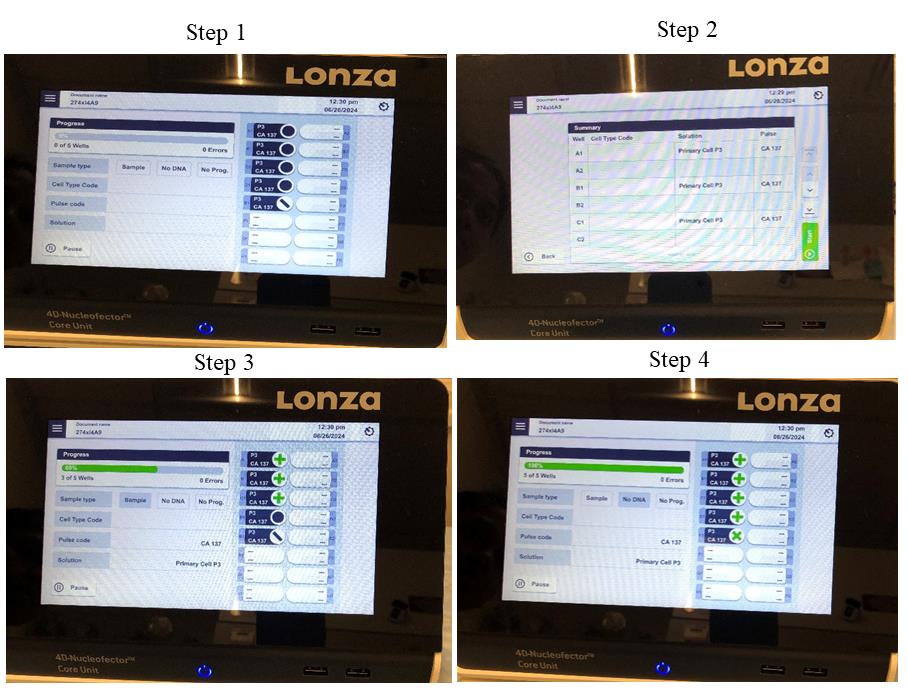
Figure 1. Pictorial presentation of setting Lonza nucleofector for gene editing. Step 1: Set the Lonza nucleofector; select pulse code, solution, and cell type. Step 2: Select the program. Step 3: Run the program after selecting all required wells. Step 4: Completion of nucleofection.Remove the Nucleocuvette from the nucleofector and incubate at 37 °C for 10–20 min.
Notes:
Do not add anything, move, or disturb the cuvette strip during this time.
This allows the lipid bilayer of the cells to reposition/organize.
After 10–20 min incubation, add 20 μL of the prewarmed nucleofection media (see Recipe 4) from the pre-prepared 24-well plate to each well in the nucleocuvette.
Using a p20 pipette, add the 40 μL nucleoporated cell suspension into the 500 μL of nucleofection media already in the Matrigel-coated 24-well plate.
Incubate cells at 37 °C; then, check and change media at 24 h with CloneR media (see Recipe 2).
Forty-eight hours after nucleofection, replace the CloneR media with fresh CloneR media. A complete change is not required. This is to compensate for the evaporation/degradation of previous CloneR.
Post-nucleofectionTwenty-four hours after nucleofection, count the percentage of GFP-expressing cells at 10× magnification. Take two images (Figure 2) with the brightfield filter and two images of the same field of view with the GFP filter per reaction (well). Repeat in 48 h. Save these images onto a USB to count using ImageJ software. After 48 h, the ability to see GFP under the microscope filter slowly dissipates.
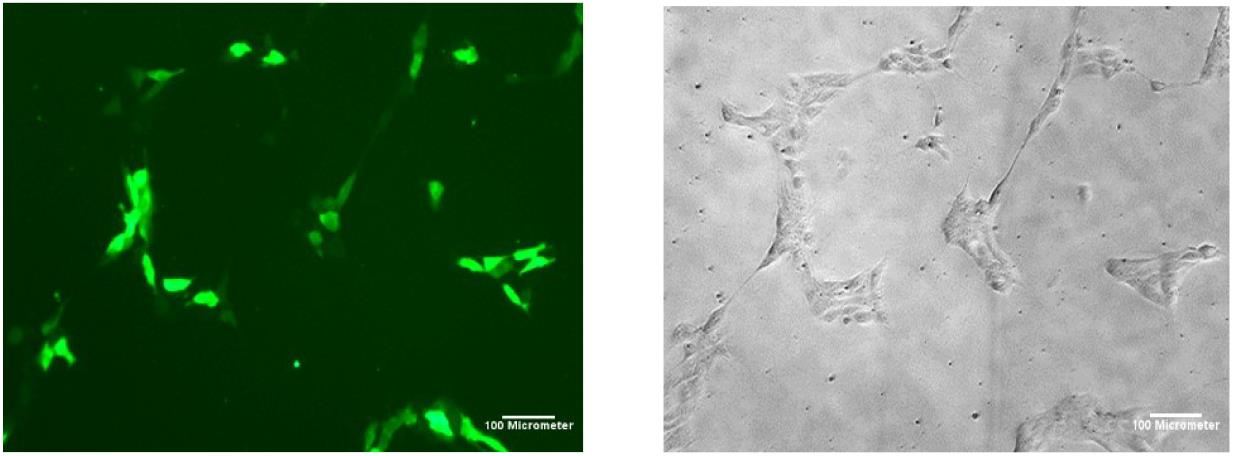
Figure 2. Induced pluripotent stem cell (iPSC) colonies with pmaxGFP 24 h post-nucleofection GFP vs. brightfield at 10× magnificationOnce cells reach 70%–80% confluency, extract genomic DNA using the Zymo quick DNATM MicroPrep kit.
Quantify genomic DNA levels using a Nanodrop and perform PCR for the desired gene.
Note: For achieving high-quality results in genomic DNA applications like PCR, it is crucial to use good-quality DNA and ensure the following practices: quantification of genomic DNA, optimization of PCR conditions, and running a gel for verification.
Run 20 μL of the PCR product on 1% agarose gel to check the quality of the PCR product.
Cut the PCR product and purify by Zymo CleanTM Gel DNA Recovery kit.
Quantify the eluate using Nanodrop to determine the optimal amount for Sanger sequencing.
Results from Sanger sequencing can be analyzed using Synthego’s ICE online software and/or gene viewer programs, e.g., SnapGene.
To analyze using ICE, download the ab.1 file for the desired sample to analyze and the corresponding control file.
Go to https://ice.synthego.com and drag and drop the ab.1 files into the corresponding control and experiment sample boxes.
Use the guide and repair ssODN sequences.
Select add samples to analysis and analyze (Figure 3).
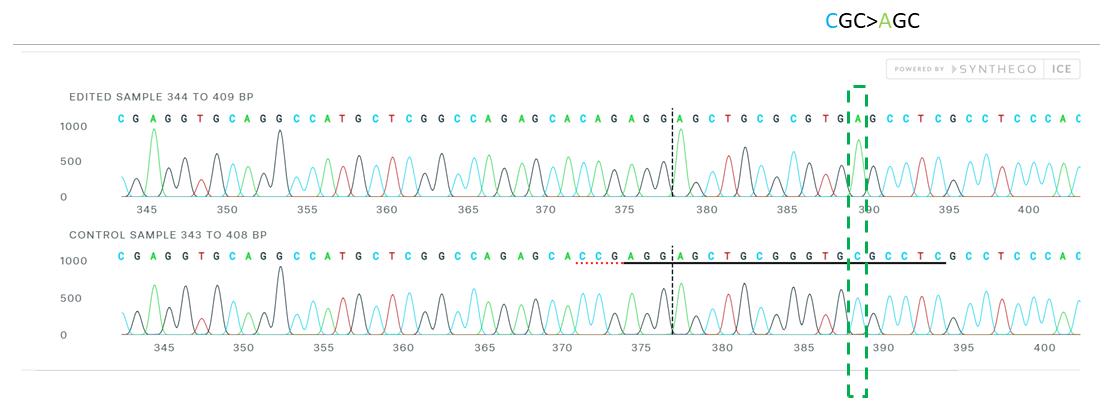
Figure 3. Sanger sequence view showing edited and control sequences in the region around the guide sequence. This shows sequence base calls from both the control and the edited sample .ab1 files. The horizontal black underlined region represents the guide sequence. The horizontal red underline is the PAM site. The vertical black dotted line represents the actual cut site.After confirmation of gene editing, cells can be dissociated and isolated into single cells.
Single clone selectionCoat a 10 cm plate with Matrigel for <1 h.
Aspirate media from bulk cell plate, wash with 2 mL of 1× D-PBS, and aspirate.
Add 0.5 mL (for a 6-well plate) and 0.125 mL (for a 24-well plate) of Accutase for <10 min at 37 °C.
Add 1 mL of DMEM/F12 to the well to wash and separate.
Transfer cell suspension to a 15 mL conical tube and top up to 10 mL with DMEM/F12.
Note: This further dilutes the dissociation reagent to ensure cell health.
Centrifuge at 100× g for 5 min.
Aspirate supernatant and flick tube to loosen cell pellet.
Add 1 mL of cloning media to the cell pellet and pipette to mix.
Count the cells by hemocytometer (File S1).
Seed two 10 cm cell culture dishes with 100 and 300 cells in 10 mL of media.
Keep the plate in the incubator at 37 °C for 48–72 h.
Examine the cells under the microscope after 48 h to see if they are single cells or not.
Cells should be ready for picking after 72–96 h of seeding (when cells form single colonies) (Figure S1).
After 72–96 h, when small colonies originating from single cells appear, prepare the microscope in the biosafety cabinet.
Prepare a 24-well plate with Matrigel media <1 h prior to picking.
With the appropriate plate under the microscope (Figure 4) at 4× magnification, use a p200 pipette on the 100 μL setting (to pick a 24-well), and pick colonies that are clearly isolated with a good size (up to 5–10 μm) and shape (Video S1).
Notes:
Cell colony may need to be lightly scraped to lift it off the plate before pipetting up.
After several colonies have been picked, the media in the plate wells may need to be topped up in order to continue picking.

Figure 4. Image of microscope setup inside the biosafety cabinet for colony pickupAllow colonies in each well to form large, good sized colonies (this can take 3–5 days post-picking).
Once the colonies are big enough to be split, prepare/prewarm a new Matrigel-coated 24-well plate with 500 μL of cloning media in each well.
Split corresponding cells into a 12-well plate from a 24-well plate by using ReLeSR as a digestive agent and extract genomic DNA from half of the cells.
After genomic DNA extraction, perform a standard PCR, send the sample for sequencing, and verify with a sequencing analysis program or perform ICE analysis (the same as steps 7 to 11).
Note: We use commercially available genomic DNA extraction kits for better DNA quality.
After confirmation, perform downstream experiments from the edited cell lines.
Validation of protocol
This protocol or parts of it has been used and validated in the following research article:
Singh et al. [11]. A high efficiency precision genome editing method with CRISPR in iPSCs. Scientific Reports.
General notes and troubleshooting
General notes
A passage number below p25 will result in better editing efficiency.
On the day of nucleofection, the media should be changed to mTeSR plus with (1%) Revitacell supplement.
Try to use the IDT resuspension calculator to resuspend the sgRNA and ssODN.
Before use, these reagents should be centrifuged for a brief amount of time.
The Cas9 enzyme should always be taken out from the -20 °C freezer directly before use such as when making the RNP complex.
If using Accutase to detach, you should use double the volume of DMEM/F12 media to dilute the Accutase.
After nucleofection, immediately keep the vessel in the incubator at 37 °C for 10–20 min.
For single-cell colony picking, seed 100 and 300 cells in two different 10 cm plates. For single-cell colony picking, seeding cells at the correct density is crucial to ensure that individual cells are spaced far enough apart to grow into isolated colonies. Our approach to seed 100 and 300 cells in two different 10 cm plates is a strategy to balance cell density and the ease of picking single colonies.
It is highly recommended to cut the desired PCR product from the gel, purify it, and send it for sequencing.
Troubleshooting
Problem 1: Low cell survival rate after nucleofection.
Possible cause: i) The cell number is low.
Solution: i) Use the appropriate cell number with a minimum of 1 million cells per reaction.
Possible cause: ii) Issue with Cas9 concentration.
Solution: ii) We recommend optimizing the Cas9 concentration by using different concentrations of Cas9 in a titration experiment.
Possible cause: iii) Harsh pipetting and centrifugation.
Solution: iii) Pipette very gently during the cell handling and perform centrifugation with centrifugation breaking methods.
Problem 2: Low editing efficiency.
Possible cause: The guide RNA or ssODN may not be working. Cas9 concentration may be incorrect.
Solution: i) Always try 2–3 different guideRNAs and ssODN.
Solution: ii) Optimize Cas9 concentration.
Problem 3: Poor sequencing results from Sanger sequencing.
Possible cause: Problems with PCR primers or the selection of the correct PCR product.
Solution: Redesign the PCR primer and always run an agarose gel, cut the expected size fragment, elute with the gel recovery kit, quantify, and send for sequencing.
Problem 4: Cells are not attaching in the 10 cm dish after plating the single cell suspension.
Possible cause: Lack of cell survival molecules.
Solution: Use CloneR in the growth media.
Acknowledgments
We acknowledge NIH grant 1R03AG070415, Veterans Administration Merit Research Award 1 I01 BX006391, University of Minnesota Faculty Research Development Grant, funds from the University of Minnesota, Institute of Translational Neuroscience (ITN) to SHY. SHY is an ITN Scholar. Figures were created with BioRender.com. We also acknowledge our previous work published in Scientific Reports (2024), DOI: https://doi.org/10.1038/s41598-024-60766-4 on which the current protocol is based [11].
Competing interests
There are no conflicts of interest or competing interests.
References
- Robb, G. B. (2019). Genome Editing with CRISPR‐Cas: An Overview. Curr Protoc Essent Lab Tech. 19(1): e36.
- Khalil, A. M. (2020). The genome editing revolution: review. J Genet Eng Biotechnol. 18(1): 68.
- Carroll, D. (2014). Genome Engineering with Targetable Nucleases. Annu Rev Biochem. 83(1): 409–439.
- Urnov, F. D., Miller, J. C., Lee, Y. L., Beausejour, C. M., Rock, J. M., Augustus, S., Jamieson, A. C., Porteus, M. H., Gregory, P. D., Holmes, M. C., et al. (2005). Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature. 435(7042): 646–651.
- Li, X. L., Li, G. H., Fu, J., Fu, Y. W., Zhang, L., Chen, W., Arakaki, C., Zhang, J. P., Wen, W., Zhao, M., et al. (2018). Highly efficient genome editing via CRISPR–Cas9 in human pluripotent stem cells is achieved by transient BCL-XL overexpression. Nucleic Acids Res. 46(19): 10195–10215.
- Budde, J. P., Martinez, R., Hsu, S., Wen, N., Chen, J. A., Coppola, G., Goate, A. M., Cruchaga, C. and Karch, C. M. (2017). Precision genome-editing with CRISPR/Cas9 in human induced pluripotent stem cells. bioRxiv. doi.org/10.1101/187377.
- Ihry, R. J., Worringer, K. A., Salick, M. R., Frias, E., Ho, D., Theriault, K., Kommineni, S., Chen, J., Sondey, M., Ye, C., et al. (2018). p53 inhibits CRISPR–Cas9 engineering in human pluripotent stem cells. Nat Med. 24(7): 939–946.
- Conti, A. and Di Micco, R. (2018). p53 activation: a checkpoint for precision genome editing? Genome Med. 10(1): 66.
- Uffelmann, E., Huang, Q. Q., Munung, N. S. (2021). Genome-wide association studies. Nat Rev Methods Primers. 1: 59.
- Israel, M. A., Yuan, S. H., Bardy, C., Reyna, S. M., Mu, Y., Herrera, C., Hefferan, M. P., Van Gorp, S., Nazor, K. L., Boscolo, F. S., et al. (2012). Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature. 482(7384): 216–220.
- Singh, A., Smedley, G. D., Rose, J. G., Fredriksen, K., Zhang, Y., Li, L. and Yuan, S. H. (2024). A high efficiency precision genome editing method with CRISPR in iPSCs. Sci Rep. 14(1): 9933.
Supplementary information
The following supporting information can be downloaded here:
- File S1. Protocol for cell counting by using hemocytometer slide
- Figure S1. Image of single cell
- Video S1. Video of colony piking under the microscope
Article Information
Publication history
Received: Aug 20, 2024
Accepted: Oct 14, 2024
Available online: Nov 5, 2024
Published: Dec 20, 2024
Copyright
© 2024 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Singh, A., Babu, S., Phan, M. and Yuan, S. H. (2024). CRISPR/Cas9-Based Protocol for Precise Genome Editing in Induced Pluripotent Stem Cells. Bio-protocol 14(24): e5141. DOI: 10.21769/BioProtoc.5141.
- Singh, A., Smedley, G. D., Rose, J. G., Fredriksen, K., Zhang, Y., Li, L. and Yuan, S. H. (2024). A high efficiency precision genome editing method with CRISPR in iPSCs. Sci Rep. 14(1): 9933.
Category
Stem Cell > Pluripotent stem cell > Cell-based analysis
Biological Sciences > Biological techniques > CRISPR/Cas9
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.