- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Chromogranin B Purification for Condensate Formation and Client Partitioning Assays In Vitro
Published: Vol 14, Iss 20, Oct 20, 2024 DOI: 10.21769/BioProtoc.5095 Views: 1563
Reviewed by: Valérian DORMOYAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Sep 2022
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols


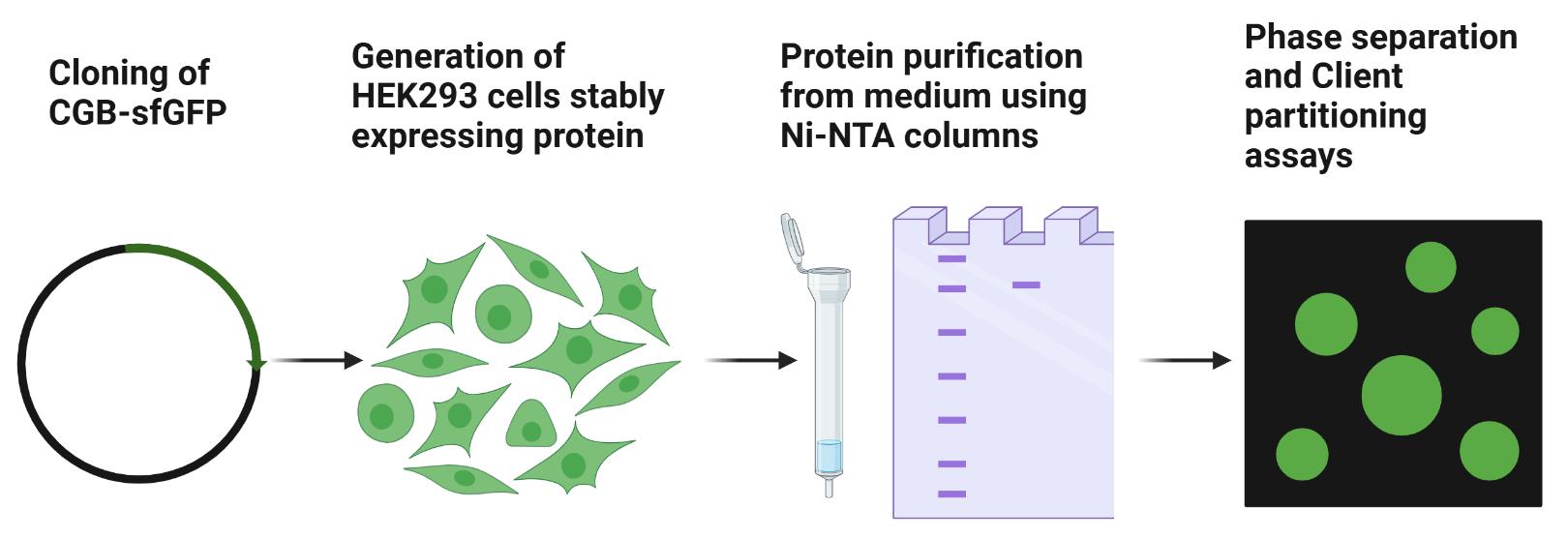
Abstract
Chromogranin B and other members of the granin protein family form condensates that recruit clients like proinsulin. The condensation in the lumen of trans-Golgi network (TGN) is critical for the biogenesis of secretory granules. Here, we describe a protocol to purify the tagged version of chromogranin B close to its native form at the TGN, which can then be utilized for microscopy-based assays to monitor condensate formation in vitro and client partitioning depending on the material properties of chromogranin B assemblies.
Key features
• First instance of purification of full-length and tagged version of members of the chromogranin family of proteins.
• Allows purification of proteins with post-translational modifications that are acquired en route in the secretory pathway, thus closely resembling their native form at the TGN.
Keywords: Phase separationGraphical overview
Background
Members of the chromogranin (CG) family of proteins are predominantly expressed in specialized secretory cells including the pancreatic beta cells and the cells of the neuroendocrine system. They play a critical role in driving secretory granule (SG) biogenesis [1–3]. Expression of either chromogranin A (CGA) and chromogranin B (CGB) or other granin proteins in non-specialized cells results in the formation of ectopic SG-like structures [2,4–6]. Experiments involving extracts or purified CGA, CGB, and secretogranin 2 (SCG2) from the adrenal or pituitary glands demonstrated aggregation of these proteins at an acidic pH (pH 5.2–5.5) and in presence of millimolar concentrations of calcium [6–10]. Since the pH used in previous studies is lower than that seen at the trans-Golgi network (TGN) and the calcium concentrations were much higher [10,11], the physiological significance of the aggregation in cargo sorting has remained elusive. Moreover, molecular mechanisms underlying higher-order assemblies of CGB and the impact of material properties of these assemblies on cargo sorting require further investigation.
We demonstrated that CGB forms condensates in milieu of the TGN, driven predominantly by the mildly acidic compartment of the TGN and independent of the requirement of divalent cations [12]. For this purpose, we developed a protocol using the PiggyBac transposon-based inducible mammalian expression system [13] to purify the superfolder GFP-tagged versions of CGB from the secreted medium. Moreover, by making different assemblies (condensates vs. aggregates) of CGB in the presence of calcium and zinc, respectively, we further demonstrate that the material properties of CGB assemblies drive client partitioning.
Materials and reagents
Materials
Plasmid for cloning human CGB gene (OriGene, RC201744L1)
RINS1 plasmid (Addgene, plasmid #107290)
2× Gibson master mix (NEB, catalog number: E2611)
Nhe1-HF (NEB, catalog number: R3131)
Not1-HF (NEB, catalog number: R3189)
Agarose (American Bio, catalog number: AB00972)
SYBR Safe DNA gel stain (Invitrogen, catalog number: S33102)
TAE Buffer (Thermo Fisher Scientific, catalog number: B49)
PB-T-PAF, PB-RN, and PBase [13]
Water, molecular biology grade (Sigma, catalog number: W4502)
Qiagen Gel Extraction kit (Qiagen, catalog number: 28704)
Mini prep kit (Takara Bio, catalog number: 740588.50)
LB ampicillin plates (Recombinant Technologies)
HEK293 cells (ATCC, catalog number: CRL-1573)
Lipofectamine 2000 (Thermo Fisher Scientific, catalog number: 11668027)
DMEM high glucose (Gibco, catalog number: 11965092)
Fetal bovine serum (FBS) (Gibco, catalog number:16000044)
Penicillin-Streptomycin-Glutamine (100×) (Gibco, catalog number: 10378016)
OptiMEM (Gibco, catalog number: 51985034)
Puromycin dihydrochloride (Thermo Fisher Scientific, catalog number: J67236.XF)
G418 disulfate (Thermo Fisher Scientific, catalog number: J63871.AB)
cOmpleteTM His-Tag purification resin (Millipore Sigma, catalog number: 5893682001)
A23187 (Millipore Sigma, catalog number: C7522)
Doxycycline monohydrate (LKT Labs, catalog number: D5898)
Aprotinin (Millipore Sigma, catalog number: A6106)
Imidazole (Spectrum, catalog number: IM105)
0.45 µm cellulose acetate filtration unit (Corning, catalog number: 430768)
Amicon® Ultra centrifugal filter (Millipore Sigma, catalog number: UFC5030)
Amicon® Ultra-4 centrifugal filter (Millipore Sigma, catalog number: UFC800324)
AmiconTM Ultra-15 centrifugal filter units (Millipore Sigma, catalog number: UFC903024)
NaOH (Sigma, catalog number: 221465-500G)
Glycerol (Fisher Scientific, catalog number: 02-002-937)
5 M NaCl solution (American Bio, catalog number: AB13198-01000)
Tris, 1 M Solution, pH 7.4 (American Bio, catalog number: AB14044-01000)
Sodium phosphate, dibasic, anhydrous (Na2HPO4) (J. T. Baker, catalog number: 3828-01)
Sodium phosphate, monobasic, monohydrate (NaH2PO4.H2O) (J. T. Baker, catalog number: 3818-01)
Cy3-labeled lysozyme (Lyz) (Nanocs, catalog number: LS1-S3-1)
Calcium chloride solution (Millipore Sigma, catalog number: 21115)
Zinc chloride (Sigma, catalog number: Z-4875)
1× DPBS (Gibco, catalog number: 14190250)
Chromatography columns (Bio-Rad, catalog number: 732-1010)
Glass-bottom imaging dishes (Cellvis, catalog number: D35-14-1.5-N)
250 mL filter system (Corning, catalog number: 430768)
PCR tubes (Thomas Scientific, catalog number: 1149K07)
1.5 mL microcentrifuge tubes (USA Scientific, catalog number: 1415-2500)
List of primers used for cloning of sfGFP and His tagged CGB using Gibson assembly (Table 1)
Table 1. List of primers
Primer name Sequence CGB_sfGFP_6XHis F1 FP ggcggccatcacaagtttgtacagctagcatgcagccaacgctgcttctcagcctc CGB_sfGFP_6XHis F1 RP caccggtggcgaccggtggatccaagcccctttggctgaatttctcagctatcttctgtagttcc CGB_sfGFP_6XHis F2 FP ggaactacagaagatagctgagaaattcagccaaaggggcttggatccaccggtcgccaccgg CGB_sfGFP_6XHis F2 RP ccagcacactggatcagttatctatgcggccgctcattagctgcccttgtacagctcgtccatgcc
Solutions
10× Na-P stock (see Recipes)
His-binding buffer (see Recipes)
Wash buffer (see Recipes)
Elution buffer (see Recipes)
Protein storage buffer (see Recipes)
Phase separation assay buffer (see Recipes)
NaOH solution for washing column (see Recipes)
Recipes
10× Na-P stock (500 mM, 500 mL)
33.1 g of Na2HPO4
2.4 g of NaH2PO4
pH 8.0
Store at 4°C
His-binding buffer (500 mL)
500 mM NaCl (14.61 g), 50 mM Na-P (50 mL stock) pH 6.8 (fresh)
Wash buffer
His-binding buffer with 10 mM imidazole: 150 mL of His-binding buffer + 0.1 g of imidazole (fresh).
Elution buffer
His-binding buffer with 250 mM imidazole: 10 mL of His-binding buffer + 0.17 g of imidazole (fresh).
Protein storage buffer (50 mL)
200 mM Tris-HCl (1 M Tris-HCl 10 mL), 500 mM NaCl (5 M NaCl 5 mL), 10% glycerol (5 mL), pH 6.8.
Store at 4 °C for a week if purifying more than one protein within a week.
Phase separation assay buffer (40 mL)
25 mM Tris-HCl (1 M Tris-HCl 1 mL), 150 mM NaCl (5 M NaCl 1.2 mL), 2.5% glycerol (1 mL), pH 6.1 (fresh).
NaOH solution for washing column
50 mL of 0.5 N NaOH (1 g)
Store at room temperature.
Equipment
Thermal cycler (Eppendorf, model: nexus eco)
Centrifuge (Eppendorf, model: 5425)
Centrifuge (Eppendorf, model: 5910 R)
Centrifuge (Thermo Scientific, model: Sorvall Legend Micro 21R)
S-4x Universal rotor
Zeiss LSM 880 Confocal with Airyscan (63×; 1.4 N.A. oil objective)
Nanodrop (Thermo Scientific, model: NANODROP ONEC)
Procedure
Cloning to generate the PB-T-PAF-CGB_sfGFP_6XHis construct
The construct for expression of sfGFP and 6× His-tagged human CGB protein under a doxycycline-inducible promoter was generated by Gibson assembly. The coding region of the human CGB gene was PCR amplified using the expression plasmid from OriGene (100 ng for a 50 µL PCR reaction). sfGFP (containing the monomerizing mutation) was amplified by PCR amplification from the RINS1 plasmid (100 ng for a 50 µL PCR reaction). PB-T-PAF vector was digested using the restriction enzymes Nhe1-HF and Not1-HF (20 units each per reaction). PCR primers were designed to ensure overlap with the vector and individual fragments. The DNA sequence encoding the 6× His-tag was also incorporated in the reverse primer while amplifying the sfGFP fragment.
Run the PCR fragments on a 1% agarose gel containing SYBR Safe DNA gel stain using TAE buffer. Excise bands of appropriate sizes and purify by gel extraction using the Qiagen gel extraction kit.
Digest the PB-T-PAF vector.
Mix cut vector and individual fragments in 1:3 molar ratios in the Gibson master mix. Make up the total reaction volume to 20 µL using molecular biology–grade water and incubate in a thermal cycler at 50 °C for 1 h.
Transform 2 µL of the Gibson reaction into competent Omni max 2 E. coli.
Extract DNA from individual colonies from Luria agar containing ampicillin plates using the mini prep kit. Confirm positive clones by sequencing individual clones.
Generating stable cell lines for protein expression
Culture and maintain HEK293 cells in DMEM high glucose supplemented with 10% FBS, 100 U/mL penicillin, and 100 µg/mL streptomycin (complete medium) in 5% carbon dioxide at 37 °C. One day before transfection, seed approximately 1 million cells in a 6-well dish in the medium mentioned above. Cell culture and transfection are done in a cell culture hood.
The next day, transfect HEK293 cells using PB-T-PAF-CGB_sfGFP_6XHis, PB-RN, and PBase (8:1:1; total DNA: 1.5 µg) and Lipofectamine 2000.
For transfection, incubate DNA in a sterile 1.5 mL microcentrifuge tube in 100 µL of OptiMEM. In a separate tube, add 3 µL of Lipofectamine 2000 to 100 µL of OptiMEM. After 5 min, mix the contents of the two tubes and incubate for 20–30 min.
In the meantime, add fresh growth medium to cells and the transfection mixture in a dropwise fashion (steps B1–4; Figure 1).
Forty-eight hours post-transfection, initiate selection using a combination of 10 µg/mL puromycin dihydrochloride and 500 µg/mL G418 disulfate.
Maintain cells under antibiotic selection for at least a week, and then expand them and freeze them at -80 °C in freezing medium (complete medium containing 10% DMSO). Store them until further use in liquid nitrogen storage.
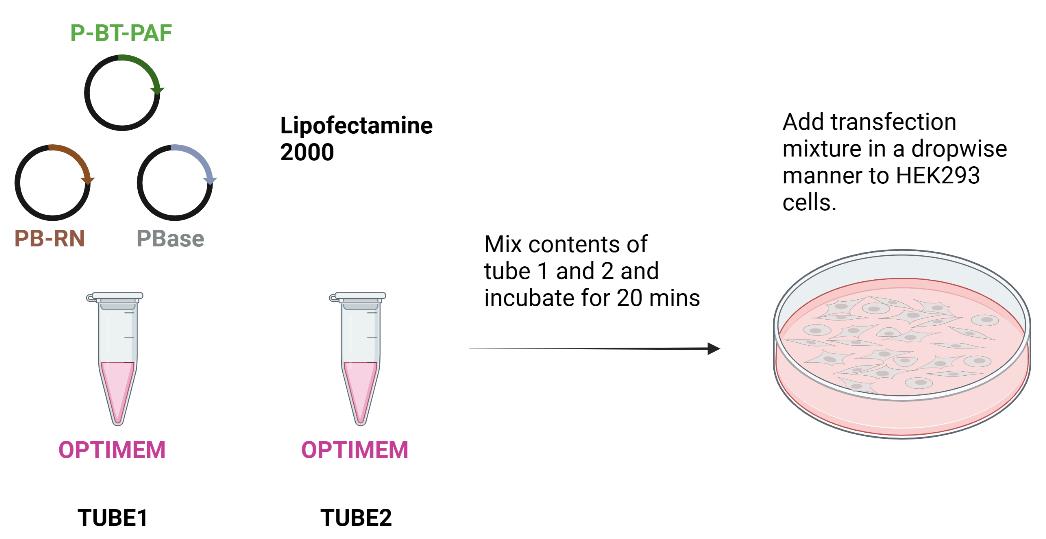
Figure 1. Scheme for transfecting HEK293 cells for generating stable lines expressing CGB-sfGFP under doxycycline-inducible promoter. Forty-eight hours after transfection, cells are subjected to antibiotic selection.
Protein expression and purification from cell culture supernatant
Harvesting cell culture supernatant
Thaw stable HEK293 cells expressing CGB-sfGFP-6X His under doxycycline-inducible promoter and expand them in at least ten 15 cm dishes. For proteins that, in pilot assays, were secreted to a lesser extent in the supernatant, we have gone up to fifteen 15 cm dishes. Additionally, when a lower percentage of cells showed us fluorescent protein expression after doxycycline induction (less than 50%; see below in notes and troubleshooting), we used 15 dishes.
When the cells are confluent, wash two times in 1× PBS and then incubate in 20 mL of serum-free DMEM containing 1 µg/mL doxycycline, 1µg/mL aprotinin, and 1µg/mL A23187 for approximately 16–20 h. Steps C1a and C1b were done in a cell culture hood.
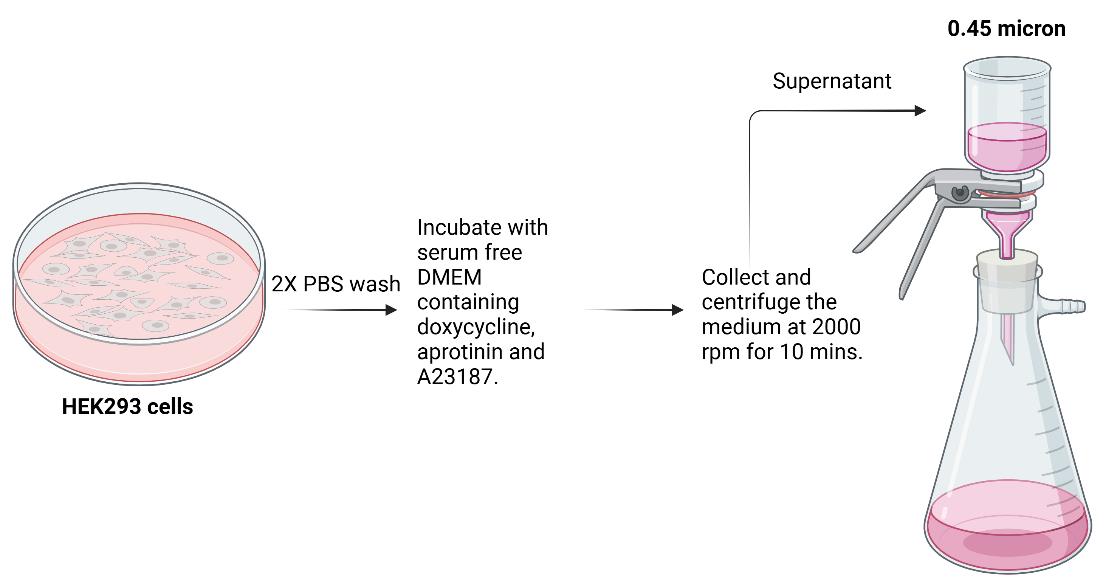
Figure 2. Scheme for collecting medium from HEK293 prior to protein purificationThe next day, collect and pool the medium from all the plates by centrifuging at 2,000 rpm (859× g) for 10 min at 4 °C. Filter using 0.45 µm filter and set aside to load on the column as described subsequently (Steps C1a–c; Figure 2).
Protein purification using Ni-NTA columns
Cut the column at the base using a scissor and then fill with PBS with 2–3 mL of slurry containing Ni-NTA beads (His tagged purification resin) and tightly pack between 1–1.5 cm of bed volume. Allow 50 mL of PBS to flow through.
Equilibrate the column with 100 mL of His-binding buffer.
Load the supernatant containing protein and allow it to run via gravity flow. Maintain a flow rate of one drop per 4–5 s. In our case, we could do this by simply adjusting the height of the reservoir from which the supernatant was loaded onto the column via the tubing.
After all the supernatant has passed through the column, wash the column in 150 mL of wash buffer.
Elute the protein in elution buffer (10 mL). Collect 1 mL fractions in Eppendorf tubes. Because of the GFP tag, visually green fractions are collected and pooled together.
To get rid of imidazole, buffer exchange the protein with protein storage buffer three times using 50 mL AmiconTM Ultra-15 centrifugal filter units with a 30 kDa cutoff by centrifugation at 4,500 rpm (4,347× g) for 15 min. Before adding the proteins, equilibrate the filter using storage buffer. In the first run, 2–3 mL of protein solution is topped with protein storage buffer to fill the tube (12 mL total volume). Concentrate the protein to 1 mL. Add 11 mL of buffer and reconcentrate the protein to 1 mL. Repeat this step two more times (Steps C2a–f; Figure 3).
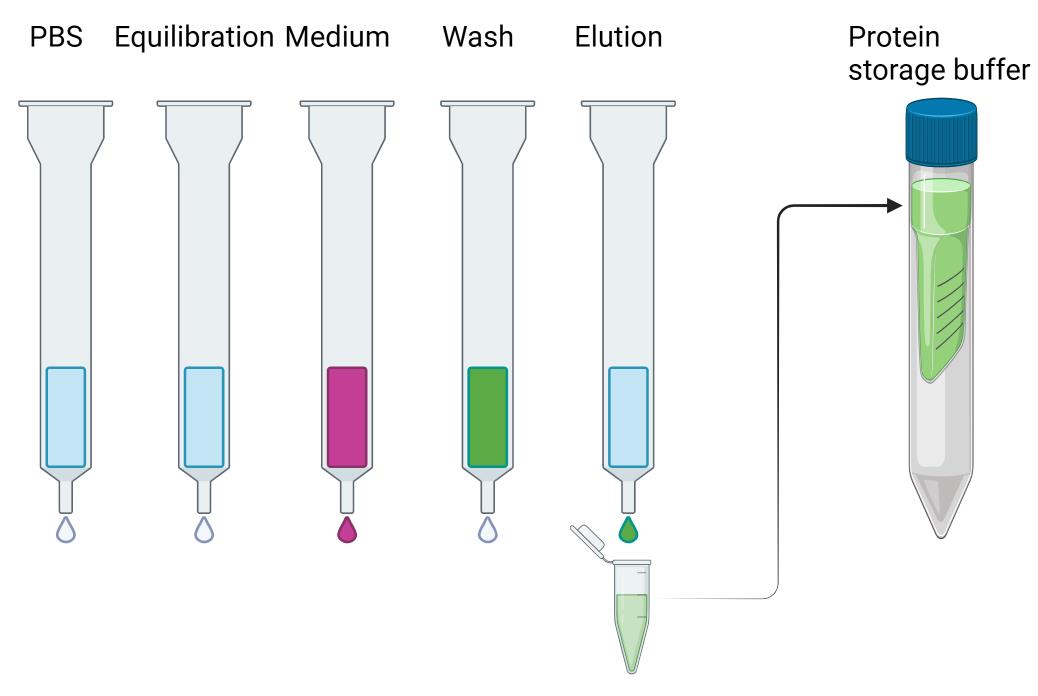
Figure 3. Scheme for protein purification using Ni-NTA columns. Protein-containing fractions (green) are pooled together and buffer exchanged into protein storage buffer and aliquoted and flash frozen using liquid nitrogen.Aliquot the protein in 100 µL fractions, flash freeze in liquid nitrogen, and store at -80 °C until further use.
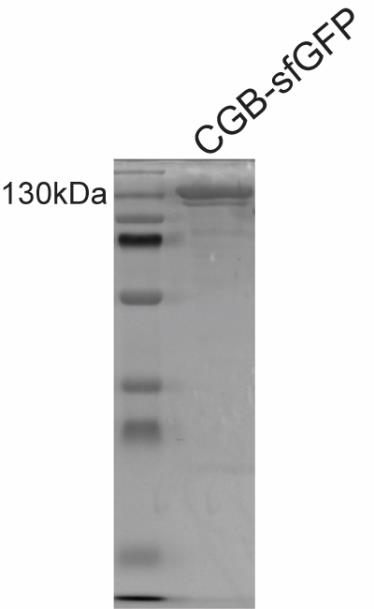
Figure 4. A typical Coomassie-stained SDS gel with purified CGB-sfGFP run on a denaturing gel. The band is seen at a size corresponding to 130 kDa.Wash the column with 50 mL of 0.5 N NaOH and then wash it with 100 mL of water. Pass 50 mL of 70% ethanol and cap and store the column for reuse at 4 °C in a cold room. We have only used the column a maximum of two times and only in instances when we were purifying the same protein within a month. We have performed protein purification in one day beginning from collection of the medium until storing the purified protein at -80 °C.
Caution:
1) Ensure there are not many dead cells at the time of collecting medium.
2) Ensure there are no air bubbles while packing the column.
Notes and troubleshooting:
1) Protein sticking while concentrating: In our hands, it was better to concentrate the protein at room temperature as it led to minimal sticking of the protein to the membrane during concentration.
2) Cells floating after incubation in serum-free medium: If the cells are alive, it should not be a problem. A solution could be plating the cells on poly-lysine-coated dishes, even though we have not tried this.
3) Cleaning impurities: Assess the quality of the protein by running 1–5 µg of protein on an SDS-PAGE gel followed by Coomassie staining. For CGB-sfGFP, we see a band at 130 kDa (Figure 4). If there are many lower-molecular-weight bands, then that indicates degradation or poor-quality purification, in which case the preparation should be repeated. Purity higher than 90% is good for biochemical experiments. We have not tried extensively with CGB-sfGFP, but we used gel filtration for purification of mCherry-tagged proinsulin using the same protocol with the only exception being that FLAG-tag was used as an affinity tag.
4) The protocol can be widely applied for the purification of mutant forms of chromogranin B and other granin proteins, provided they are secreted from HEK293 cells. Importantly, proteins purified using this method retain the post-translational modifications acquired during their transit through the secretory pathway (Figure 5). After the generation of stable lines, we purified proteins when 50% of the population was fluorescent after doxycycline addition. In some instances, when we felt that the number of fluorescent cells was much lower, we resorted to increasing the number of dishes (starting material) and, in some instances, redoing the transfection and selection, expanding the cell line, and purifying the protein right away before freezing cells.
5) A pilot secretion assay must be performed before large-scale purification. Grow cells to confluency in a 10 cm dish and then induce protein production in serum-free medium as described above. Concentrate the medium and run on SDS-PAGE gels followed by Coomassie staining; see if there is a strong band at the desired size. Alternatively, one can also use Ni-NTA beads to incubate with the concentrated medium. Since the proteins are GFP tagged, beads would turn green post-binding in a cold room for 1 h by rotating it end-to-end. The protein can be eluted after washing the beads with 1× PBS (three times) and boiling in SDS sample buffer. Run samples on SDS-PAGE gels followed by Coomassie staining to monitor the band at the appropriate size.
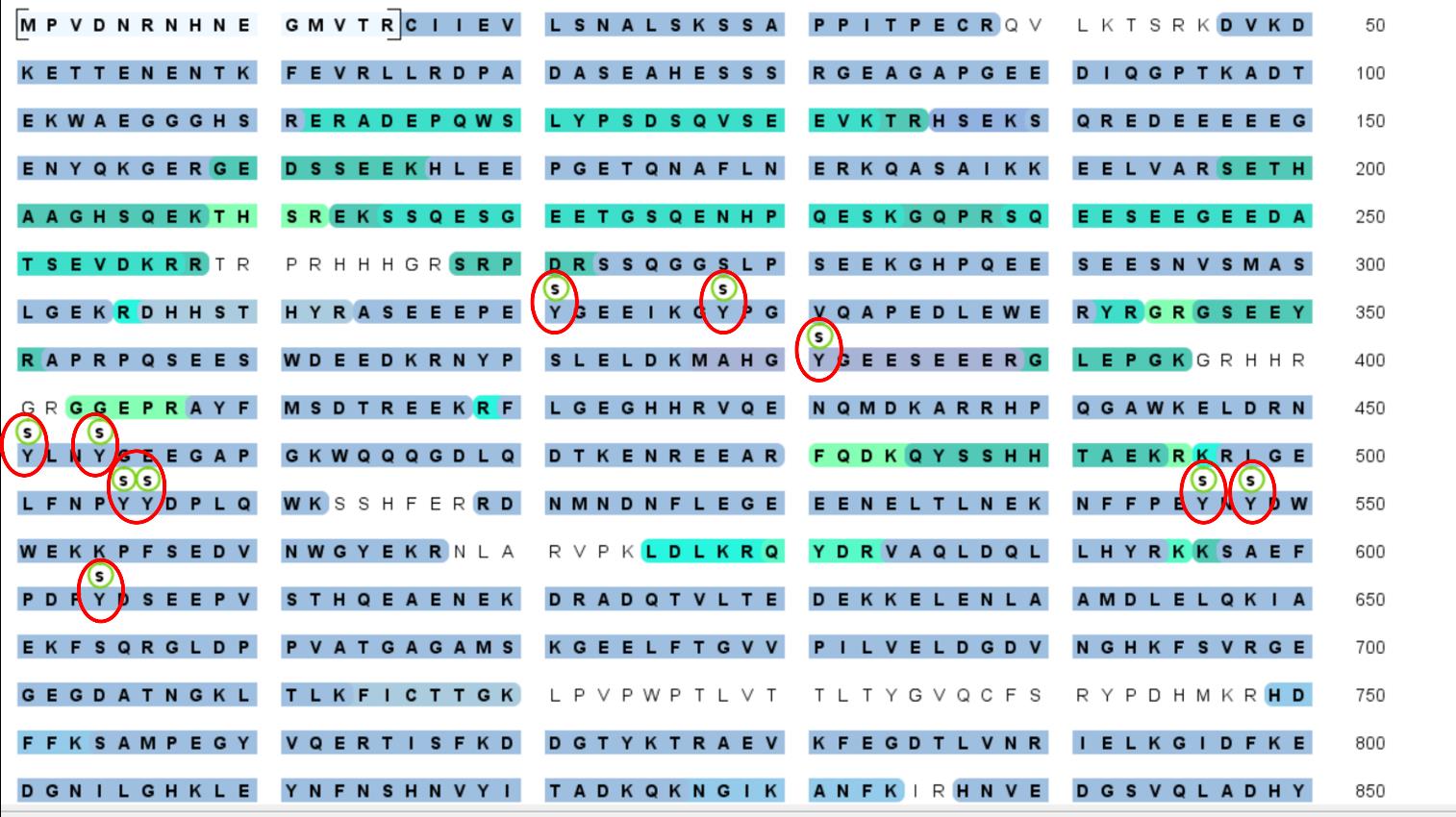
Figure 5. Purified CGB-sfGFP was subjected to mass spectrometry analysis to detect post-translational modifications at the facility at Yale university. Labeled in red are the sulfation on tyrosine residues. The presence of the post-translational modifications confirms that the purified protein passed through the secretory pathway where it acquires these modifications and suggests that the protein purified is close to the native form at the TGN. This is critical considering that post-translational modifications have been shown to play an important role in governing condensate formation in many different contexts.
Condensate formation and client partitioning assay
Buffer exchanging protein in phase separation buffer at pH 6.1
Thaw the protein solution on ice.
Buffer exchange the protein to phase separation buffer using 0.5 mL Amicon® Ultra centrifugal filter with a 30 kDa cutoff. Repeat the process at least three times. Take 200 µL of protein and fill the tube with 300 µL of buffer. Spin at 12,000 rpm (13,523× g) and concentrate to 100 µL. Repeat the process two more times. In case the initial yields are low, start with a larger volume, bring it down to 100 µL, and then proceed to three buffer exchanges.
Measure the protein concentration using Nanodrop by reading the absorbance at 280 nm. Since CGB-sfGFP forms condensates at pH 6.1, spin down the buffer-exchanged protein solution at full speed (21,100× g) in a tabletop microcentrifuge at 4 °C for 20 min.
Collect the supernatant in a separate Eppendorf tube and store it on ice. Immediately proceed to set up client partitioning assays.
Client partitioning assay using Cy3-tagged Lyz in the presence of calcium and zinc
Buffer exchange Cy3-tagged Lyz in the phase separation buffer by using 0.5 mL Amicon® Ultra-4 centrifugal filter with a 3 kDa cutoff three times.
Dilute calcium chloride and zinc chloride solution with phase separation buffer to a final concentration of 20 mM in two separate tubes. To each tube, add Cy3-tagged Lyz to a final concentration of 1 µM. To each tube, add CGB-sfGFP to a final concentration of 2.5 µM. Gently tap the tubes and incubate at room temperature for 5 min.
Plate the solution from each tube on an imaging dish from Cellvis.
After 5 min, image the samples on a Zeiss 880 confocal microscope using a 63×/1.4 oil objective at room temperature.
Acquire images in both GFP and Cy3 channels for droplets formed using calcium and aggregates formed using zinc (Figure 6). A 488 nm laser was used at 0.2% and a 561 nm laser was used at 5%. The pixel dwell time was 0.67 µs.
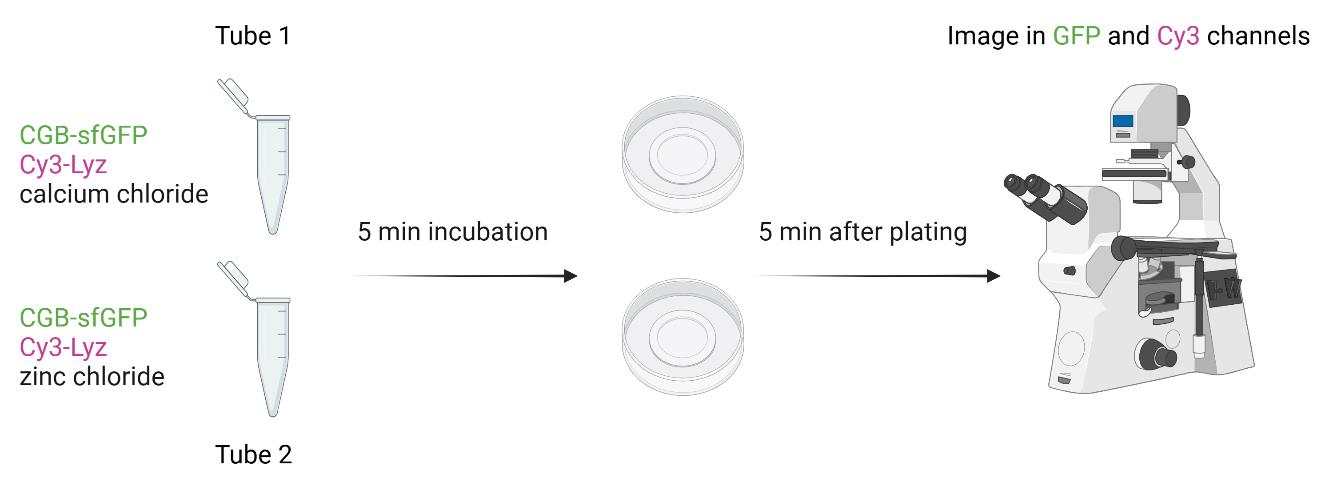
Figure 6. Scheme for client partitioning assay form monitoring differential client recruitment to calcium-induced condensates and zinc-induced CGB aggregatesNote: Client partitioning assays can also be set by mixing different client proteins including Cy3-tagged proinsulin by directly mixing the protein with CGB-sfGFP or by inducing phase separation using PEG 8000.
Data analysis for measuring relative client partitioning
Open images acquired in the .czi format on Zeiss 880 in Fiji.
Draw a region of interest (ROI) on the condensate image using the GFP channel (green).
In Fiji, go to Analyze > Tools > ROI manager > Add[t].
Click on measure and you will get intensity values in the results tab. Open the Cy3 channel, click on the ROI in the ROI manager, and again click on measure.
Follow the same procedure for the images of CGB aggregates using zinc (Figure 7).
Copy the values in an Excel sheet and normalize the values (mean intensity) in the Cy3 channel to GFP channel for both calcium and zinc conditions. The normalized values represent the amount of client (Cy3-Lyz) that partitions into condensate or aggregate relative to the amount of CGB-sfGFP.
Do this for multiple ROIs.
Analyze the differences in the relative partitioning from multiple ROIs using a t-test and a two-tailed distribution in Prism.
The data can also be analyzed by quantifying the signals of CGB-sfGFP or Cy3-LyZ from the assemblies; that from the background and absolute partitioning can be quantified, which is the ratio of signals within the condensate/aggregate to that outside. The data can also be represented as the amount of signal in condensate/aggregate to total signal (inside + outside). However, we have only performed the relative partitioning analysis in the manuscript where the client portioning assay was used [12].
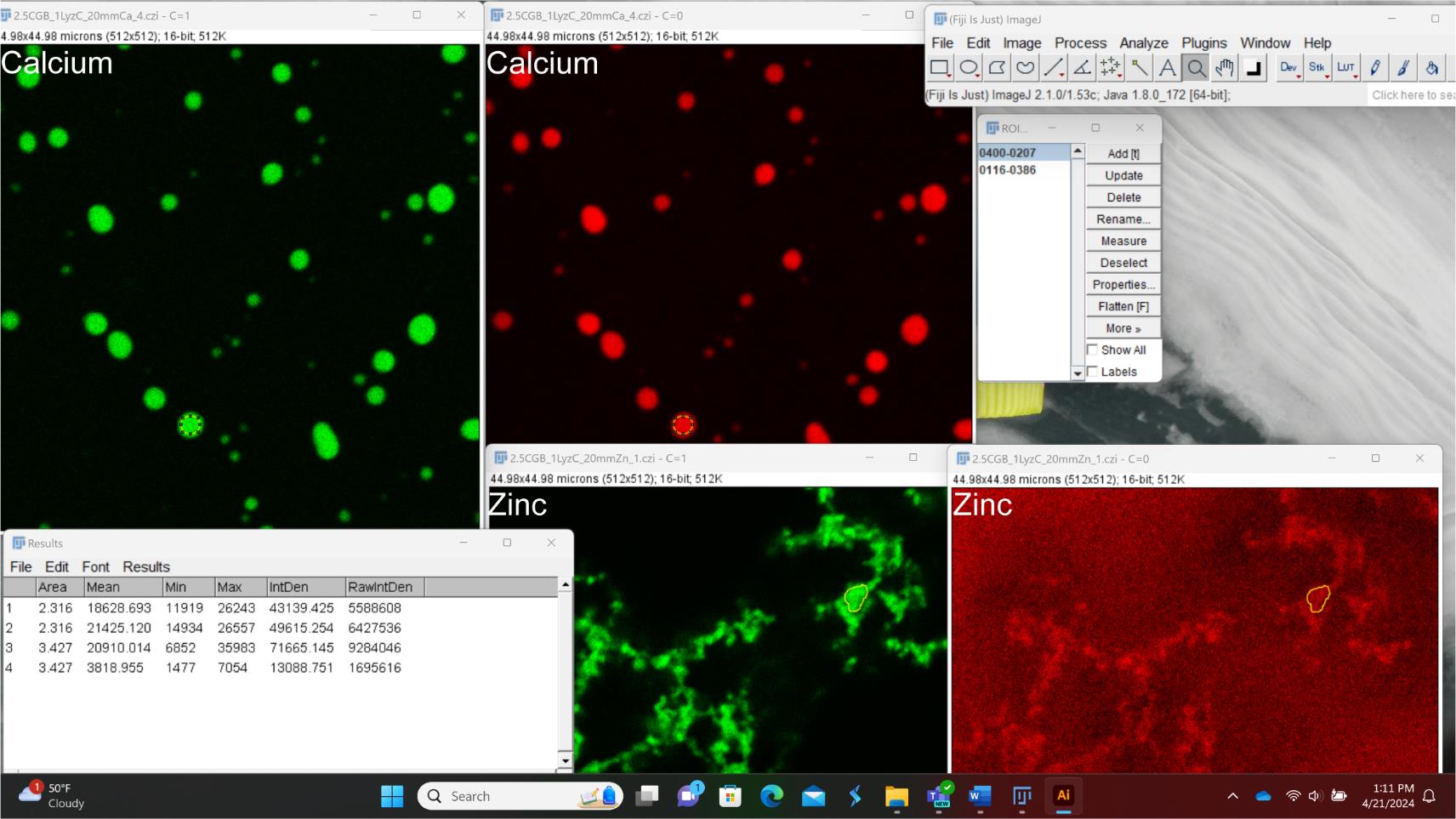
Figure 7. Screenshot explaining data analysis in Fiji to measure relative client (Cy3-Lyz; red) partitioning into CGB-sfGFP (green) assemblies generated in the presence of either zinc or calcium. All images have the same size; however, for the purpose of the screenshot, they have been adjusted such that only a part of some of the images is seen. Yellow regions depict ROI. Labeling was done in Adobe Illustrator and Microsoft Power Point.
Validation of protocol
This protocol or parts of it has been used and validated in the following research article(s):
Parchure et al. [12]. Liquid–liquid phase separation facilitates the biogenesis of secretory storage granules. Journal of Cell Biology (Figure 1B, Figure 5A, B).]
Acknowledgments
Anup Parchure is funded by a Project and Feasibility Award from the Yale Diabetes Research Center (P30 DK04573) and would like to acknowledge the support from R01DK129466 awarded to Jonathan S. Bogan. Julia von Blume is funded by a MIRA grant from NIGMS (1R35GM149293-01) and the NIDDK Innovative Science Accelerator Program (ISAC) Award (DK128851) and would like to acknowledge support from a Project and Feasibility award from Yale Diabetic Research Center (GR112420). We would like to acknowledge support from The Mass Spectrometry (MS) & Proteomics Resource of the W.M. Keck Foundation Biotechnology Resource Laboratory for detection of post-translational modifications on purified CGB-sfGFP. The protocol was adapted from Li et al. [13] PNAS. We used BioRender for making figures for this manuscript. The licenses for all the figures are listed below.
Graphical overview: Created in BioRender. Parchure, A. (2024) BioRender.com/z45l203
Figure 1: Created in BioRender. Parchure, A. (2024) BioRender.com/o81v327
Figure 2: Created in BioRender. Parchure, A. (2024) BioRender.com/a76y056
Figure 3: Created in BioRender. Parchure, A. (2024) BioRender.com/c76z345
Figure 6: Created in BioRender. Parchure, A. (2024) BioRender.com/t30m682
Competing interests
There are no conflicts of interest or competing interest.
References
- Bearrows, S. C., Bauchle, C. J., Becker, M., Haldeman, J. M., Swaminathan, S. and Stephens, S. B. (2019). Chromogranin B regulates early-stage insulin granule trafficking from the Golgi in pancreatic islet β-cells. J Cell Sci. 132(13): e231373.
- Kim, T., Tao-Cheng, J. H., Eiden, L. E. and Loh, Y. (2001). Chromogranin A, an “On/Off” Switch Controlling Dense-Core Secretory Granule Biogenesis. Cell. 106(4): 499–509.
- Tooze, S. A. and Huttner, W. B. (1990). Cell-free protein sorting to the regulated and constitutive secretory pathways. Cell. 60(5): 837–847.
- Rustom, A., Bajohrs, M., Kaether, C., Keller, P., Toomre, D., Corbeil, D. and Gerdes, H. (2002). Selective Delivery of Secretory Cargo in Golgi‐Derived Carriers of Nonepithelial Cells. Traffic. 3(4): 279–288.
- Wacker, I., Kaether, C., Krömer, A., Migala, A., Almers, W. and Gerdes, H. H. (1997). Microtubule-dependent transport of secretory vesicles visualized in real time with a GFP-tagged secretory protein. J Cell Sci. 110(13): 1453–1463.
- Huh, Y. H., Jeon, S. H. and Yoo, S. H. (2003). Chromogranin B-induced Secretory Granule Biogenesis. J Biol Chem. 278(42): 40581–40589.
- Colomer, V., Kicska, G. A. and Rindler, M. J. (1996). Secretory Granule Content Proteins and the Luminal Domains of Granule Membrane Proteins Aggregate in Vitro at Mildly Acidic pH. J Biol Chem. 271(1): 48–55.
- Gerdes, H. H., Rosa, P., Phillips, E., Baeuerle, P. A., Frank, R., Argos, P. and Huttner, W. B. (1989). The primary structure of human secretogranin II, a widespread tyrosine-sulfated secretory granule protein that exhibits low pH- and calcium-induced aggregation. J Biol Chem. 264(20): 12009–12015.
- Chanat, E. and Huttner, W. B. (1991). Milieu-induced, selective aggregation of regulated secretory proteins in the trans-Golgi network. J Cell Biol. 115(6): 1505–1519.
- Paroutis, P., Touret, N. and Grinstein, S. (2004). The pH of the Secretory Pathway: Measurement, Determinants, and Regulation. Physiology. 19(4): 207–215.
- Pizzo, P., Lissandron, V., Capitanio, P. and Pozzan, T. (2011). Ca2+ signalling in the Golgi apparatus. Cell Calcium. 50(2): 184–192.
- Parchure, A., Tian, M., Stalder, D., Boyer, C. K., Bearrows, S. C., Rohli, K. E., Zhang, J., Rivera-Molina, F., Ramazanov, B. R., Mahata, S. K., et al. (2022). Liquid–liquid phase separation facilitates the biogenesis of secretory storage granules. J Cell Biol. 221(12): e202206132.
- Li, Z., Michael, I. P., Zhou, D., Nagy, A. and Rini, J. M. (2013). Simple piggyBac transposon-based mammalian cell expression system for inducible protein production. Proc Natl Acad Sci USA. 110(13): 5004–5009.
Article Information
Publication history
Received: Apr 22, 2024
Accepted: Sep 1, 2024
Available online: Sep 25, 2024
Published: Oct 20, 2024
Copyright
© 2024 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Parchure, A. and von Blume, J. (2024). Chromogranin B Purification for Condensate Formation and Client Partitioning Assays In Vitro. Bio-protocol 14(20): e5095. DOI: 10.21769/BioProtoc.5095.
- Parchure, A., Tian, M., Stalder, D., Boyer, C. K., Bearrows, S. C., Rohli, K. E., Zhang, J., Rivera-Molina, F., Ramazanov, B. R., Mahata, S. K., et al. (2022). Liquid–liquid phase separation facilitates the biogenesis of secretory storage granules. J Cell Biol. 221(12): e202206132.
Category
Biochemistry > Protein > Isolation and purification
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.