- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Improving Stability of Spiroplasma citri MreB5 Through Purification Optimization and Structural Insights

Published: Vol 14, Iss 20, Oct 20, 2024 DOI: 10.21769/BioProtoc.5086 Views: 1959
Reviewed by: Joana Alexandra Costa ReisAnonymous reviewer(s)
Original research article
The authors used this protocol in:

May 2022
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
MreB is a prokaryotic actin homolog. It is essential for cell shape in the majority of rod-shaped cell-walled bacteria. Structural and functional characterization of MreB protein is important to understand the mechanism of ATP-dependent filament dynamics and membrane interaction. In vitro studies on MreBs have been limited due to the difficulty in purifying the homogenous monomeric protein. We have purified MreB from the cell-wall-less bacteria Spiroplasma citri, ScMreB5, using heterologous expression in Escherichia coli. This protocol provides a detailed description of purification condition optimization that led us to obtain high concentrations of stable ScMreB5. Additionally, we have provided a protocol for detecting the presence of monovalent ions in the ScMreB5 AMP-PNP-bound crystal structure. This protocol can be used to obtain a high yield of ScMreB5 for carrying out biochemical and reconstitution studies. The strategies used for ScMreB5 show how optimizing buffer components can enhance the yield and stability of purified protein.
Key features
• The protocol is a useful approach to standardize purification of nucleotide-dependent cytoskeletal filaments and other nucleotide-binding proteins.
• The mechanistic basis of how different ions could stabilize a protein, and hence improve yield in purification, has been demonstrated.
• The change in buffer conditions/salt enabled us to get sufficient yield for biochemical and structural characterization.
Keywords: MreBGraphical overview
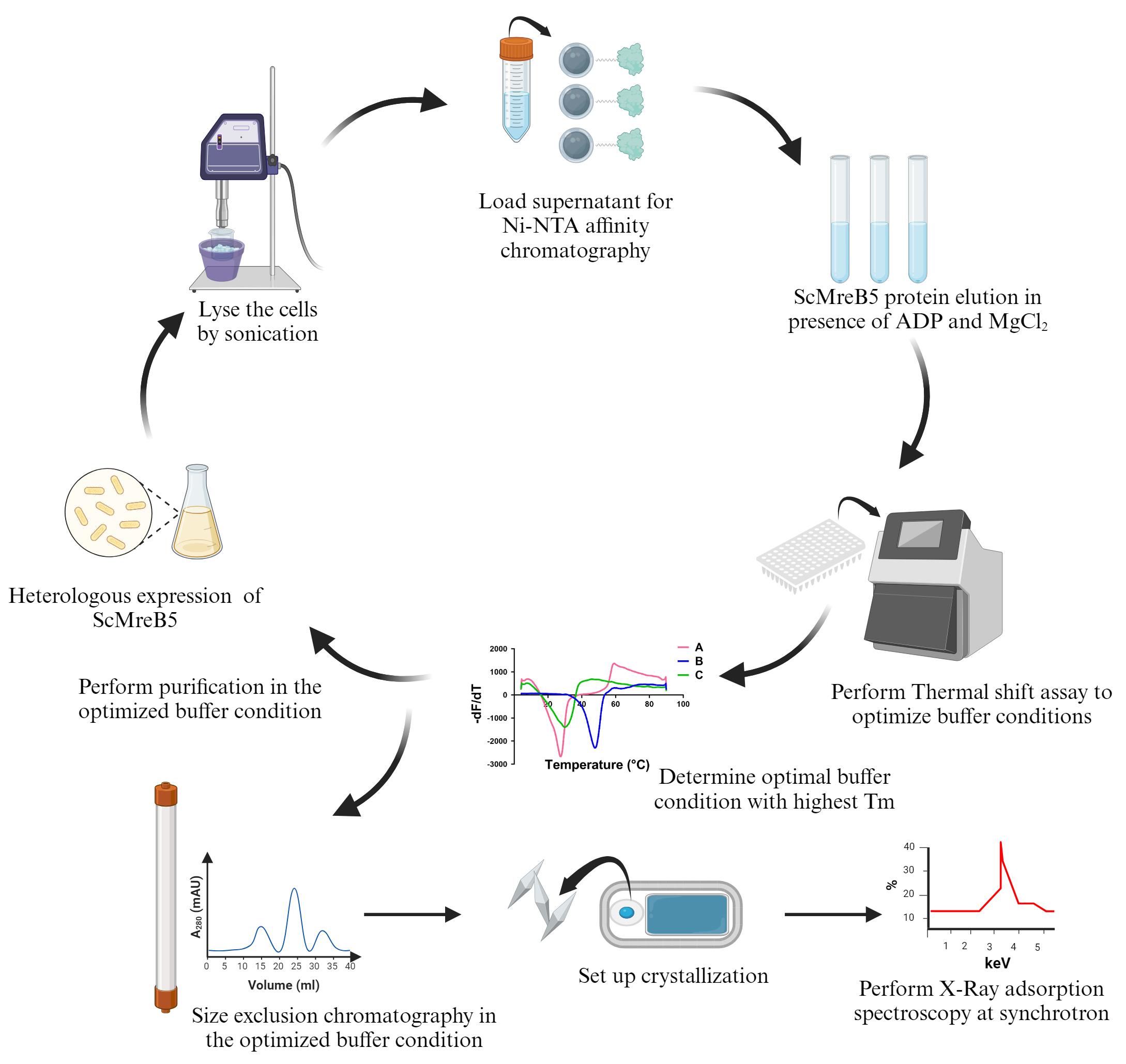
Background
There is an array of prokaryotic filamentous proteins that play a pivotal role in maintaining overall cellular functioning [1]. In the majority of bacteria, complex cell wall synthesis machinery plays a major role in cell shape maintenance. Studies have extensively analyzed how shape is regulated in E. coli and B. subtilis. Genetic studies show that deleting mreB causes loss of rod shape and eventual lysis, indicating the pivotal role of the MreB protein in bacterial cell shape and maintenance [2–4].
MreB forms antiparallel double protofilaments in the presence of nucleotide, ATP, or ADP that align parallel to the long axis of the rod-shaped cells [5]. The processive movement of MreB along with the other components of cell wall synthesis machinery drives the insertion of peptidoglycan, thus enforcing the cell shape [6,7].
MreB5 is responsible for determining helicity and motility in the cell-wall-less bacteria Spiroplasma citri [8]. In our recent work, we reported the structure and biochemical characterization of ScMreB5 and its mutants [9]. We showed that the nucleotide state of ScMreB5 determines the filament dynamics and membrane interaction. MreBs are notoriously challenging to purify because they tend to aggregate when overexpressed. Obtaining monomeric and homogeneous proteins for biochemical and structural studies has been challenging, resulting in few MreBs being characterized in vitro.
For in vitro characterization and structure determination, the scmreb5 gene was cloned into the pHis17 expression vector, under the T7 promoter, between NdeI and BamHI restriction endonuclease site. The cloned gene was codon-optimized by mutating the TGA codon to TGG, since in S. citri, UGA is recognized as tryptophan [10]. A restriction-free cloning strategy [11] was chosen for cloning, and the clone was confirmed by DNA sequencing. The cloned plasmid was transformed into the BL21-AI expression strain, which has a T7 RNA polymerase tightly regulated by the araBAD promoter and induced with arabinose for overexpression.
This protocol paper describes optimizing ScMreB5 protein purification, where precipitation in NaCl-containing buffer was prevented by adding excess ADP and MgCl2 to stabilize the protein. This addition prevented any further biochemical characterization of ScMreB5. To eliminate this issue, we optimized the buffer conditions for protein purification using a thermal shift assay [12,13]. The optimized buffer containing KCl removed the necessity for ADP and MgCl2 addition during the purification process. To understand the structural significance of KCl in stabilizing the protein, we performed X-ray adsorption spectroscopy on ScMreB5-AMP-PNP crystals. This technique is widely used for detecting metal ions within a protein crystal. Since the X-ray absorption edges of different elements occur at specific energies, it allows the identification of specific metal ions within the crystal. The presence of potassium ion bridging the protein with the nucleotide provided a structural basis for why the stability of ScMreB5 improved in the presence of KCl.
Materials and reagents
For transformation and large culture expression
100 mm Petri dish (Himedia, catalog number: PW1305-1x100NO)
Luria Bertani agar, LB agar (Himedia, catalog number: M1151-1KG)
Luria Bertani broth powder (Himedia, catalog number: M1245-1KG)
BL21-AI chemically competent E. coli cells (Invitrogen, catalog number: C6070-03)
ScMreB5 cloned pHis17 plasmid (Hexa His tag at the C-terminus)
Ampicillin (Sigma-Aldrich, catalog number: A9518-25G)
100 mL conical flask (Borosil)
2 L conical flask (Borosil)
0.22 µm syringe filter (Cytiva, catalog number: WHA-9914-2502)
10 mL syringe for single use (Dispovan, catalog number: JK-10ml-0110)
L-Arabinose (Himedia, catalog number: GRM037-25G)
For purification
150 mL beaker (Borosil, catalog number: 1000D18)
0.5 mL centrifuge tubes (Tarsons, catalog number: 500000)
1.5 mL centrifuge tubes (Tarsons, catalog number: 500010)
0.2 mL PCR tubes (Tarsons, catalog number: 510051)
50 mL conical centrifuge tubes (Tarsons, catalog number: TAR-500041)
100 mL conical flasks (Borosil, catalog number: 5020016)
500 mL conical flasks (Borosil, catalog number: 5020024)
2 L conical flasks (Borosil, Catalog number: 5020030)
Test tubes, 15 × 125 mm (Borosil, catalog number: 9800U04)
HiIndicator pH paper, pH range 6.5–9.0 (Himedia, catalog number: LA321-1PK)
5 mL His-Trap column (Cytiva, catalog number: 17524801)
Bradford 1× dye reagent (Bio-Rad, catalog number: 5000205)
Bovine serum albumin (BSA) (Himedia, catalog number: TC548-5G)
Durapore membrane filter, 0.22 µm, hydrophilic PVDF, 47 mm membrane (Sigma-Aldrich, catalog number: GVWP04700)
Superdex 200 Increase 10/300 GL (Cytiva, catalog number: 28990944)
Oak Ridge centrifuge tubes, 50 mL (Thermo Scientific, catalog number: 3118-0050)
Thick-walled Polycarbonate tubes, 11 × 34 mm, 1 mL (Beckman Coulter, catalog number: 343778)
Hydrochloric acid (Qualigens, catalog number: Q29145)
Tris, free base, for molecular biology (Himedia, catalog number: MB029-5KG)
Glycine (Sigma-Aldrich, catalog number: G8898-1KG)
Sodium dodecyl sulphate (Sigma-Aldrich, catalog number: 436143)
TEMED (Tetramethylethylenediamine) (Sigma-Aldrich, catalog number: T9281-25ML)
Ammonium persulphate (Sigma-Aldrich, catalog number: A3678)
Acrylamide (Sigma-Aldrich, catalog number: A8887-500G)
Bis-acrylamide (Sigma-Aldrich, catalog number: 146072-500G)
0.75 mm spacer plate (Bio-Rad, catalog number: 1653310)
Short plates (Bio-Rad, catalog number: 1653308)
10-well comb, 0.75 mm (Bio-Rad, catalog number: 1653354)
15-well comb, 0.75 mm (Bio-Rad, catalog number: 1653355)
Coomassie Brilliant Blue (R-250) (Sigma-Aldrich, catalog number: 27816)
2× Laemmli sample buffer (Bio-Rad, catalog number: 1610737)
DTT (Sigma-Aldrich, catalog number: D9779-25G)
Ethanol for molecular biology (Omnis)
Glacial acetic acid (Qualigens, catalog number: Q11007)
Sodium chloride (NaCl) (Qualigens, catalog number: Q15918)
Potassium chloride (KCl) (Qualigens, catalog number: Q13305)
Magnesium chloride (MgCl2) (Sigma-Aldrich, catalog number: M8266-100G)
ADP adenosine 5'-diphosphate sodium salt (Sigma-Aldrich, catalog number: A2754-500MG)
Imidazole (Sigma-Aldrich, catalog number: I2399-500G)
Glycerol (Fisher Scientific, catalog number: 15457)
Dialysis tubing 10 kDa MWCO (Thermo Fisher Scientific, catalog number: 68100)
Dialysis tubing clamps (Sigma-Aldrich, catalog number: Z371092-10EA)
Vivaspin® Turbo 15 PES 10 kDa MWCO (Sartorius, catalog number: VS15T01)
Precision Plus Protein Dual Color Standards (Bio-Rad, catalog number: 1610394)
Micro test plate, flat Bottom (Tarsons, catalog number: 941196)
Liquid nitrogen
For thermal shift assay
MultiplateTM 96-well PCR plates, low profile, unskirted, white (Bio-Rad, catalog number: MLL9651)
Microseal 'B' PCR plate sealing film, adhesive, optical (Bio-Rad, catalog number: MSB1001)
SYPRO orange (Sigma-Aldrich, catalog number: S5692)
For crystallization
48-well crystallization plate (Hampton Research, catalog number: HR3-180)
Sodium phosphate monobasic monohydrate (Sigma-Aldrich, catalog number: 71507-250G)
Sodium phosphate dibasic (Sigma-Aldrich, catalog number: S9763-100G)
AMP-PNP Adenosine 5'-(β, γ-imido) triphosphate lithium salt hydrate (Sigma-Aldrich, catalog number: 10102547001)
Magnesium chloride (details mentioned above)
PEG 3350 (Sigma-Aldrich, catalog number: 202444-500G)
Glycerol (Sigma-Aldrich, catalog number: G5516)
CrystalCap SPINE HT, 0.05-0.1 mm CryoLoop (Hampton Research, catalog number: HR8-120)
Stock solutions (see Recipes)
100 mg/mL ampicillin
1 mg/mL BSA
2 M MgCl2
1 M unbuffered Tris
100 mM ADP
100 mM AMP-PNP
20% Arabinose (w/v)
5 M NaCl
3 M KCl
2 M Tris pH 8
2 M Imidazole
30% Acrylamide solution (w/v)
10% Ammonium persulphate (w/v)
10% SDS
1.5 M Tris pH 6.8
1.5 M Tris, pH 8.8
1 M sodium phosphate monobasic monohydrate
1 M sodium phosphate dibasic
1 M sodium phosphate buffer, pH 7.8
40% PEG 3350 (w/v)
20% Glycerol (v/v)
50× SYPRO Orange
Solutions and buffers for purification (see Recipes)
10× TGS buffer for SDS-PAGE gels
SDS-PAGE staining solution
SDS-PAGE destaining solution
5% SDS-PAGE stacking gel
12% SDS-PAGE resolving gel
Lysis buffer (Na)
Buffer A (Na)
Buffer B (Na)
Storage buffer
Lysis buffer (K)
Buffer A (K)
Buffer B (K)
Crystallization conditions
Recipes
Materials to be autoclaved
LB Agar
Dissolve 4 g of LB Agar in RO (reverse osmosis) water to a final volume of 100 mL in a 500 mL conical flask. Autoclave at 121 °C for 15 min at 2.8 bar pressure for sterilization. Once cooled enough to touch, add 100 µL of 100 mg/mL ampicillin solution to the LB agar and gently stir. Pour 25 mL of LB agar into the Petri plates. Store the plates at 4 °C after the LB agar solidifies.
1× LB Broth, 30 mL
Dissolve 0.75 g of Luria Bertani broth powder in RO water to a final volume of 30 mL in a 100 mL conical flask. Autoclave at 121 °C for 15 min at 2.8 bar pressure for sterilization.
1× LB Broth, 1,000 mL
Dissolve 25 g of Luria Bertani broth powder in RO water to a final volume of 1,000 mL in a 2 L conical flask. Autoclave at 121 °C for 15 min at 2.8 bar pressure for sterilization.
15 × 12.5 mm test tubes
Autoclave 20–25 clean test tubes at 121 °C for 15 min at 2.8 bar pressure for sterilization.
Stock solutions
100 mg/mL Ampicillin
Dissolve 0.5 g of ampicillin powder in ultrapure water to a final volume of 5 mL. Sterilize the solution using a 0.22 µm syringe filter.
1 mg/mL BSA
Dissolve 1 mg of BSA powder in ultrapure water to a final volume of 1 mL. Use this solution to prepare standards (0.1–1.0 mg/mL) in ultrapure water for protein concentration estimation using Bradford reagent.
2 M MgCl2
Dissolve 1.90 g of MgCl2 powder in ultrapure water to a final volume of 10 mL. Filter the solution using a 0.22 µm syringe filter.
1 M unbuffered Tris
Dissolve 1.21 g of unbuffered Tris-base powder in ultrapure water to a final volume of 10 mL. Filter the solution using a 0.22 µm syringe filter.
100 mM ADP
Dissolve 23.5 mg of ADP powder in 100 µL of ultrapure water. Adjust the pH to 7–7.5 with 1 M unbuffered Tris using a pH strip. Make up the volume to 250 µL.
100 mM AMP-PNP
Dissolve 5 mg of ADP powder in 30 µL of ultrapure water. Adjust the pH to 7–7.5 with 1 M unbuffered Tris using a pH strip. Make up the volume to 100 µL.
20% (w/v) Arabinose
Dissolve 10 g of arabinose in RO water to a final volume of 50 mL. Filter the solution using a 0.22 µm syringe filter.
5 M NaCl
Dissolve 146.1 g of NaCl powder in RO water to a final volume of 500 mL. Filter the solution using a vacuum filter assembly with a 0.22 µm membrane filter.
3 M KCl
Dissolve 111.8 g of KCl powder in RO water to a final volume of 500 mL. Filter the solution using a vacuum filter assembly with DuraporeTM membrane filter (0.22 µm, hydrophilic PVDF, 47 mm membrane).
2 M Tris, pH 8
Dissolve 60.7 g of Tris-base in 120 mL of RO water. Adjust the pH to 8 using concentrated hydrochloric acid using a pH meter. Make up the volume to 250 mL. Filter the solution using a vacuum filter assembly with DuraporeTM membrane filter (0.22 µm, hydrophilic PVDF, 47 mm membrane).
Note: Follow the required lab safety procedures while handling hydrochloric acid.
2 M Imidazole
Dissolve 68.07 g of imidazole powder in 500 mL of RO water. Filter the solution using a vacuum filter assembly with DuraporeTM membrane filter (0.22 µm, hydrophilic PVDF, 47 mm membrane).
30% Acrylamide solution
Dissolve 146 g of acrylamide and 3.9 g of bis-acrylamide powders in 500 mL of RO water. Filter the solution using a vacuum filter assembly with DuraporeTM membrane filter (0.22 µm, hydrophilic PVDF, 47 mm membrane).
10% Ammonium persulphate
Dissolve 5 g of ammonium persulphate powder in RO water to a final volume of 50 mL. Filter the solution using a 0.22 µm syringe filter.
10% SDS
Dissolve 5 g of SDS powder in RO water to a final volume of 50 mL.
1.5 M Tris pH 6.8
Dissolve 45.5 g of Tris-base in 120 mL of RO water. Adjust the pH to 6.8 using concentrated hydrochloric acid and a pH meter. Make up the volume to 250 mL. Filter the buffer using a 0.22 µm syringe filter.
1.5 M Tris, pH 8.8
Dissolve 45.5 g of Tris-base in 120 mL of RO water. Adjust the pH to 8.8 using concentrated hydrochloric acid and a pH meter. Make up the volume to 250 mL. Filter the buffer using a 0.22 µm syringe filter.
1 M sodium phosphate monobasic monohydrate
Dissolve 1.37 g of sodium phosphate monobasic monohydrate powder in ultrapure water to a final volume of 10 mL. Filter the solution using a 0.22 µm syringe filter.
1 M sodium phosphate dibasic
Dissolve 1.42 g of sodium phosphate dibasic powder in ultrapure water to a final volume of 10 mL. Filter the solution using a 0.22 µm syringe filter.
1 M sodium phosphate buffer, pH 7.8
Titrate sodium phosphate monobasic monohydrate using 1 M sodium phosphate dibasic to obtain pH 7.8. Use HiIndicator pH Paper, pH range 6.5–9.0 for adjusting the pH.
40% PEG 3350
Dissolve 20 g of PEG 3350 powder in ultrapure water to a final volume of 50 mL. Filter the solution using a 0.22 µm syringe filter. Use this stock to make 16% PEG 3350.
20% Glycerol
Mix 2 mL of glycerol with 8 mL of ultrapure water. Filter the solution using a 0.22 µm syringe filter.
50× SYPRO Orange
Mix 1 µL of 5,000× SYPRO Orange in 9 µL of DMSO to make 500× SYPRO orange. Dilute 500× SYPRO orange to 50× by mixing 2 µL of 500× SYPRO orange in 18 µL in ultrapure water.
Buffers and working stock solutions
10× TGS Buffer
Dissolve 30 g of Tris-base, 144 g of glycine, and 10 g of SDS in 1,000 mL of RO water. Use it to make 1× TGS buffer for running the SDS-PAGE gels.
Coomassie staining solution
Dissolve 2.5 g of Coomassie Brilliant Blue (R-250) in 500 mL of ethanol by stirring. Add 100 mL of glacial acetic acid and 400 mL of RO water to make the final volume of 1,000 mL.
De-staining solution
Mix 100 mL of glacial acetic acid and 200 mL of ethanol with 700 mL of RO water.
12% SDS-PAGE resolving gel (5 mL)
Mix the following solutions and pour onto the 0.75 mm clamped gel setup.
RO water (1.6 mL)
30% acrylamide solution (2 mL)
Tris-HCl, pH 8.8 (1.5 mL)
10% SDS (50 µL)
10% ammonium persulphate (50 µL)
TEMED (3 µL)
Add 100 µL of 100% isopropanol to even out the layer of resolving gel. Once solidified, carefully tilt the gel setup to one side and use tissue paper to remove isopropanol.
5% SDS-PAGE stacking gel (1 mL)
Mix the following solutions and pour over the solidified resolving gel.
RO water (680 µL)
30% acrylamide solution (170 µL)
Tris-HCl, pH 6.8 (130 µL)
10% SDS (10 µL)
10% ammonium persulphate (10 µL)
TEMED (1 µL)
After pouring the stacking solution, place a 10- or 15-well 0.75 mm comb.
Lysis buffer (Na)
200 mM NaCl (10 mL from 5 M NaCl)
50 mM Tris-HCl pH 8 (6.25 mL from 2 M Tris-HCl pH 8)
10% glycerol (12.5 mL from 100% glycerol)
Make up the volume to 250 mL with RO water. Filter and degas the solution using a vacuum filter assembly with DuraporeTM membrane filter (0.22 µm, hydrophilic PVDF, 47 mm membrane). Pre-chill the buffer at 4 °C for purification.
Buffer A (Na)
Used as binding buffer in Ni-NTA chromatography.
200 mM NaCl (20 mL from 5 M NaCl)
50 mM Tris-HCl pH 8 (12.5 mL from 2 M Tris-HCl pH 8)
Make up the volume to 500 mL with RO water. Filter and degas the solution using a vacuum filter assembly with DuraporeTM membrane filter (0.22 µm, hydrophilic PVDF, 47 mm membrane). Pre-chill the buffer at 4 °C for purification.
Buffer B (Na)
Used as elution buffer in Ni-NTA chromatography.
200 mM NaCl (20 mL from 5 M NaCl)
50 mM Tris-HCl pH 8 (12.5 mL from 2 M Tris-HCl pH8)
500 mM imidazole (125 mL from 2 M imidazole)
Make up the volume to 500 mL with RO water. Filter and degas the solution using a vacuum filter assembly with DuraporeTM membrane filter (0.22 µm, hydrophilic PVDF, 47 mm membrane). Pre-chill the buffer at 4 °C before using for purification.
Storage buffer
Used for buffer exchange and protein storage.
50 mM NaCl (1 mL from 5 M NaCl)
50 mM Tris-HCl pH 8 (2.5 mL from 2 M Tris-HCl pH 8)
Make up the volume to 100 mL with RO water. Filter and degas the solution using a vacuum filter assembly with DuraporeTM membrane filter (0.22 µm, hydrophilic PVDF, 47 mm membrane). Pre-chill the buffer at 4 °C for purification.
Lysis buffer (K)
300 mM KCl (25 mL from 3 M KCl)
50 mM Tris-HCl pH 8 (6.25 mL from 2 M Tris-HCl pH 8)
10% glycerol (12.5 mL from 100% glycerol)
Make up the volume to 250 mL with RO water. Filter and degas the solution using a vacuum filter assembly with DuraporeTM membrane filter (0.22 µm, hydrophilic PVDF, 47 mm membrane). Pre-chill the buffer at 4 °C for purification.
Buffer A (K)
Used as binding buffer in Ni-NTA chromatography and buffer for size exclusion chromatography.
300 mM KCl (100 mL from 3 M KCl)
50 mM Tris-HCl pH 8 (25 mL from 2 M Tris-HCl pH 8)
Make up the volume to 1000 mL with RO water. Filter and degas the solution using a vacuum filter assembly with DuraporeTM membrane filter (0.22 µm, hydrophilic PVDF, 47 mm membrane). Pre-chill the buffer at 4 °C for purification.
Buffer B (K)
Used as elution buffer in Ni-NTA chromatography.
300 mM KCl (50 mL from 3 M KCl)
50 mM Tris-HCl pH 8 (12.5 mL from 2 M Tris-HCl pH 8)
500 mM imidazole (125 mL from 2 M imidazole)
Make up the volume to 500 mL with RO water. Filter and degas the solution using a vacuum filter assembly with DuraporeTM membrane filter (0.22 µm, hydrophilic PVDF, 47 mm membrane). Pre-chill the buffer at 4 °C before using for purification.
Crystallization condition for X-ray fluorescence scanning
0.15 M sodium phosphate buffer, pH 7.8 (150 µL from 1 M sodium phosphate buffer, pH 7.8)
16% PEG 3350 (400 µL from 40% PEG 3350)
Make up the volume to 1000 µL with ultrapure water. Sterilize using a 0.22 µm syringe filter.
Equipment
Shaker incubator (Thermo Scientific, model: MaxQTM 6000)
Eppendorf® ThermoMixer® F2.0 (EP5387000013)
Sonicator (Sonics VibraCell)
E-GelTM imager system (Fisher Scientific, catalog number: 4466611)
pH meter (Thermo Scientific, model: ORION STAR A111)
Vacuum filter assembly (Millipore, model: XX1014720)
Centrifuge (Avanti J26S-XP Non-IVD)
Tabletop ultracentrifuge (Beckman Coulter Optima MAX-XP)
High-speed centrifuge (Eppendorf, model: 5430 R)
Benchtop centrifuge (Eppendorf, model: 5810 R)
Centrifuge (Eppendorf MiniSpin®)
ÄKTA PrimeTM Plus FPLC system
Bio-Rad NGCTM chromatography system QuestTM 10
SDS-PAGE apparatus (Bio-Rad, catalog number: 1658002FC)
CLARIOstar® microplate reader
Bio-Rad CFX96 Touch Real-Time PCR Detection System
Software and datasets
GraphPad Prism (for analyzing and plotting thermal shift assay and size exclusion chromatography)
Microsoft Excel (for exporting thermal shift assay results)
ImageJ (for SDS-PAGE gel)
Procedure
Large culture expression
Take 100 ng of the plasmid and add into pre-thawed 50 µL of BL21-AI competent cells under sterile conditions. Incubate for 30 min on ice.
Give a heat shock at 42 °C for 90 s and immediately place the tube on ice for 5 min.
Add 200 µL of LB media into the tube and incubate for 40 min at 37 °C for recovery in shaking condition.
Plate 100 µL of the culture in LB agar plate containing 100 µg/µL of ampicillin antibiotic. Incubate the plate for 10–12 h at 37 °C.
Note: Follow the manufacturer’s protocol when using the commercially available competent BL21-AI cells.
Primary culture:
Next day, inoculate 10–20 colonies in 30 mL of LB media containing 30 µL (100 µg/µL) of ampicillin antibiotic in a 100 mL conical flask.
Grow the culture until OD600 reaches 0.8 at 37 °C in a shaker incubator (180 rpm).
Secondary culture:
Inoculate 10 mL of primary culture into two 2 L conical flasks containing 1 L of LB media.
Add 1,000 µL (100 µg/µL) of ampicillin antibiotic into the media.
Grow the secondary culture until OD600 reaches 0.8–1.0 at 37 °C in a shaker incubator (180 rpm).
Note: It should take 2.5–3 h to reach OD600 of 0.8.
Induction:
Change the shaker incubator temperature to 18 °C.
To induce the secondary cultures, add 10 mL of 20% arabinose to each flask under sterile conditions.
Incubate the culture flasks at 18 °C in the shaker incubator for 10–12 h for protein overexpression.
Cell pelleting:
Pellet down the cells from 2 L culture in JLA 9.1 rotor using an Avanti J26S-XP. Centrifuge at 7,500–8,000 RCF for 20 min at 4 °C.
Discard the supernatant, leaving 10 mL of supernatant for resuspending the pellet.
Transfer the resuspended pellet into 50 mL centrifuge tubes, spin down the cells at 3,200 RCF using a benchtop centrifuge, and discard the supernatant.
Plunge freeze the cell pellet containing the centrifuge tubes in liquid nitrogen and store at -80 °C until further use.
Note: Handle liquid nitrogen with caution. Wear safety glasses and cryo-gloves during use.
Protein purification in NaCl-containing buffer
Cell pellet lysis
Thaw 2 L of culture cell pellet on ice.
Resuspended the cell pellet in 50 mL lysis buffer (Na).
Sonicate the cell suspension using a probe sonicator. Keep the following settings: 1 s ON, 3 s OFF, total time = 3 min, amplitude = 60%. Perform sonication twice with a 5 min resting time between the two cycles.
Cell debris removal
Centrifuge the lysate at 45,000 RCF for 45 min at 4 °C using JA 25.5 rotor in Avanti J26S-XP centrifuge to remove the cell debris.
Transfer the supernatant into a pre-chilled 50 mL centrifuge tube on ice.
Ni-NTA purification
Wash and equilibrate the Akta Prime plus system with Buffer A (Na).
Equilibrate the 5 mL His-Trap column by passing five column volumes of Buffer A (Na).
Load the supernatant at a flow rate of 2.5 mL/min onto the column. The Hexa-His tag at the C-terminus facilitates the binding of the protein to the Ni-NTA beads.
Collect the flowthrough in a 100 mL conical flask.
After loading the supernatant, wash the column with 15 column volumes of Buffer A (Na).
Collect 10 column volumes each of 2% and 5% Buffer B (Na) in 100 mL conical flasks.
Perform fraction collection (5 mL fractions, 5 column volumes) for 10%, 20%, 50%, and 100% Buffer B (Na) in 15 × 12.5 mm autoclaved test tubes.
Observe the UV280 peak on the chromatogram. As the protein elution peak is observed, add 1 mM ADP and 1 mM MgCl2 to the fractions immediately to prevent precipitation.
Run a 12% SDS-PAGE gel for the lysate, supernatant, wash, flowthrough, 2%, 5%, and the fractions along with a protein ladder.
The protein elutes from the last fraction of 20% and all fractions of 50% Buffer B (Na).
Pool the fractions (after analyzing them on the gel) that contain a single band of the protein corresponding to 37 kDa relative to the protein ladder.
Note: Ensure that the fraction has 1 mM ADP and 1 mM MgCl2 added to avoid protein precipitation.
Centrifuge the pooled protein fractions at 45,000 RCF for 30 min at 4 °C using JA 25.5 rotor in Avanti J26S-XP centrifuge to remove any protein precipitants.
Concentration and buffer exchange
Wash and equilibrate a 10 kDa molecular weight cutoff concentrator with Buffer A (Na). Concentrate the pooled fractions in the concentrator at 3,200 RCF in an Eppendorf benchtop centrifuge 5810 R at 4 °C.
As the protein concentrates, observe for any visible precipitation. If precipitation occurs, centrifuge the protein at 21,000 RCF at 4 °C in an Eppendorf high-speed centrifuge.
Add 1 mM ADP and 1 mM MgCl2 to the supernatant to avoid any further precipitation.
Simultaneously, clean the concentrator using Buffer A (Na). Add the supernatant back to the concentrator.
Once the protein volume reaches ~1 mL, dilute it with ~10 mL of storage buffer, 1 mM ADP, and 1 mM MgCl2 for buffer exchange. Concentrate the protein until the concentration reaches ~2 mg/mL. Estimate the protein concentration by performing the Bradford assay as described below.
In the Eppendorf high-speed centrifuge, spin the concentrated protein at 21,000 RCF for 20 min at 4 °C in a 1.5 mL centrifuge tube.
Make 10 µL protein aliquots in 0.2 mL PCR tubes. Plunge freeze the protein aliquots in liquid nitrogen and store them at -80 °C until further use.
Notes:
i. This protein will be used to perform thermal shift assay for optimizing the purification conditions for increasing protein stability.
ii. Wear safety glasses and cryo-gloves while handling liquid nitrogen.
Bradford assay
Make BSA standards from 0.1 to 1.0 mg/mL using 1 mg/mL of BSA stock (see Recipes).
In a clear 96-well microplate, aliquot 5 µL of BSA standards, 5 µL of ultrapure water (blank for the standards), and 5 µL of storage buffer/buffer A (blank for the protein).
Prepare 20 µL of 5×, 10×, and 20× dilutions of purified protein in Buffer A.
Aliquot 5 µL of dilutions into the 96-well microplate in triplicates.
Add 250 µL of Bradford 1× dye reagent to the wells.
Measure the absorbance at 595 nm in a plate reader.
Estimate the protein concentration using Microsoft Excel as follows:
i. Subtract the readings of the standards and the protein dilutions with the respective readings of the blank.
ii. Average the triplicate readings for the protein dilutions.
iii. Plot a standard curve in Microsoft Excel using the scatterplot option.
iv. Format the trendline as “linear” and set the intercept.
v. Calculate the slope and R-squared value.
vi. Using the slope value (m) and absorbance reading (y), calculate the protein concentration (×) using the formula:
Note: The Bradford assay kit can also be used for protein concentration estimation. Follow the instructions from the kit for estimating the concentration.
Thermal shift assay
Protein concentration estimation
Thaw the protein aliquots (purified as described above) on ice. Transfer the aliquots to a thick-walled polycarbonate tube.
Ultracentrifuge at 100,000 RCF for 20 min at 4 °C in the OptimaTM Max-XP table-top ultracentrifuge.
Estimate the protein concentration of the supernatant by performing the Bradford assay as discussed in section B.
Performing a thermal shift assay
Note: Optimization experiments for ScMreB5 included testing various buffering agents (Tris, HEPES), pH ranges (5–10), salts (NaCl, KCl), and various additives (CaCl2, MgCl2, ADP, AMP-PNP, GDP, GTP, EDTA).
The protocol outlined below is a generalized protocol that was also used during ScMreB5 purification optimization. Different conditions can be tested in a similar manner. The table below is an example of a reaction setup for comparing NaCl and KCl salts at a concentration of 500 mM.
Prepare the reaction given in Table 1 in a 0.5 mL centrifuge tube on ice.
Table 1. Thermal shift assay reaction composition
Components (stock concentration) Reaction (µL) NaCl condition KCl condition Protein (~ 50 µM) 1.25 µL 1.25 µL Tris-HCl, pH 8 (100 mM) 12.5 µL 12.5 µL Salt: NaCl (5 M) 2.5 µL Salt: KCl (3 M) 4.2 µL Ultrapure water 6.25 µL 4.55 µL SYPRO Orange (50×) 2.5 µL 2.5 µL Total reaction volume 25 µL 25 µL Replace KCl/NaCl with any other salt condition (with different concentrations) to be tested from the above table.
Transfer the reaction to a MultiplateTM 96-well PCR plate and seal the reaction well with Microseal 'B' PCR plate sealing film.
Spin the reaction in the Eppendorf centrifuge 5810 R at 2,500 RCF for 1 at 4 °C min to bring the reaction mix to the bottom of the well.
Place the plate in a Bio-Rad CFX96 Real-Time System.
Set up the protocol as follows:
i. Open CFX Maestro software and create a new Protocol by going to File > New tab.
ii. In the Protocol Editor window, select Before in the Insert Step tab and click on Insert Melt Curve.
iii. Remove the step 2–4 by clicking on Delete Step. Only step 1 is required for the assay.
iv. Set the temperature range to 4–90 °C with a rise of 0.4 °C every 20 s in the step 1.
v. Set the sample volume to 25 µL.
vi. Save the protocol by clicking OK.
vii. Select the Next option and click on Create New.
viii. The Plate Editor window will open. Select the wells to be analyzed by dragging the cursor across the wells.
ix. In the Scan Mode tab, select FRET (excitation: 470 nm; emission: 569 nm).
x. In the Sample Type menu, choose unknown option.
xi. Under Target Names tab, tick the Load Box.
xii. Click on OK when finished.
Use the Run tab to initiate the reaction. (It should take around 1 h 20 min with the above settings.)
After run completion, export the Melt Curve Derivative Results as an Excel file and proceed for analysis.
Open GraphPad Prism and paste the readings in an XY format. The readings of the conditions will be in (-(dRFU)/dT) format.
The first derivative of the florescence (-(dRFU)/dT) is plotted against the temperature. The dip in the curve determines the Tm of the protein for a given condition.
A representative image of an expected melting curve for the above reaction is provided in Figure 1. The original data for 300 mM KCl buffer, used for purifying ScMreB5, has been provided in the original publication as Figure S1, C and D [9].
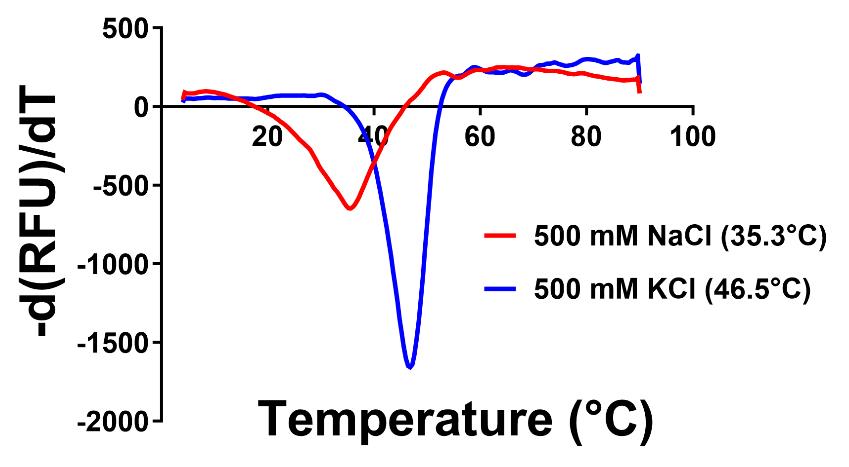
Figure 1. Representative thermal shift assay for purification condition optimization. Melting curve for ScMreB5 showing Tm for 500 mM NaCl (red) and 500 mM KCl (blue). The buffer used in both conditions is 50 mM Tris, pH 8. The first derivative of the fluorescence (-(dF)/dT) is plotted against the temperature. The dip in the curve determines the Tm of the protein in the given condition. RFU is the relative florescence units.Protein purification in KCl-containing buffer
After condition optimization, the following changes were made to the protocol in Section B “Protein Purification in NaCl containing buffer”:
A. Lysis buffer (Na) is replaced with lysis buffer (K).
B. Buffer A (Na) is replaced with Buffer A (K).
C. Buffer B (Na) is replaced with Buffer B (K).
D. ADP and MgCl2 were not added at any stage during purification.
E. Purified protein is stored in Buffer A (K)
Cell pellet lysis (as described previously)
Cell debris removal (as described previously)
Ni-NTA purification (as described previously) (Figure 2)
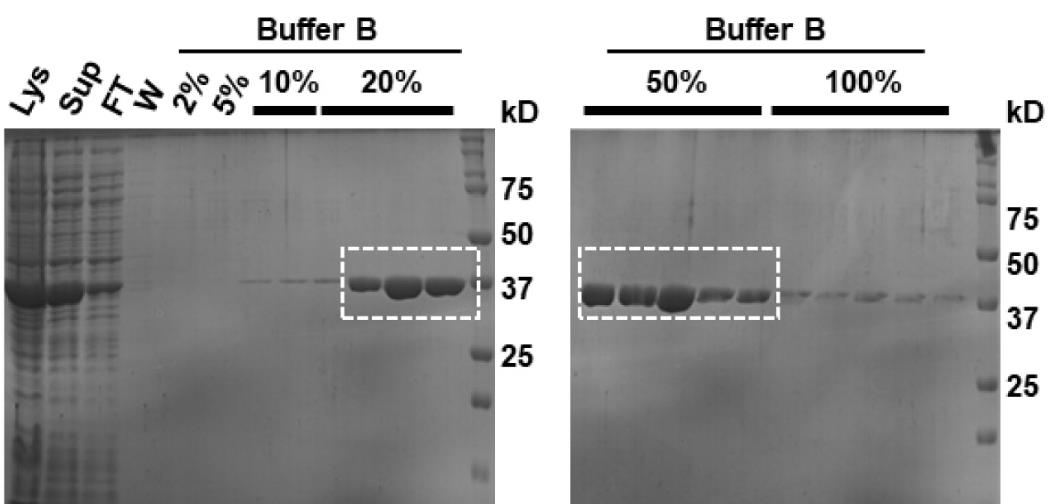
Figure 2. Ni–NTA affinity chromatography of ScMreB5 purified in the optimized buffer condition containing KCl. Representative 12% SDS-PAGE gel of the purified protein after Ni-NTA chromatography; 5 µL of sample mixed with 5 µL of 2× Laemmli buffer is loaded in the gel. Purest fractions (within the dotted rectangular box) are pooled and concentrated to ~1 mL for size exclusion chromatography. Lys: lysate, Sup: supernatant, FT: flowthrough, W: wash. Percentage values denote Buffer B (K) percentage going through the Ni-NTA column along with Buffer A (K).Size exclusion chromatography
Concentrate the protein to 800 µL in 10 kDa molecular weight cut-off concentrators.
Pre-equilibrate the Superdex 200 column with two column volumes of Buffer A (K).
Inject the concentrated protein into the system using a 1 mL loop.
Elute the protein in 0.5 mL fractions across one column volume of Buffer A (K). The protein will elute at around 14.5 mL, corresponding to the monomeric size of the protein (Figure 3A).
Load the samples from the monomeric protein fractions corresponding to the protein on a 12% SDS-PAGE gel (Figure 3B).
Pool the fractions containing the pure protein after confirming from the gel.
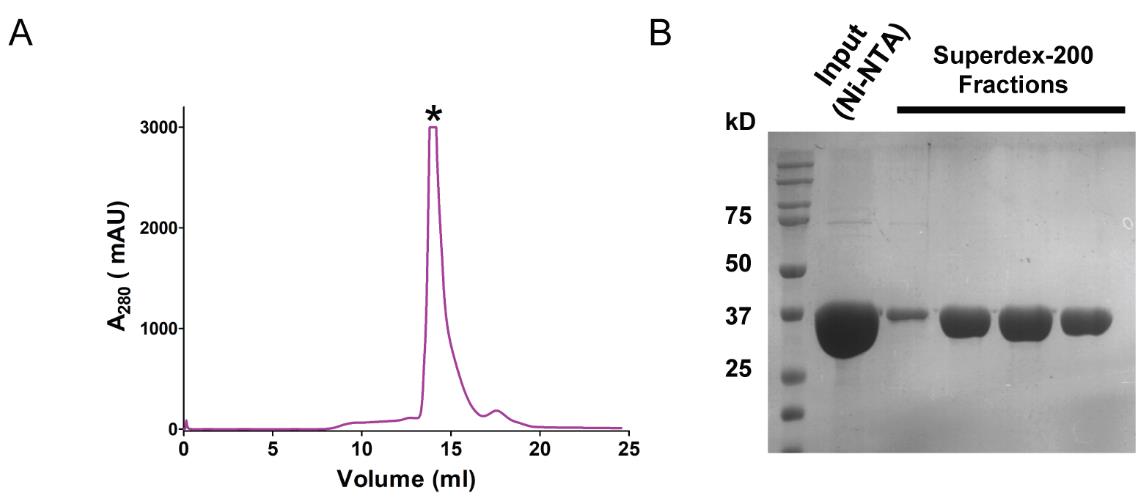
Figure 3. Size-exclusion chromatography of ScMreB5 purified in the optimized buffer condition. (A) Elution profile of the protein eluted during size exclusion chromatography from Superdex-200. Protein starts eluting from ~14.3 mL. The y-axis in the graph represents the milli absorbance unit (mAU). (B) Representative 12% SDS-PAGE gel of the fractions containing monomeric protein from the size exclusion chromatography. “Input (Ni-NTA)” is the concentrated protein from Ni-NTA chromatography; 5 µL of the sample mixed with 5 µL of 2× Laemmli buffer is loaded in the gel. Purest fractions are concentrated and stored at -80 °C.Concentrate the protein to ~500 µL in a pre-equilibrated 10 kDa molecular weight cut-off concentrator at 3,200 RCF in Eppendorf benchtop centrifuge 5810 R at 4 °C.
Estimate the protein concentration using Bradford assay. The overall yield of the protein will be around 10 mg/mL from a 2 L culture pellet.
In the Eppendorf high-speed centrifuge, spin the concentrated protein at 21,000 RCF for 20 min at 4 °C in a 1.5 mL centrifuge tube.
Prepare 10 µL protein aliquots in 0.2 mL PCR tubes. Plunge freeze the protein aliquots in liquid nitrogen and store at -80 °C until further use.
Running 12% SDS-PAGE Gel
Run SDS-PAGE gel after affinity and size exclusion chromatography as follows:
Mix 10 µL of sample with 10 µL of 2× Laemmli buffer. Heat the sample for 5–10 min at 95 °C in a thermomixer.
Give a quick spin to the sample in a MiniSpin® centrifuge.
Place 12% SDS-PAGE gel in the gel running tank containing 1× TGS.
Load 10 µL of the mix into 12% SDS-PAGE gel along with the protein ladder.
Run the gel at 200 V for 40 min or until the dye front runs out from the gel.
Stain the gel with the staining solution for 5 min.
Destain the gel using the destaining solution for 15 min.
Determining the presence of potassium ion
Crystallization in the absence of K+ in the condition:
Estimate protein concentration using the Bradford assay as described in section B.
Set up crystallization for ScMreB5-AMPPNP as given in the Table 2:
Table 2. Crystallization conditions for ScMreB5 – AMPPNP
Protein ScMreB5 – AMPPNP Method Sitting drop Vapour diffusion Plate type 48 well Temperature (K) 291 Protein concentration 4 mg/mL Buffer composition of protein 300 mM KCl, 50 mM Tris, pH 8, 2 mM AMPPNP, 2 mM MgCl2 Composition of reservoir solution 0.15 M sodium-phosphate buffer and 16% PEG 3350, pH 7.8 Volume and ratio of drop (µL: µL) 1:1 Volume of the reservoir (mL) 0.1 mL Add 100 µL of reservoir solution to the reservoir of a 48-well sitting drop crystallization plate.
Prepare a 20 µL reaction containing Buffer A (K), 2 mM AMP-PNP, 2 mM MgCl2, and 4 mg/mL of ScMreB5.
Aliquot 1 µL of reservoir solution into the well and add 1 µL of protein reaction. Mix the reservoir solution with the protein reaction mix.
Prepare 10 such wells for crystallization.
Incubate the plate at 18 °C undisturbed.
After three days, check for needle-like crystals under the microscope.
Pick thick individual needles or thin bundles of needle-shaped crystals using 0.05 or 0.1 mm cryo loops.
Wash out the remaining potassium ion (coming from the protein-containing buffer A) from the crystal by successfully passing the loop through 20% glycerol cryo-protectant drops (made in parent condition). Pass the loop containing crystal at least through three drops.
Freeze the crystal in liquid nitrogen and perform X-ray adsorption spectroscopy at the synchrotron facility.
A peak corresponding to 3.5 keV corresponds to the K+ ion in the scan. The scan plot can be found in the original publication as supplementary Figure S1 A.
Data analysis
Details pertaining to data analysis can be found in the material and methods section of the original research paper [9].
Validation of protocol
This protocol has been validated in the original publication: https://doi.org/10.1083/jcb.202106092
Pande et al. [9]. Filament organization of the bacterial actin MreB is dependent on the nucleotide state. Journal of Cell Biology [(Figure 1 (A–D) and Figure S1 (A–E)].
General notes and troubleshooting
The plasmid used for expressing ScMreB5, pHis17, does not have the lacI repressor gene; therefore, IPTG is not required for expression in BL21-AI cells. This plasmid has been used previously for expressing other MreBs [5,14]. Alternatively, ScMreB5 orthologs can also be cloned pET-derived vectors, having the lacI repressor, as demonstrated for Spiroplasma eriocheris MreB5 [15]. Expression of protein from vectors having the lacI repressor requires the addition of IPTG for expression in BL21-AI, as detailed in the manufacturer’s protocol.
It is important that the entire purification of ScMreB5 be at 4 °C or in ice. For affinity purification, one can use the manual setup in a cold room. It is important to run SDS-PAGE gel after every step of purification to ensure that only fractions that have a single band corresponding to ScMreB5 are taken forward. Before setting up crystallization, it is advisable to perform an ATPase assay to ensure that the protein is catalytically active. The protocol for the ATPase assay is available in the original publication.
This protocol can serve as a reference for purifying other ScMreB5 orthologs. Heterologous expression of MreB5 in E. coli could be toxic for the cells due to the presence of native E. coli MreB. It is recommended to optimize protein expression in various E. coli strains suitable for toxic heterologous proteins. Protein insolubility may be encountered while performing expression check, for which mild detergents can be used in the lysis buffer.
During purification, issues like low protein yield due to protein precipitation may arise. The thermal shift assay can be performed to screen for optimal conditions (salts, buffers, detergents, ligands). The condition with the highest melting temperature can be selected as the optimized condition for further purification trials. Size exclusion chromatography is recommended to assess the polymeric state of the protein, followed by performing crystallography with monomeric protein.
The protocol might be applicable for standardizing other MreBs as well as other proteins that often get stabilized upon the addition of a ligand, most often either the substrate or the product in the case of enzymes. Ligand-induced stability helps us to obtain sufficient protein for carrying out the stability assays using thermal shift. This could lead us to identify other stabilizing additives, which can be used for purifying the protein in the absence of the initial ligand. The presence of excess stabilizing ligand, such as the substrate of the product, precludes us from performing many biochemical experiments such as ligand affinity studies as well as enzyme kinetic assays. Hence, the identification of conditions that stabilize the protein without the excess substrate/ligand is highly beneficial.
Acknowledgments
We thank Saket R. Bagde, Nivedita Mitra and Shrikant Harne for their contributions to the study of ScMreB5 characterization. We thank the macromolecular crystallography facility at IISER Pune and synchrotron facilities at European Synchrotron Radiation Facility (ESRF), Grenoble, Diamond Light Source (MX22637) and Department of Biotechnology India for facilitating data collection at ID29, ESRF. We thank the IISER Pune for the infrastructure to perform experiments. P. Gayathri acknowledges the support from the following funding agencies: Department of Science and Technology INSPIRE Faculty Fellowship (IFA12/LSBM-52), Innovative Young Biotechnologist Award (BT/07/IYBA/2013) and Department of Biotechnology Membrane Structural Biology Program grant (BT/PR28833/BRB/10/1705/2018) and IISER Pune. V. Pande thanks IISER Pune for the fellowship and the Infosys Travel award. Graphical overview created with BioRender.com.
Competing interests
The authors declare that they have no competing interests.
References
- Wagstaff, J. and Löwe, J. (2018). Prokaryotic cytoskeletons: protein filaments organizing small cells. Nat Rev Microbiol. 16(4): 187–201.
- Kawai, Y., Asai, K. and Errington, J. (2009). Partial functional redundancy of MreB isoforms, MreB, Mbl and MreBH, in cell morphogenesis of Bacillus subtilis. Mol Microbiol. 73(4): 719–731.
- Kruse, T., Bork‐Jensen, J. and Gerdes, K. (2004). The morphogenetic MreBCD proteins of Escherichia coli form an essential membrane‐bound complex. Mol Microbiol. 55(1): 78–89.
- Errington, J. (2015). Bacterial morphogenesis and the enigmatic MreB helix. Nat Rev Microbiol. 13(4): 241–248.
- van den Ent, F., Izoré, T., Bharat, T. A., Johnson, C. M. and Löwe, J. (2014). Bacterial actin MreB forms antiparallel double filaments. eLife. 3: e02634.
- Salje, J., van den Ent, F., de Boer, P. and Löwe, J. (2011). Direct Membrane Binding by Bacterial Actin MreB. Mol Cell. 43(3): 478–487.
- Ursell, T. S., Nguyen, J., Monds, R. D., Colavin, A., Billings, G., Ouzounov, N., Gitai, Z., Shaevitz, J. W. and Huang, K. C. (2014). Rod-like bacterial shape is maintained by feedback between cell curvature and cytoskeletal localization. Proc Natl Acad Sci USA. 111(11): E1025–E1034.
- Harne, S., Duret, S., Pande, V., Bapat, M., Béven, L. and Gayathri, P. (2020). MreB5 Is a Determinant of Rod-to-Helical Transition in the Cell-Wall-less Bacterium Spiroplasma. Curr Biol. 30(23): 4753–4762.e7.
- Pande, V., Mitra, N., Bagde, S. R., Srinivasan, R. and Gayathri, P. (2022). Filament organization of the bacterial actin MreB is dependent on the nucleotide state. J Cell Biol. 221(5): e202106092.
- Citti, C., Maréchal-Drouard, L., Saillard, C., Weil, J. H. and Bové, J. M. (1992). Spiroplasma citri UGG and UGA tryptophan codons: sequence of the two tryptophanyl-tRNAs and organization of the corresponding genes. J Bacteriol. 174(20): 6471–6478.
- van den Ent, F. and Löwe, J. (2006). RF cloning: A restriction-free method for inserting target genes into plasmids. J Biochem Bioph Methods. 67(1): 67–74.
- Cimmperman, P. and Matulis, D. (2011). Protein Thermal Denaturation Measurements via a Fluorescent Dye. In: Podjarny, A., Dejaegere, A. P. and Kieffer, B. (Eds.). Biophysical Approaches Determining Ligand Binding to Biomolecular Targets: Detection, Measurement and Modelling. The Royal Society of Chemistry, p.0.
- Kozak, S., Lercher, L., Karanth, M. N., Meijers, R., Carlomagno, T. and Boivin, S. (2016). Optimization of protein samples for NMR using thermal shift assays. J Biomol NMR. 64(4): 281–289.
- van den Ent, F., Amos, L. A. and Löwe, J. (2001). Prokaryotic origin of the actin cytoskeleton. Nature. 413(6851): 39–44.
- Takahashi, D., Fujiwara, I., Sasajima, Y., Narita, A., Imada, K. and Miyata, M. (2022). ATP-dependent polymerization dynamics of bacterial actin proteins involved in Spiroplasma swimming. Open Biol. 12(10): e220083.
Article Information
Publication history
Received: Apr 18, 2024
Accepted: Aug 23, 2024
Available online: Sep 28, 2024
Published: Oct 20, 2024
Copyright
© 2024 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Pande, V. and Gayathri, P. (2024). Improving Stability of Spiroplasma citri MreB5 Through Purification Optimization and Structural Insights. Bio-protocol 14(20): e5086. DOI: 10.21769/BioProtoc.5086.
- Pande, V., Mitra, N., Bagde, S. R., Srinivasan, R. and Gayathri, P. (2022). Filament organization of the bacterial actin MreB is dependent on the nucleotide state. J Cell Biol. 221(5): e202106092.
Category
Biochemistry > Protein > Isolation and purification
Cell Biology > Cell structure > Plasma membrane
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.