- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Development and Characterization of Primary Brain Cultures from Japanese Quail Embryos

(*contributed equally to this work) Published: Vol 14, Iss 18, Sep 20, 2024 DOI: 10.21769/BioProtoc.5071 Views: 1778
Reviewed by: Sébastien GillotinSilvia Olivera-BravoHong Lian
Original research article
The authors used this protocol in:

Mar 2023
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
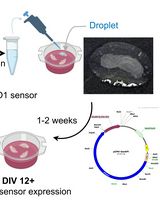
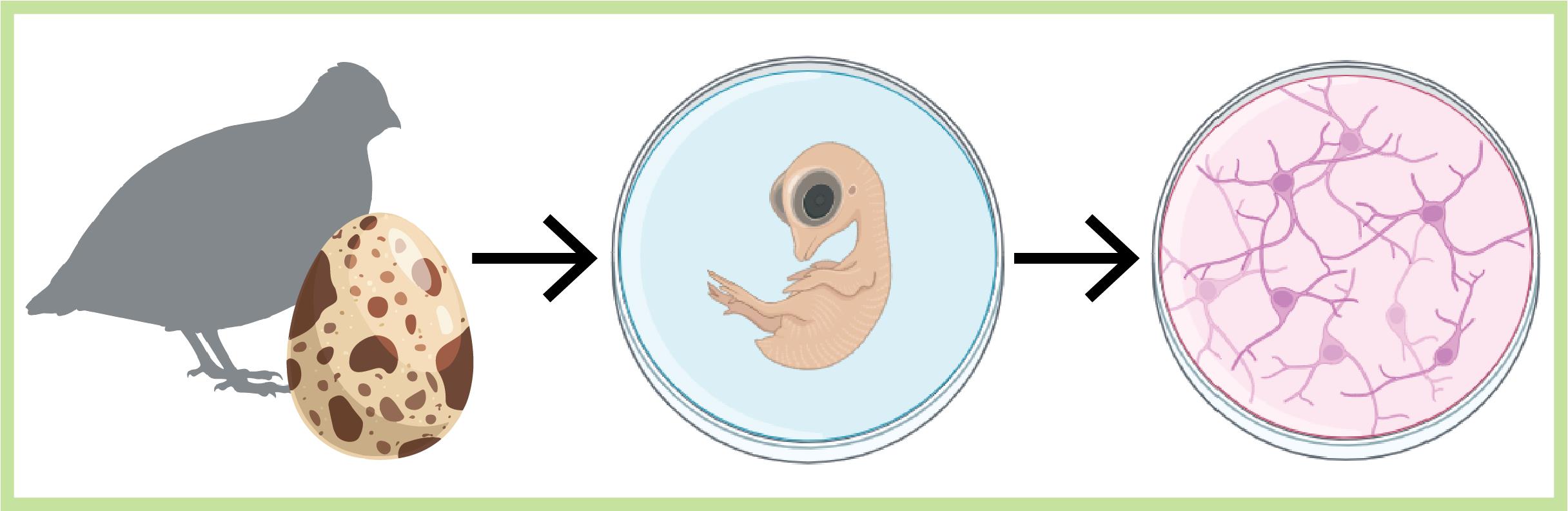
Abstract
Cell cultures play a crucial role in neuroscience research, facilitating the elucidation of the complexities of cellular physiology and pathology. The relative simplicity in producing cultures and the accessibility to cells that the cultures provide, in contrast to in vivo settings, allow users to manipulate and monitor cells more easily at higher throughputs and lower costs. These are ideal for screening purposes and electrophysiological characterizations. Despite the prevalence of methodologies for producing brain cultures from various animal models, rodents in particular, approaches for culturing neurons (and glia) from birds are less established or completely absent as in the case of the Japanese quail model. Here, we present a unique culturing protocol for brain cells (e.g., neurons at different maturation levels, such as progenitor cells, excitatory and inhibitory neurons, microglia, and endothelial cells) from entire forebrains of Japanese quail embryos for high-throughput screening of viral vectors in vitro and other various purposes. Following dissection and digestion methods uniquely suited for avian brains, we tailored the growth media and culturing surface to allow the survival of quail brain cultures for more than three weeks in vitro.
Key features
• We introduce a detailed protocol for producing primary brain cultures from quail embryos' forebrains for up to 30 days.
• We show that the cultures support in vitro viral transfections effectively.
• We demonstrate the use of the cultures for rapid (days) screening for suitable viruses for quail brain cells, electrophysiological characterizations, and single mRNA sequencing.
Keywords: Primary cell cultureGraphical overview
Background
Bird species offer intriguing prospects for neuroscience research due to their sophisticated cognitive abilities and specialized behaviors [1–6]. Despite differences in neuroarchitecture and neuronal densities in avian brains when compared with mammals [4,7], birds demonstrate remarkable and distinctive behavioral performances, including long-distance navigation [8], imprinting, homing, food-caching, and song-learning [9,10], to name a few. Consequently, these provide unique opportunities for comparative studies aiming to elucidate the cellular mechanisms underlying these capabilities [6]. Indeed, these realizations are reflected by the growing interest in avian neuroscience [1,3,9,11–18]. Among the different avian species explored, our focus centers on the domestic Japanese quail (Coturnix japonica). The quail offers several advantages over other birds due to its small size, rapid sexual maturation, and frequent egg-laying; advantages that make this species highly suitable for routine experimentations [17]. Importantly, the ground-dwelling nature of quails makes them ideal for studying birds’ spatial navigation by reducing dimensionalities (from 3D to 2D) [19]. In spite of these advantages and the extensive use of quails in the field of developmental biology [17,20], this species remains relatively underexplored in neuroscience [5,19]. This is partly attributed to the absence of methods and tools to manipulate and examine quail neurons [21], in particular, the lack of efficient viral vectors for neuronal transduction [5,17]. To screen for suitable viral vectors, we choose not to pursue in vivo exploration, as is common practice, due to the considerably large number of animals and time this task necessitates (to overcome animal-to-animal variability, variable injection performances, and other technical limitations). Instead, we envisioned that primary brain cultures would present a more rapid and cost-effective approach for examining multiple viruses in parallel [22].
Primary brain cell cultures constitute a valuable tool in neuroscience [23,24]. Brain cultures faithfully preserve many complex cellular behaviors, electrophysiological properties, and phenotypic traits, as observed in vivo, despite the loss of the precise network architecture and organization in the tissue, as is exclusively found in vivo [23-28]. Nevertheless, the alteration in tissue architecture, which includes the removal of the blood-brain barrier (BBB) and extracellular matrix, proves to be advantageous as it enhances cellular accessibility and reduces time to expression. Indeed, cultures are ideal for high-throughput screening of pharmacology, which should likely translate toward the screening of different viruses [22].
We have initially attempted to produce primary cultures based on standard protocols for culturing cells from the brains of neonatal rodents (such as rats and mice, e.g., [29]) or chicken embryos (e.g., [30,31]). However, these attempts were unsuccessful. Consequently, we decided to optimize the procedure by experimenting with embryos collected from different days in ovo (DIO), modifying the culturing media, and assessing cell growth directly on plastic dishes instead of glass coverslips. After successfully establishing suitable conditions for maintaining viable cultures for several weeks (up to 30 days), we leveraged them for screening several commonly employed viruses in neuroscience research, including adeno-associated viruses (AAV), lentiviruses, and baculovirus. We further harnessed the cultures for the rational engineering of a novel AAV variant, specifically tailored for Japanese quail neurons (denoted AAV1*) [22]. Lastly, we characterized the diverse cellular populations in the cultures through use of electrophysiology, calcium imaging, and single-cell mRNA sequencing [22].
Materials and reagents
Biological materials
Fertilized Japanese quail eggs (Coturnix japonica). The eggs we employ are obtained from our quail colony. Eggs are collected from cages with male and female quails. Nevertheless, there are multiple commercial providers of fertilized quail eggs; however, the number of farms and thereby commercial availability may vary from one country to another.
Reagents
B-27 serum-free supplement (50×) liquid (Gibco, catalog number: 17504-044)
Pen/Strep solution (Sartorius, catalog number: 03-031-1B)
L-glutamine (GlutaMAX) (Gibco, catalog number: 35050-038)
Neurobasal-A medium (Gibco, catalog number: 10888-022)
Papain from papaya latex (Sigma-Aldrich, catalog number: 9001-73-4)
Phosphate buffered saline (PBS) (Sartorius, catalog number: 02-023-5A)
Poly-D-lysine hydrobromide (PDL) (Sigma-Aldrich, catalog number: 27964-99-4)
DNase I set (Geneaid, catalog number: DNS300)
Sylgard 184 silicone elastomer (Sigma-Aldrich, catalog number: 761036-5EA)
DMEM/F-12 (Ham) (Sartorius, catalog number: 01-170-1A)
Solutions
0.2% PDL (see Recipes)
Quail neurobasal medium (QNBM) (see Recipes)
Digestion solution (see Recipes)
Papain stock (see Recipes)
Recipes
Note: Prepare all solutions in a laminar flow cell culture hood.
0.2% PDL (store at -20 °C)
Reagent Final concentration Amount PDL 2 mg/mL 100 mg Double-distilled water (DDW) n/a 50 mL Total n/a 50 mL Quail neurobasal medium (NBM) (store at 4 °C)
Reagent Final concentration Amount B-27 2% 5 mL Pen/Strep 1% 2.5 mL L-glutamine 0.25% 625 µL Neurobasal-A medium n/a Complete to 250 mL Total n/a 250 mL Digestion solution (prepare on the day of extraction)
Reagent Final concentration Amount Papain stock 33 U/mL 1 mL PBS n/a 2 mL DNase I 57 U/mL 85 µL Total n/a 3.085 mL Papain stock
Reagent Final concentration Amount Papain 10 U/mg 100 mg PBS n/a 10 mL Total n/a 10 mL
Laboratory supplies
Spray bottle with 70% ethanol (Bio-Lab Chemicals, catalog number: 000521020500)
Tissue culture dish (60 mm × 15 mm) (Corning, catalog number: 430166)
Tissue culture dish (100 mm × 20 mm) (Corning, catalog number: 430167)
Microtubes, 1.7 mL (Corning, catalog number: MCT-175-C)
Conical bottom tubes, 15 ml (Greiner Bio-One, catalog number: 188261)
Conical bottom tubes, 50 ml (Greiner Bio-One, catalog number: 227270)
Cell strainer 40 μm (LifeGene, catalog number: LG-CSS010040S)
Parafilm (Sigma-Aldrich, catalog number: P7543)
Delicate task Kimwipes (Kimberly-Clark professional, catalog number: 34120)
Minutien Pins, 1 cm, 0.0175 mm tip (Fisher Scientific, catalog number: 2600215)
Equipment
Stereomicroscope system (Zeiss Stemi 2000, Fisher Scientific, catalog number: 10331390)
Microbiological safety cabinet class II (Thermo Scientific, catalog number: 51025757)
Isotemp water bath (PolyScience, catalog number: E12020004)
HERAcell 150i CO2 incubator (Thermo Fisher Scientific, catalog number: 50116047)
Gilson (or equivalent) pipettes and tips (P20/P200/P1000) (Fisher Scientific)
Dumont #5 forceps (Roboz surgical store, catalog number: RS-4905) (Figure 1, #1)
Curved dissecting scissors (Stoelting, catalog number: 52132-11P) (Figure 1, #2)
Plastic Pasteur pipette (blunt tip obtained by trimming pipette ~1 cm from tip) (Sigma-Aldrich, catalog number: 747775 (Figure 1, #3)
Vannas spring scissors (Fine Science Tools, catalog number: 15000-08) (Figure 1, #4)
Iris forceps (Fine Science Tools, catalog number: 11064-07) (Figure 1, #5)
Fine-angled forceps (Stoelting, catalog number: 52102-02P) (Figure 1, #6)
Egg incubator, 37 °C (DMP Engineering, model: INCA 100)
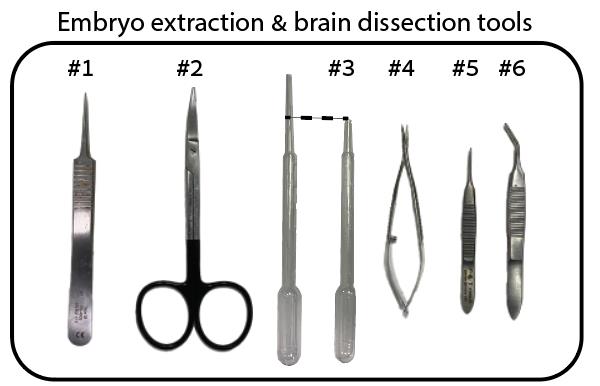
Figure 1. Fine surgical tools required for the removal of eggshell and embryo from egg
Procedure
PDL plate coating
Note: Do this in a sterile laminar flow cell culture hood.
Fill 60 mm Corning dishes with 5 mL of 0.2% PDL solution (see Recipes).
Place the plates in a 37 °C incubator overnight.
Critical: Non-satisfactory coating of the plates can lead to cell dissociation from the surface of the plate and neuronal death causing unhealthy, nonviable cultures.
Collect the PDL solution using a pipette (the solution can be recycled) until plates are empty.
Wash the plates three times using sterile DDW.
Allow residual DDW to evaporate and the plates to dry (leave in the laminar flow cell culture hood).
Seal each plate with parafilm to keep sterile.
Keep coated plates at 4 °C for later use. Plates can be stored for several weeks.
Silicone plate coating
Note: Do this in a sterile laminar flow cell culture hood.
Prepare a 60 mm Corning dish coated with 7 mm silicone, using the Sylgard 184 silicone elastomer kit. The manufacturer’s instructions are included and simple to follow.
Keep overnight at room temperature.
Embryo extraction and brain dissection from quail eggs
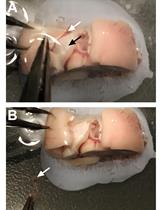
Obtain fresh fertilized eggs. Fertilized eggs cannot be distinguished from unfertilized ones at this stage (Figure 2A).
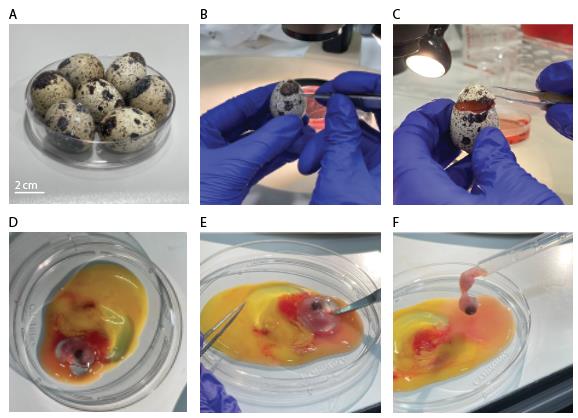
Figure 2. Egg shell removal and embryo extraction from quail eggs. A. Clean quail eggs post-incubation. B. Eggshell penetration. C. Circular eggshell crack, letting the “cap” loose. D. Quail embryo in dish, together with yolk sac and blood vessels. E. Separating embryo from yolk sac and blood vessels using scissors. F. Embryo picked up for transfer with plastic Pasteur pipette.Store eggs in a suitable incubator at 37 °C with 70%–80% humidity for 7–9 days.
Spray eggshells with 70% ethanol and wipe dry (gently, using Kimwipes), before placing inside the laminar hood.
Note: From this point onward, all procedures are performed in the laminar hood.
Using Dumont #5 forceps (Figure 1, #1), gently penetrate the eggshell in its upper quarter (Figure 2B).
Crack the eggshell with a circular forceps movement, letting the eggshell’s “cap” loose (Figure 2C).
Pour egg content into a 100 mm Corning dish.
Using curved dissection scissors (Figure 1, #2), separate the embryo from the yolk sac and cut the interacting blood vessels (Figure 2D, E).
Transfer the embryo with a blunt plastic Pasteur pipette (Figure 1, #3; Figure 2F) into a 100 mm Corning dish filled with DMEM (Figure 3A).
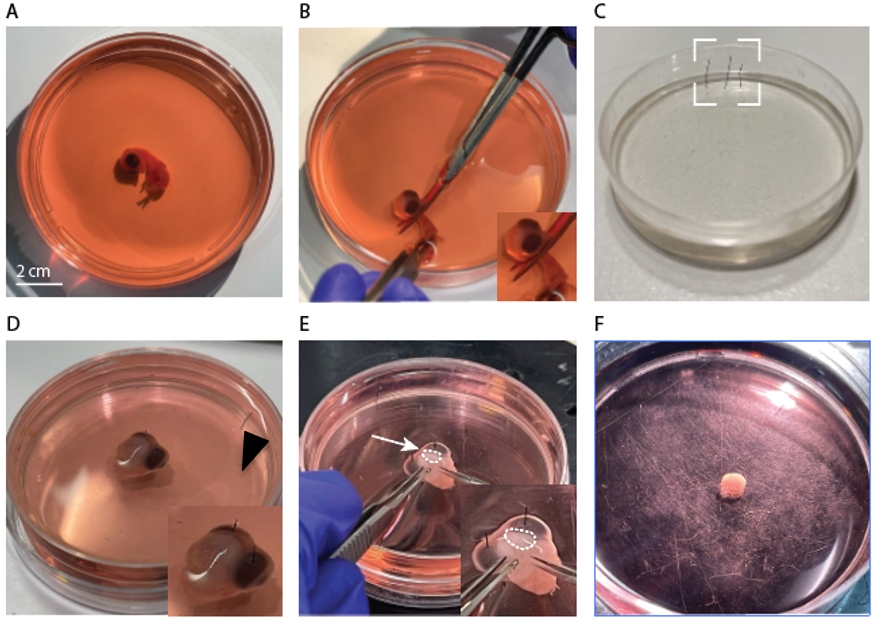
Figure 3. Embryo forebrain extraction. A. Isolated embryo in a Petri dish, filled with DMEM. B. Embryo decapitation using dissection scissors. C. Silicone-coated Petri dish and fixation pins (dashed box). D. Isolated embryo head in DMEM, pinned to the silicone surface. Inset: closer view of pins (arrowhead). E. Forebrain dissection using forceps and scissors. Inset: closer view of the right forebrain (dashed region). F. One isolated forebrain (without meninges).Decapitate the embryo with curved dissection scissors (Figure 3B).
Transfer the head of the embryo using a plastic Pasteur pipette to a 60 mm Petri dish coated with silicone and filled with DMEM.
Immobilized the head using Minutien Pins inserted in each eye (Figure 3C, D) and place under a light microscope.
With the Vannas spring scissors (Figure 1, #4), create a mid-sagittal incision from the nape to the eye line (Figure 3E). Peel the soft tissues overlaying the brain with Iris forceps (Figure 1, #5).
Slip the Vannas spring scissors under the forebrain and gently cut through the tissues that are connected to it (Figure 3E, inset).
Using Iris forceps and fine-angled forceps (Figure 1, #5 & #6, respectively), separate the forebrain from the head.
Clean the two hemispheres gently from residual connective tissue and meninges by pealing the meningeal layers with the Iris and the fine-angled forceps (Figure 1, #5 & #6).
Critical: Partial pealing and removal of the meninges can cause fibroblast overgrowth and dominate the culture.
Transfer the clean hemispheres to a 35 mm Petri dish filled with 2 cm of DMEM (Figure 3F) using a plastic Pasteur pipette (Figure 1, #3).
Repeat steps C4–16 until the desired number of hemispheres is obtained. See Troubleshooting.
Tissue dissociation
Place the isolated forebrains in a 15 mL conical tube containing the digestion solution (3.085 mL; see Recipes) using a plastic Pasteur pipette (Figure 1, #3).
Incubate at 37 °C for 1 h (no shaking is needed).
Critical: Incubation of cells for less than 1 h may lead to insufficient dissociation of the tissue, which will result in the appearance of cell clumps in the culture, whereas longer incubations (>1 h) may lead to excessive cell (especially neuronal) death.
Gently remove the solution with a 5 mL pipette without disturbing the precipitated forebrains.
Remove the residual solution with a 1,000 μL plastic tip.
Wash the forebrains by adding PBS to the tube. Repeat three times.
After the third wash, remove the PBS completely using a 1,000 μL plastic tip and replace it with 2 mL of quail neuro-basal medium (NBM) (see Recipes).
Manually dissociate the tissue by gentle trituration with a large, 5 mL plastic pipette tip (×15 times).
Then, triturate the solution gently with a smaller 1,000 μL plastic pipette tip until a homogenous solution is obtained (with no visible tissue fragments).
Place a 40 μm cell strainer on a 50 mL conical tube and prewash it with 1 mL of quail NBM.
Critical: Prewashing the cell strainer prevents cells from sticking to the strainer.
Transfer the entire solution containing the dissociated brains onto the cell strainer placed with a 1,000 μL plastic tip.
After passing the entire solution through the cell strainer, wash the strainer with additional quail NBM to elute the remaining cells from the strainer. The final volume of the filtered (strained) solution should be ~5 mL of quail NBM per one quail’s hemisphere. The optimal density for seeding is 5 × 105 cells/cm2. See Troubleshooting.
Plating
Transfer 5 mL of strained solution onto a sterile 60 mm tissue culture dish that has been pre-coated with 2 mg/mL PDL.
Note: Cells do not adhere to glass coverslips even if the coverslips have been pre-coated with PDL. They clump after several hours and die.
Place the seeded plates in a standard biological cell culture incubator at 37 °C and 5% CO2 for 1 h to ensure cell attachment to the surface of the plates.
After 1 h, gently remove the medium by aspiration and replace it with fresh and pre-warmed quail NBM.
Critical: Aggressive handling and removal of the medium can cause cells to detach from the dish.
Place the plates back in the incubator for 72 h.
After 72 h, replace half the medium with fresh and pre-warmed quail NBM.
Repeat step E4 every other day.
Individual cells should be easily discernible in cultures after one day in vitro (Figure 4A and B, left top and bottom panels). Cells should begin sprouting processes within this time frame. Cellular clumps are an indication of a dying culture (Figure 4A, right panel compared to right panel in B).
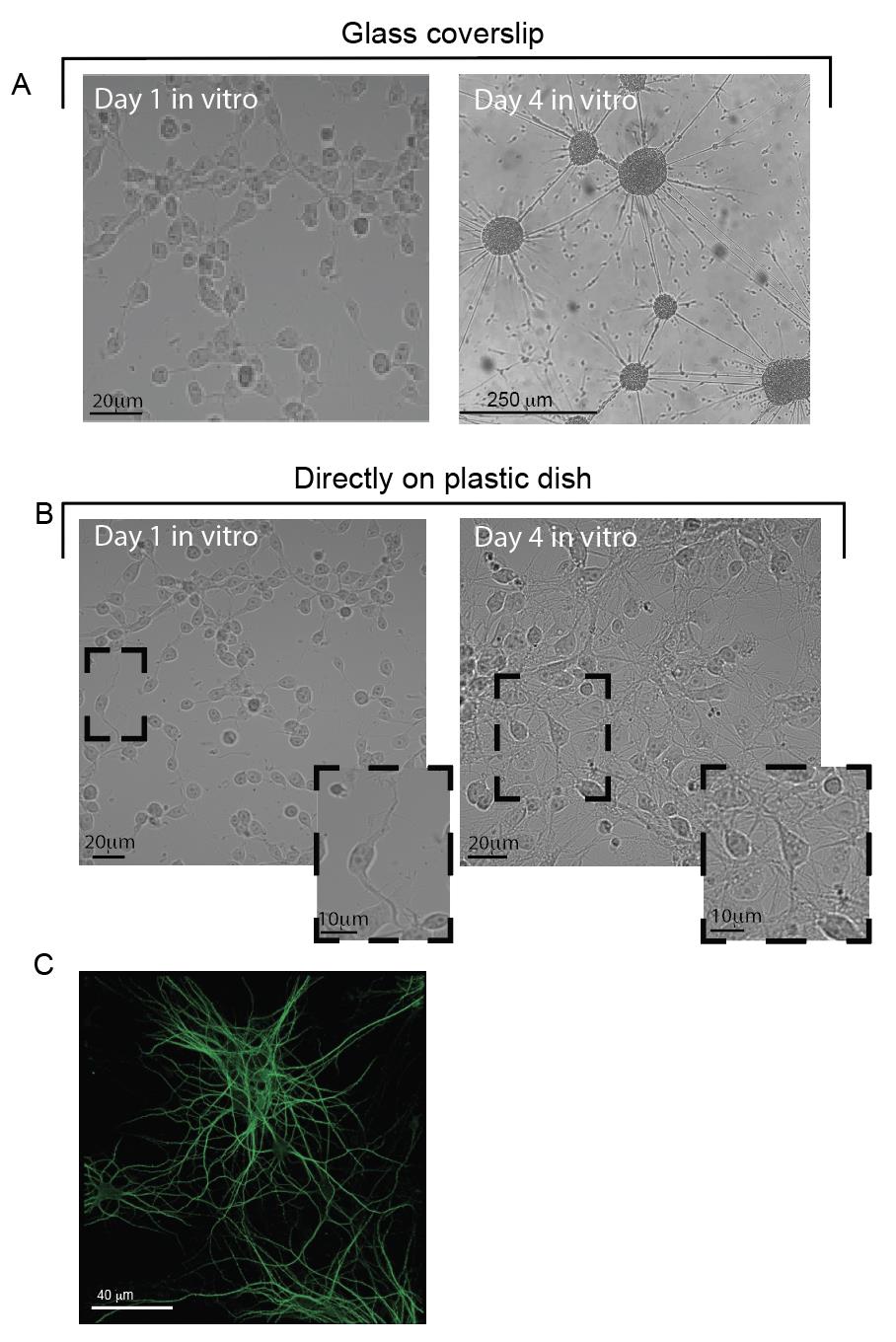
Figure 4. Viability of primary cultures grown on glass or plastic after one week in vitro. A, B. Cells grown in vitro at days 1 and 4 post-dissection, on PDL-coated glass coverslips in Petri dishes (A) or directly on the Petri dish (also coated with PDL) (B). The top-right image in A shows a clumping of cells, which does not appear in B. Insets in B show a closer view of the cells’ morphology in culture. Note the development of processes (bottom left) and the appearance of neurons (bottom right). C. MAP2-immunostaining of quail neurons at 14 days in vitro (DIV).
Validation of protocol
This protocol is robust and reproducible. Number of replicates done is >30. Primary neuronal cultures were employed for electrophysiological recordings (patch clamp), calcium imaging, high-throughput viral infection screening, immunohistochemical staining, and single-cell mRNA sequencing experiments (see Figures 1, 2, 3, 4, 6e–g, Suppl. Figure 2 [3]).
General notes and troubleshooting
General notes
This protocol was also found to be suitable for producing primary brain cultures from the forebrains of age-matched chicken embryos (see suppl. Figure 8 in [3]). We therefore assume it may also be suitable for different bird species.
Sources of variability between different cultures may arise from the initial conditions and age of the embryos. Embryos undergoing healthy development are likely to produce more viable cultures, compared to embryos with delayed development. Cultures prepared from poorly developing embryos may yield sparser cultures.
Troubleshooting
Cell density in culture: We recommend dissociating and plating one hemisphere per plate (i.e., one hemisphere per 5 mL of culture medium) (Figure 4A, B; left panels). We noted that this yields favorable conditions for cellular growth, without excessive density of cells. We find that the optimal seeding density for viability of the culture is 5 × 105 cells/cm2. To increase cell density, simply increase the number of hemispheres per plate, for instance plating 1.5 hemispheres per plate. For sparser cultures, dilute the cell suspension in a larger volume of medium. These adjustments may offer strategic means to tailor cell density to experimental requirements, thereby influencing cellular interactions, proliferation rates, and experimental outcomes.
Acknowledgments
Funding: Support was provided by the Rappaport Family Thematic grant (S.B. and Y.G.).
The protocol was first described and validated in Zoabi et al. [3]. A custom-made AAV1 variant (AAV1-T593K) enables efficient transduction of Japanese quail neurons in vitro and in vivo. Communications Biology, 6(1): 337.
Competing interests
The authors declare no competing interests.
Ethical considerations
Animal experiments were approved by the Technion Institutional Animal Care and Use Committee (permit no. IL-157-11-17 and IL-19-10-143) and all experiments strictly followed the approved guidelines.
References
- Clayton, N. S. and Emery, N. J. (2015). Avian Models for Human Cognitive Neuroscience: A Proposal. Neuron. 86(6): 1330–1342.
- Lormant, F., Cornilleau, F., Constantin, P., Meurisse, M., Lansade, L., Leterrier, C., Lévy, F. and Calandreau, L. (2020). Research Note: Role of the hippocampus in spatial memory in Japanese quail. Poult Sci. 99(1): 61–66.
- Nieder, A., Wagener, L. and Rinnert, P. (2020). A neural correlate of sensory consciousness in a corvid bird. Science. 369(6511): 1626–1629.
- Olkowicz, S., Kocourek, M., Lučan, R. K., Porteš, M., Fitch, W. T., Herculano-Houzel, S. and Němec, P. (2016). Birds have primate-like numbers of neurons in the forebrain. Proc Natl Acad Sci USA. 113(26): 7255–7260.
- Seidl, A. H., Sanchez, J. T., Schecterson, L., Tabor, K. M., Wang, Y., Kashima, D. T., Poynter, G., Huss, D., Fraser, S. E., Lansford, R., et al. (2012). Transgenic quail as a model for research in the avian nervous system: A comparative study of the auditory brainstem. J Comp Neurol. 521(1): 5–23.
- Yartsev, M. M. (2017). The emperor’s new wardrobe: Rebalancing diversity of animal models in neuroscience research. Science. 358(6362): 466–469.
- Stacho, M., Herold, C., Rook, N., Wagner, H., Axer, M., Amunts, K. and Güntürkün, O. (2020). A cortex-like canonical circuit in the avian forebrain. Science. 369(6511): eabc5534.
- Mouritsen, H. (2018). Long-distance navigation and magnetoreception in migratory animals. Nature. 558(7708): 50–59.
- McCabe, B. J. (2019). Visual Imprinting in Birds: Behavior, Models, and Neural Mechanisms. Front Physiol. 10: e00658.
- Brodin, A. (2010). The history of scatter hoarding studies. Philos Trans R Soc Lond B Biol Sci. 365(1542): 869–881.
- Cohen, Y., Shen, J., Semu, D., Leman, D. P., Liberti, W. A., Perkins, L. N., Liberti, D. C., Kotton, D. N. and Gardner, T. J. (2020). Hidden neural states underlie canary song syntax. Nature. 582(7813): 539–544.
- Lipkind, D., Zai, A. T., Hanuschkin, A., Marcus, G. F., Tchernichovski, O. and Hahnloser, R. H. R. (2017). Songbirds work around computational complexity by learning song vocabulary independently of sequence. Nat Commun. 8(1): 1247.
- Daou, A. and Margoliash, D. (2020). Intrinsic neuronal properties represent song and error in zebra finch vocal learning. Nat Commun. 11(1): 952.
- Ben-Yishay, E., Krivoruchko, K., Ron, S., Ulanovsky, N., Derdikman, D. and Gutfreund, Y. (2020). Head-direction coding in the hippocampal formation of birds. bioRxiv. doi.org/10.1101/2020.08.31.274928.
- Thiele, N., Hildebrandt, K. J. and Köppl, C. (2020). Gene delivery to neurons in the auditory brainstem of barn owls using standard recombinant adeno-associated virus vectors. Curr Res Neurobiol. 1: 100001.
- Ben-Tov, M., Duarte, F. and Mooney, R. (2021). A neural hub that coordinates learned and innate courtship behaviors. bioRxiv. doi.org/10.1101/2021.09.09.459618.
- Serralbo, O., Salgado, D., Véron, N., Cooper, C., Dejardin, M. J., Doran, T., Gros, J. and Marcelle, C. (2020). Transgenesis and web resources in quail. eLife. 9: e56312.
- Martinho, A. and Kacelnik, A. (2016). Ducklings imprint on the relational concept of “same or different”. Science. 353(6296): 286–288.
- Ben-Yishay, E., Krivoruchko, K., Ron, S., Ulanovsky, N., Derdikman, D. and Gutfreund, Y. (2021). Directional tuning in the hippocampal formation of birds. Curr Biol. 31(12): 2592–2602.e4.
- Huss, D., Benazeraf, B., Wallingford, A., Filla, M., Yang, J., Fraser, S. E. and Lansford, R. (2015). Transgenic quail to dynamically image amniote embryogenesis. Development. 142: 2850–2859.
- Finkelstein, A., Derdikman, D., Rubin, A., Foerster, J. N., Las, L. and Ulanovsky, N. (2014). Three-dimensional head-direction coding in the bat brain. Nature. 517(7533): 159–164.
- Zoabi, S., Andreyanov, M., Heinrich, R., Ron, S., Carmi, I., Gutfreund, Y. and Berlin, S. (2023). A custom-made AAV1 variant (AAV1-T593K) enables efficient transduction of Japanese quail neurons in vitro and in vivo. Commun Biol. 6(1): 337.
- Egger, B., van Giesen, L., Moraru, M. and Sprecher, S. G. (2013). In vitro imaging of primary neural cell culture from Drosophila. Nat Protoc. 8(5): 958–965.
- Kaech, S. and Banker, G. (2006). Culturing hippocampal neurons. Nat Protoc. 1: 2406–2415.
- Mueller-Klieser, W. (1997). Three-dimensional cell cultures: from molecular mechanisms to clinical applications. Am J Physiol, Cell Physiol. 273(4): C1109–C1123.
- Abbott, A. (2003). Cell culture: Biology’s new dimension. Nature. 424: 870–873.
- Gordon, J., Amini, S. and White, M. K. (2013). General Overview of Neuronal Cell Culture. Neuronal Cell Culture. 1–8.
- Kim, S. U. (1983). Neuronal aging in tissue and cell cultures: A review. In Vitro. 19(2): 73–82.
- Berlin, S. and Isacoff, E. Y. (2017). Optical Control of Glutamate Receptors of the NMDA-Kind in Mammalian Neurons, with the Use of Photoswitchable Ligands. Neuromethods.: 293–325.
- Matsui, R., Tanabe, Y. and Watanabe, D. (2012). Avian Adeno-Associated Virus Vector Efficiently Transduces Neurons in the Embryonic and Post-Embryonic Chicken Brain. PLoS One. 7(11): e48730.
- Kumar, A. and Mallick, B. N.(2016). Long-term primary culture of neurons taken from chick embryo brain: A model to study neural cell biology, synaptogenesis and its dynamic properties. J Neurosci Methods. 263: 123–133.
Article Information
Publication history
Received: May 15, 2024
Accepted: Aug 11, 2024
Available online: Aug 21, 2024
Published: Sep 20, 2024
Copyright
© 2024 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Zoabi, S., Blau, A. and Berlin, S. (2024). Development and Characterization of Primary Brain Cultures from Japanese Quail Embryos. Bio-protocol 14(18): e5071. DOI: 10.21769/BioProtoc.5071.
Category
Neuroscience > Cellular mechanisms > Tissue isolation and culture
Cell Biology > Model organism culture
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.