- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Imaging Assays to Detect DNA Damage in Trypanosome Parasites Using γH2A
Published: Vol 14, Iss 13, Jul 5, 2024 DOI: 10.21769/BioProtoc.5026 Views: 2007
Reviewed by: Marcelo S. da SilvaAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jun 2023
Advertisement





Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Diseases caused by trypanosomatid parasites remain a significant unmet medical need for millions of people globally. Trypanosomatid parasites such as Trypanosoma cruzi and subspecies of Trypanosoma brucei cause Chagas disease and human African trypanosomiasis (HAT), respectively. Although efforts to find novel treatments have been successful for HAT, Chagas disease is still treated with decades-old therapies that suffer from long treatment durations and severe safety concerns. We recently described the identification and characterization of the cyanotriazole compound class that kills trypanosomes, in vitro and in vivo, by selective inhibition of the trypanosome nuclear topoisomerase II enzyme. To evaluate whether inhibition of the topoisomerase II enzyme led to parasite death due to lethal double-strand DNA breaks, we developed assays for detecting DNA damage in both intracellular amastigotes of T. cruzi and bloodstream-form T. brucei by using the canonical DNA damage marker γH2A. Herein, this article describes the protocols for detecting DNA damage using an immunofluorescence assessment of γH2A by microscopy in trypanosome parasites.
Key features
• Immunofluorescence-based assay to detect the γH2A response in T. brucei and T. cruzi parasites.
• Robust DNA damage pathway–based cellular assays to evaluate topoisomerase II poisons’ ability to cause DNA damage.
• A 384-well plate–based T. cruzi protocol allows high-resolution and high-throughput evaluation of compounds that cause DNA damage by measuring γH2A in intracellular parasites.
• This assay could be modifiable for evaluation of DNA damage responses in various intracellular and extracellular eukaryotic pathogens.
Keywords: DNA damageBackground
Trypanosomatid parasites cause various medically important diseases including Chagas disease and human African trypanosomiasis, mainly affecting people living in Latin America and sub-Saharan Africa, respectively. Chagas disease is caused by the protozoan parasite Trypanosoma cruzi, which is transmitted to humans through the triatomine bug [1]. Human African trypanosomiasis (HAT) is caused by T. brucei gambiense and T. b. rhodesiense, which are transmitted through the bite of the tsetse fly [2]. Recent efforts have led to the discovery and development of fexinidazole as a novel oral treatment for HAT [3,4], whilst Chagas disease is still treated with nitroheterocyclics that have poor tolerability [1]. New efforts are needed to develop novel therapies to treat Chagas disease that are safer, efficacious, and have the potential for shortened treatment durations.
We described the identification and characterization of a novel cyanotriazole (CT) class of compounds showing promising parasiticidal activity in murine models of Chagas and HAT diseases [5]. Extensive mode of action studies showed that CTs act as trypanosome-specific topoisomerase II (Topo II) poisons. Topoisomerase II poisons act by stabilizing the enzyme in the Topo II-DNA complex after the topoisomerase II enzyme has cleaved the double-stranded DNA. The accumulation of these double-strand DNA breaks due to Topo II poisons ultimately results in cell death (Reviewed in Vann et al. [6]).
In eukaryotes, several DNA repair pathways are known to respond to DNA damage. One of the early responses to double-strand DNA breaks is a phosphorylation in the C-terminal region of histone H2A (H2AX in humans) to generate γH2A(X) [7]. In humans and non-human primates, this phosphorylation occurs on serine-139 of the conserved SQ motif. However, in T. brucei and T. cruzi, the phosphorylation occurs on threonine-130 and threonine-131, respectively. This phosphorylation is involved in signaling cascades to recruit DNA repair machinery to the site of damage. The H2A response can be detected with phospho-antibodies that recognize the specific γH2A-associated phosphorylation of histone H2A(X). Previous work by Glover and Horn identified this response in T. brucei [8], and the antibody was reported to be weakly cross-reactive against T. cruzi epimastigotes by western blot [9]; however, we were unsuccessful in detecting γH2A in T. cruzi using the T. brucei antibody in either epimastigotes or intracellular amastigotes by western blot or immunofluorescence microscopy. Therefore, we designed a new antibody to specifically detect γH2A in T. cruzi. This new antibody, specific for the T. cruzi epitope (Figure 1B), can detect T. cruzi γH2A in both western blots and immunofluorescence assays. Herein, we describe protocols for imaging-based γH2A assays for intracellular T. cruzi and bloodstream-form T. brucei.
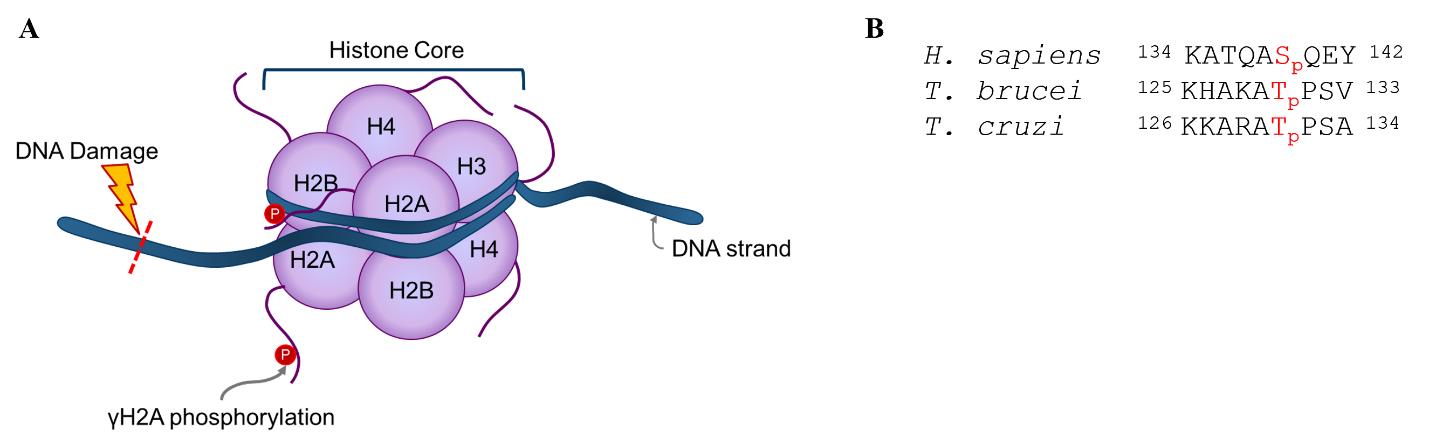
Figure 1. Overview of γH2A response to DNA damage. (A) Graphical representation of γH2A response to DNA damage. The graphic is not to scale. (B) Alignment of γH2A epitope between human (Homo sapiens), Trypanosoma brucei, and Trypanosoma cruzi. Sequences obtained from UniProt (P16104) [10] and TriTrypDB (Tb927.7.2820, TcCLB.508321.21) [11]. Numbering does not include initiator methionine.
Materials and reagents
Biological materials
Vero cells (ATCC CCL-81)
Trypanosoma cruzi CL Brener parasites expressing tdTomato (Tc) [12]
Bloodstream form (BSF) Trypanosoma brucei Plimmer and Bradford (ATCC PRA-382)
Reagents
RPMI 1640 (HyClone, catalog number: SH30027.02)
0.25% Trypsin-EDTA (Gibco, catalog number: 25200056)
Fibronectin powder (Corning, catalog number: 354008)
IMDM powder (Gibco, catalog number: 12200036)
Sodium bicarbonate (Sigma-Aldrich, catalog number: S5761)
Hypoxanthine (Sigma-Aldrich, catalog number: H9636)
Sodium hydroxide (Sigma-Aldrich, catalog number: S8045)
Sodium pyruvate (100 mM) (Gibco, catalog number: 11360070)
Thymidine (Sigma-Aldrich, catalog number: T1895)
L-cysteine (Sigma-Aldrich, catalog number: C7352)
Bathocuproine disulfonic acid (Sigma-Aldrich, catalog number: 146625)
Beta-mercaptoethanol (Millipore-Sigma, catalog number: M6250)
Heat-inactivated fetal bovine serum (FBS) (Gibco, catalog number: 10082147)
Penicillin-Streptomycin (Gibco, catalog number: 15070063)
Penicillin-Streptomycin-Glutamine (100×) (Gibco, catalog number: 10378016)
Phosphate buffered saline (PBS) (Gibco, catalog number: 20012027)
Paraformaldehyde 16% aqueous solution (Electron Microscopy Sciences, catalog number: 15710-S)
Triton X-100 (Sigma, catalog number: X100)
Tween-20 (Sigma, catalog number: P9416)
Bovine serum albumin (BSA) (Sigma, catalog number: A7906-500G)
Primary antibodies:
Rabbit anti-T. cruzi γH2A polyclonal antibody (custom-made by Thermo Fischer Scientific)
i. Epitope: KKARATpPSA
ii. Bleed 3, affinity purified
Rabbit anti-T. brucei γH2A polyclonal antibody (custom-made by Thermo Fischer Scientific)
i. Epitope: KHAKATpPSV
ii. Bleed 2, affinity purified
Secondary antibody: Goat anti-rabbit Alexa Fluor 488 (Abcam, catalog number: 150081)
Hoechst 33258 (Anaspec, catalog number: AS-83219)
VECTASHIELD Vibrance® antifade mounting medium with DAPI (Vector Laboratories, catalog number: H-1800-2)
Dimethyl sulfoxide (DMSO) (Sigma-Aldrich, catalog number: D2650-100ML)
Phleomycin D1 (Zeocin) (Life Technologies, catalog number: R25001)
Compounds (as described in Rao et al. [5]):
GNF6702 (kinetoplastid proteasome inhibitor)
Benznidazole (Sigma-Aldrich, catalog number: 419656)
CT0
CT1
CT3
Solutions
All solutions are stored at 4 °C. Solutions that are made fresh or need to be protected from light are indicated below each recipe.
RPMI complete medium (for Vero cells and T. cruzi parasites) (see Recipes)
8% fixative solution (see Recipes)
0.5% permeabilizing solution (see Recipes)
Blocking buffer 1 (see Recipes)
IFA wash buffer (see Recipes)
T. cruzi primary antibody solution (see Recipes)
T. cruzi secondary antibody solution (see Recipes)
Hoechst dye solution (see Recipes)
HMI-9 complete medium (for T. brucei) (see Recipes)
4% fixative solution (see Recipes)
0.25% permeabilizing solution (see Recipes)
Blocking buffer 2 (see Recipes)
T. brucei primary antibody solution (see Recipes)
T. brucei secondary antibody solution (see Recipes)
Recipes
RPMI complete medium (for Vero cells and T. cruzi parasites)
Reagent Final concentration Amount RPMI 1640 1× 1,000 mL FBS 9% 100 mL Penicillin-Streptomycin 0.9% 10 mL Total n/a 1,110 mL Filter medium through a 0.22 µm, low-protein binding filter.
8% fixative solution
Reagent Final concentration Amount 1× PBS - 15 mL Paraformaldehyde (16%) 8% 15 mL Total n/a 30 mL Make fresh on the day of the experiment.
0.5% permeabilizing solution
Reagent Final concentration Amount 1× PBS 1× 500 mL Triton X-100 0.5% 2.5 mL Total n/a 502.5 mL Blocking buffer 1 (for T. cruzi)
Reagent Final concentration Amount 1× PBS 1× 100 mL BSA 4% 4 g Tween-20 0.1% 0.1 mL Total n/a 100 mL IFA wash buffer
Reagent Final concentration Amount 1× PBS 1× 1,000 mL Tween-20 0.1% 1 mL Total n/a 1,000 mL T. cruzi primary antibody solution
Reagent Final concentration Amount Blocking buffer 1 1× 25 mL Primary antibody 1:250 0.1 mL Total n/a 25 mL Make fresh on the day of the experiment.
T. cruzi secondary antibody solution
Reagent Final concentration Amount Blocking buffer 1 1× 25 mL Secondary antibody 1:1,000 0.025 mL Total n/a 25 mL Make fresh on the day of the experiment, protect from light.
Hoechst dye solution
Reagent Final concentration Amount 1× PBS 1× 25 mL BSA 1% 0.25 g Hoechst 1:250 0.1 mL Total n/a 25 mL Make fresh on the day of the experiment, protect from light.
HMI-9 complete medium for T. brucei
Reagent Final concentration Amount Water n/a 730 mL IMDM powder 1× 1 packet Sodium bicarbonate 0.036 mM 3.02 g Hypoxanthine* 1.0 mM 0.136 g dissolved in 10 mL 0.1 M NaOH Sodium pyruvate 1.0 mM 10 mL Thymidine* 0.160 mM 0.039 g dissolved in 10 mL water L-cysteine* 1.0 mM 0.12 g dissolved in 10 mL water β-mercaptoethanol** 0.2 mM 14 µL diluted in 10 mL water Bathocuproine disulfonic acid* 0.050 mM 0.028 g dissolved in 10 mL water FBS 10% 100 mL Serum plus 10% 100 mL Penicillin-Streptomycin-Glutamine 1% 10 mL Total n/a 1,000 mL *Solid salts are dissolved in 10 mL of water (or 0.1 M NaOH) before adding to medium.
**β-mercaptoethanol is diluted in 10 mL of water before adding to medium. Combine ingredients and mix well. Filter medium through a 0.22 µm, low-protein binding filter.
4% fixative solution
Reagent Final concentration Amount 1× PBS - 12 mL Paraformaldehyde (16%) 4% 4 mL Total n/a 16 mL Make fresh on the day of the experiment.
0.25% permeabilizing solution
Reagent Final concentration Amount 1× PBS 1× 25 mL Triton X-100 0.25% 0.063 mL Total n/a 25 mL Blocking buffer 2 (for T. brucei)
Reagent Final concentration Amount 1× PBS 1× 100 mL BSA 3% 3 g Total n/a 100 mL T. brucei primary antibody solution
Reagent Final concentration Amount Blocking buffer 2 1× 25 mL Primary antibody 1:250 0.1 mL Total n/a 25 mL Make fresh on the day of the experiment.
T. brucei secondary antibody solution
Reagent Final concentration Amount Blocking buffer 2 1× 25 mL Secondary antibody 1:1,000 0.025 mL Total n/a 25 mL Make fresh on the day of the experiment, protect from light.
Laboratory supplies
384-well black, clear and flat-bottom microplates (Greiner, catalog number: 781097)
Tissue culture–treated cell culture flasks with vented caps in T-25, T-75, T-175 sizes (Corning, catalog numbers: 430639, 430641U, and 431306, respectively)
Micropipettes of various sizes and compatible tips (e.g., Rainin, catalog number 30456871)
Neubauer Improved hemocytometer (SKC, Inc. INCYTO C-ChipTM, catalog number: DHC-N015)
0.22 µm vacuum filter and storage bottle (Corning, catalog number: 431205)
15 mL and 50 mL conical tube (VWR, catalog numbers: 21008-216, 21008-242, respectively)
WypAll tissue wipes (WYPALL, catalog number: 34770)
24-well plate (Corning, catalog number: 3524)
1.7 mL microcentrifuge tube (Axygen, catalog number: MCT-175-C-S)
Poly-L-Lysine–coated coverslips, #1 thickness, 12 mm diameter (Corning, catalog number: 354085)
Parafilm (Amcor, catalog number: PM996)
Microscope slides (Propper, catalog number: 15400100)
Sharp-angled tweezers (Excelta, catalog number: 89411-808)
Blunt-end tweezers (Burkle, catalog number: 5386-0104)
KimTech Science KimWipes (Kimberley-Clark Professional, catalog number: 34120)
Immersion oil (Type F, Index = 1.518) (Nikon, catalog number: MXA22168)
Optional: Nalgene wash bottles (Thermo Fischer Scientific, catalog number: 2402-0500)
Optional: Benchtop vacuum aspirator
Equipment
Humidified incubator set at 37 °C, 5% CO2
NucleoCounter (ChemoMetec, NC-200) and counting cassettes: Via1-Cassette (ChemoMetec, model: 941-0012)
Microplate reagent dispenser (Thermo Fisher Scientific Multidrop) and cassettes:
Multidrop standard cassette (Thermo, catalog number: 24072670)
Multidrop small tube cassette (Thermo, catalog number: 24073295)
Benchtop light microscope with 20× (0.3 NA) objective (Zeiss, Primovert, catalog number: 491206-0005-000)
Microplate orbital shaker (Perkin Elmer DELFIA PlateShake, catalog number: 1296-004)
Plate sealer (Agilent PlateLoc Thermal Microplate Sealer)
Fluorescence microscope
ImageXpress Micro Confocal High-Content Imaging System (Molecular Devices)
Ti2-E epifluorescence microscope (Nikon)
Software and datasets
Nucleoview (ChemoMetec, version 1.2.0.0)
MetaXpress (Molecular Devices, LLC., version 6.7.0.211, 8 March 2021)
Prism v9.5.1 (GraphPad, January 26, 2023)
Excel (Microsoft, v2304, 25 April 2023)
NIS Elements AR (Nikon, version 5.20.02)
Procedure
Intracellular T. cruzi
Notes:
Culture Vero cells and parasites in RPMI complete media at 37 °C and 5% CO2 in a humidified incubator.
Utilize Vero cells between passage 2 and 30 for experiments and passage between 70% and 90% confluence.
Viability of Vero cells should be 92%–100% (determined by NucleoCounter).
A1. Vero cell cultureRemove RPMI complete medium from flask.
Add 2.5 mL of trypsin-EDTA and incubate for 6 min at 37 °C and 5% CO2 in a humidified incubator.
Tap the sides of the flask to ensure cell detachment.
Gently pipette up and down to de-clump cells and add to 10 mL of RMPI complete medium in a 15 mL conical tube to collect cells.
Spin down in the conical tube at 130× g for 7 min at room temperature.
Decant off supernatant and loosen pellet by flicking the bottom of the tube or tapping on the biosafety cabinet (BSC) work surface.
Resuspend Vero cells in 5 mL of fresh RPMI complete media.
Count cells on ChemoMetec NucleoCounter and normalize count for viability.
Add cells to prewarmed media. Cell numbers for T-75 flasks (T-25 flask will require 1/3, and T-175 flask will require three times more):
1-day passage: 6 × 106 cells
2-day passage: 3 × 106 cells
3-day passage: 1 × 106 cells
4-day passage: 7 × 105 cells—avoid using these directly for experiments.
A2. T. cruzi parasite culture
Passage T. cruzi parasites every 4 or 5 days in RPMI complete medium.
Seed 3 × 106 Vero cells in a T-175 flask (5-day passage) or T-75 flask (4-day passage) in 40 mL or 20 mL of media, for 5 and 4-day passages, respectively.
Allow cells to adhere for at least 30–60 min at 37 °C and 5% CO2 in a humidified incubator before infection.
Check parasites on the benchtop light microscope; parasites should have burst with plenty of agile, moving trypomastigotes in the culture supernatant.
Holding the flask flat on the BSC work surface, shake flask vigorously 2–3 times and collect supernatant in a 50 mL conical tube by decanting.
Count parasites by hemocytometer, with a 1:10–1:50 dilution of parasites in RPMI complete medium.
For routine passage, infect the T-175 flask containing Vero cells at a multiplicity of infection (MOI) of 5 for 5-day passage or a T-75 flask at MOI of 10 for 4-day passage.
Two to three days post-infection, rinse infected flasks with 40 mL of PBS, and add 25 mL of fresh RPMI complete medium.
A3. Cell seeding, infection, and compound treatment
Standard: tissue culture–treated plastic plates.
Using the standard cassette for a MultiDrop reagent dispenser, seed 5,000 Vero CCl-81 cells in 40 µL of RPMI complete mediuma per well of a 384-well microplate.
Allow Vero cells to adhere to microplate for at least 30–60 min in the incubator.
Note: Make sure all wells have a similar number of cells and that the cells have adhered.
Collect parasites from source flask and count parasite density on hemocytometer ensuring to only count healthy-looking trypomastigotes that are actively moving and no potential amastigotes or other rounded/ unhealthy parasites in the culture.
Infect Vero cells with trypomastigotes at MOI 5 in 10 µL of RPMI complete medium per well using small tube cassette with a MultiDrop reagent dispenser.
Incubate infected microplate for 48 h at 37 °C, 5% CO2.
Add compound to desired concentration.
Appropriate vehicle control should be used. The volume of compound or vehicle added to cultures should be kept consistent.
DMSO tolerability has been tested up to 0.6%. It is recommended to keep DMSO concentrations at or below 0.5%, but 0.6% can be tolerated.
We have typically used compounds dissolved in 100% DMSO at 10 mM stock concentration.
Return microplate to incubator at 37 °C, 5% CO2 and incubate for 24 h.
Alternative: Fibronectin-coated glass plates for high-resolution imaging adapted from Jumani et al. [13].
Add 1 mL (5 mL) of deionized (or MilliQ) water to 1 mg (5 mg) of solid-form fibronectin and leave for 30 min at room temperature to obtain a final concentration of 1 mg/mL. Allow the powder to dissolve on the bench, without disturbing it. Make aliquots and store at -20 °C. Avoid freeze-thaw of the frozen aliquots.
Dilute stock of fibronectin in PBS (without calcium and magnesium) at a dilution of 1:40 to achieve 25 µg/mL. For 384-well plates, coat wells by adding at least 20 µL per well (and at least 50 µL per well for a 96-well plate), making sure that the complete surface area of the plate is covered by the liquid.
Centrifuge for 2–4 mins at 400× g, making sure the liquid covers the surface and there are no air bubbles at the bottom of the plate.
Incubate for 2 h at room temperature.
Keep at 4 °C overnight up to 1 week for longer term storage.
For cell seeding, dump out fibronectin and immediately seed cells as above without allowing wells to dry (less than 5 min); after seeding, centrifuge plates for 2 min at 130× g to ensure proper settling.
Infect, incubate, and add compound to plate as above. All subsequent steps can be carried out in the same manner for both plastic and glass plates, below.
A4. Fixation, permeabilization, and stainingAdd 50 µL of 8% fixative solution to each well.
Incubate plate with fixative solution for at least 15 min at room temperature (or at 4 °C for up to 2 weeks).
Remove fixative solution by dumping microplate contents onto absorbent, disposable wipe (WypAll wipes).
Add 35 µL of 0.5% permeabilization solution.
Centrifuge microplate at 130× g for 3 min to ensure permeabilization solution is fully in contact with cell monolayer.
Incubate microplate for 20 min with permeabilization solution at 37 °C, 5% CO2.
Remove permeabilization buffer and add 40 µL of blocking buffer 1 per well.
Incubate plate for 1 h at room temperature with gentle shaking on a plate shaker (approximately 225 rpm).
Remove blocking buffer 1 and add 40 µL of T. cruzi primary antibody solution per well.
Incubate plate for 1 h at room temperature with gentle shaking on a plate shaker (approximately 225 rpm).
Wash microplate twice with 100 µL of IFA wash buffer per well and add 40 µL of T. cruzi secondary antibody solution per well.
Incubate as above but add an opaque cover to protect the microplate from light.
Wash microplate twice with 100 µL of IFA wash buffer and then add 40 µL of Hoechst dye solution per well.
Incubate plate for 15 min at room temperature with gentle shaking as above and protected from light.
Remove Hoechst dye solution and add 75 µL of 1× PBS.
Dry any potential liquid on top of microplate with a KimWipe. Optional: seal with a light protective seal for storage.
A5. Imaging and analysisAcquire images using an ImageXpress (or similar) confocal microscope with a confocal Acquisition Mode of “Confocal: 50 µm slit.” Select the correct plate type and confirm the dimensions. This should set the initial focus of each well to approximately the center of the well. If not, adjust the plate type and dimensions until this is achieved.
Under Acquisition settings enable Autofocus by selecting Enable laser-based focusing option.
In the Autofocus section, select Configure Laser Setting and set Well to well autofocus to Focus on well bottom option.
Under the Image-based Focusing section, select Algorithm to Standard and a Binning of 1.
For the Initial well for finding sample option, select First well acquired, and set the Number of wells to attempt initial find sample to 3.
Under the Set Autofocus option, select All sites.
For wavelengths, under Number of wavelengths add 3, with TL Legacy Shading Correction Refinement Level of 2. Select wavelengths W1 as DAPI, W2 as FITC, and W3 as TexasRed; the microscope includes standard filter sets for these wavelengths. Determine exposures using positive and negative controls for each wavelength and objective with a Target max intensity set to 33000 and making sure the highest intensity is well within the intensity maximum as visualized by the intensity histogram. Adjust the focus for each wavelength using Calculate Offset function and select the Use Z-stack. For Shading Correction select FL Shading Only option.
Acquire 3 × 3 with a 20 µm spacing (total 9 images per well) around the center of the well in an automated fashion using a 20× (0.95 NA) objective with water immersion. Higher-resolution images can be acquired using a 60× (1.2 NA) objective with water immersion. The higher-resolution images are easier to visualize and quantify.
For higher resolution, glass-bottom plates can be imaged using 60× water or oil or 100× oil objectives.
Bloodstream-form T. brucei
This protocol is modified from Kumar et al. [14] and is adapted from Glover and Horn [8].
B1. Compound treatment
Culture parasites in a humidified incubator at 37 °C, 5% CO2. Maintain parasites in logarithmic growth phase between 5 × 105 and 1 × 106 /mL.
Count parasites by hemocytometer and adjust density to 1 × 106 parasites/mL in HMI-9 complete medium.
Add 1 mL of 1 × 106/mL parasite culture per well of 24-well plates.
Add compound to desired final concentration.
Appropriate vehicle control should be used. The volume of compound or vehicle added to cultures should be kept consistent. If using DMSO, final concentration of DMSO should not exceed 0.5%.
We have typically used compounds dissolved in 100% DMSO at 10 mM stock concentration.
Incubate plates in humidified incubator at 37 °C, 5% CO2 for 4 h.
B2. Fixing and staining
Note: Once parasites are adhered to the coverslips, all dispensing and aspirating steps should be done away from direct contact with the coverslip. Dispense solutions and buffers along the well wall and aspirate from gaps between the coverslip and well walls. Orbital shaker is set at approximately 225 rpm for all shaking steps.
After compound treatment, collect entire well of treated parasites in a microcentrifuge tube.
Pellet parasites at 3,000× g for 5 min at room temperature.
Aspirate supernatant and resuspend pellet in 500 µL of 4% fixative solution.
Incubate in fixative solution for at least 15 min at room temperature or at 4 °C for up to 1 week.
Pellet samples at 3,000× g for 5 min at room temperature.
Carefully aspirate all but approximately 10 µL of the supernatant.
Resuspend parasite pellet in remaining supernatant.
Dot 10 µL of sample onto parafilm fixed to benchtop.
Place poly-L-lysine coverslip on sample, ensuring full coverage of the coverslip and minimizing bubbles. We recommend marking the coverslip with a thin marker before placing it on the sample to differentiate which side has the sample.
Allow sample to adhere to the coverslip for 15 min at room temperature.
Pick up the coverslip and place it sample-side facing up in a new 24-well plate for subsequent staining steps.
Rinse coverslips briefly with PBS, two times. We recommend using a squeeze bottle to dispense PBS; the volume is not particularly important, but it should be enough to completely cover the coverslip.
Add 500 µL of 0.25% permeabilizing solution to the well, shaking gently at room temperature on an orbital shaker.
Aspirate permeabilizing solution and add 500 µL of blocking buffer 2; incubate for 1 h at room temperature with gentle shaking (or overnight at 4 °C).
Aspirate blocking buffer and add T. brucei primary antibody solution; incubate for 1 h at room temperature with gentle shaking.
Wash sample 3× with PBS with 5 min of shaking in-between washing.
Add 500 µL of secondary antibody solution; incubate for 1 h at room temperature with gentle shaking.
Wash sample 3× with PBS with 5 min of shaking in-between washing.
Remove coverslip from 24-well plate. We recommend using sharp-angled tweezers to pry up the coverslip and a second pair to grab the coverslip once it is lifted from the bottom of the plate.
Gently dab off excess liquid from the edges of the coverslip with a Kimwipe.
Dot 3 µL of hard-set DAPI in Vectashield on a microscope slide and place the coverslip sample-side facing down.
Image using the Nikon Eclipse Ti-2E epifluorescence microscope (or equivalent).
Locate parasites in transmitted light in XY and Z-focus using a 40× (0.6 NA) objective. Turn on perfect focus and adjust to bring parasites into focus. Find location of parasites in XY plane and save 3–10 locations of XY.
For the multipoint XY locations selected, set up automated imaging using standard DAPI and FITC filters. Set exposures using DMSO vehicle control and phleomycin positive control. Make sure to not saturate the camera.
Higher-resolution images can be acquired using a 60× (1.4 NA) objective with oil immersion to improve signal-to-background.
Note: Although not necessary, Z-stacks and image deconvolution can be used to further improve resolution and signal for 40× (0.6 NA) and 60× (1.4 NA) objectives.
Data analysis
Data analysis for T. cruzi and T. brucei
Quantify the total number of parasites by counting the number of nuclei for T. brucei and tdTomato-positive parasites for T. cruzi. Perform analysis for at least 200 T. brucei nuclei or T. cruzi parasites per replicate.
For T. cruzi assay, it is essential to make sure host cell monolayers are intact by analyzing nuclei staining. A reduction in host nuclei counts could suggest cytotoxicity and/or technical error (for example, cells being washed off during the staining procedure).
Using the nuclei or parasites as a mask, count the total number of parasites that are positive for γH2A signal. The threshold/background of fluorescence intensity can be set using DMSO treated parasites. Consider parasites with any signal (foci or nuclear-wide) as positive for γH2A.
The above counts can be done manually or using an image analysis software. For manual counting, it is critical that the images are blinded. For example, image acquisition can be done by one person and then image file names changed to a generic name with codes. Image counting will then be performed by another person who only has access to the generic name without information of treatment. The first person can then unblind and analyze the data.
It is critical to run DMSO vehicle (negative control) and phleomycin for T. brucei and CT for T. cruzi as positive controls for each experiment; use these to set exposures.
Anticipated results are shown in Figures 2 and 3 with chemical structure of compounds used shown in Figure 4.
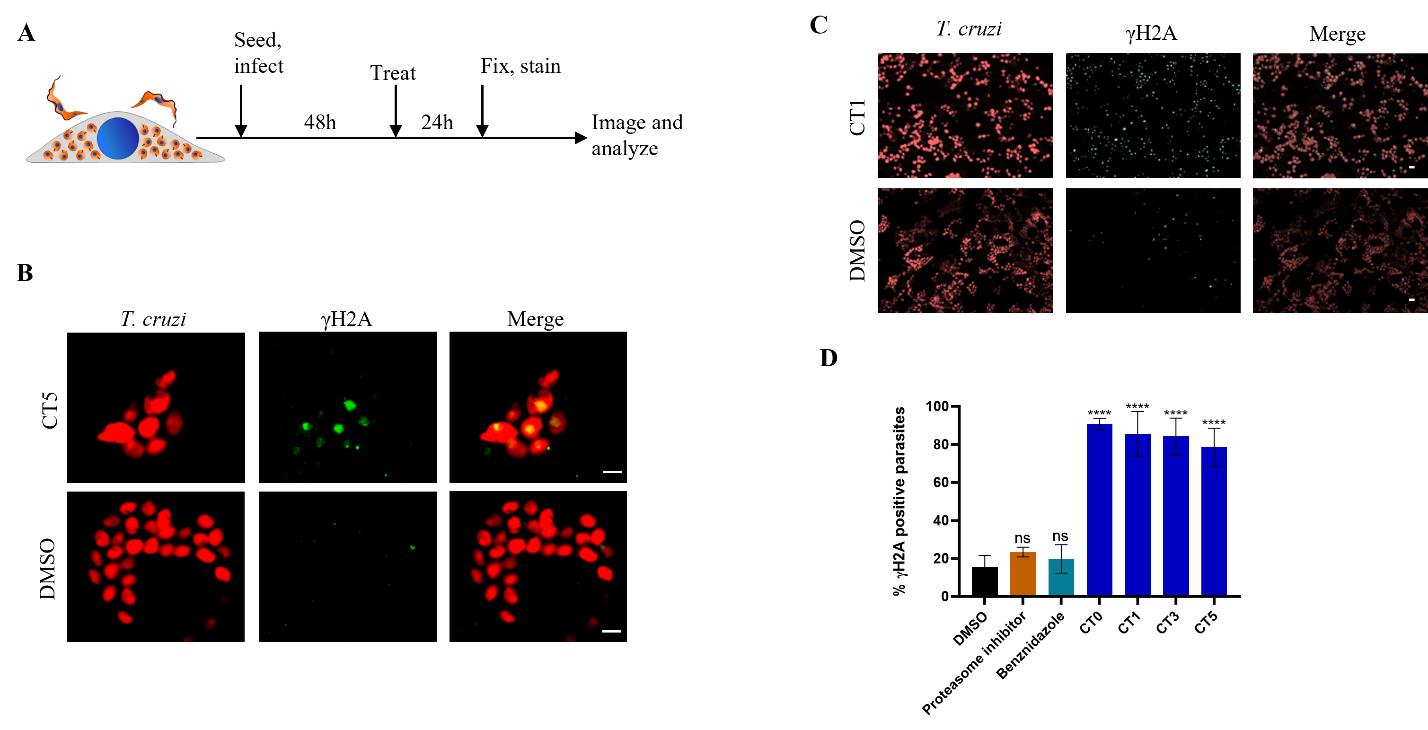
Figure 2. Detection of γH2A response in intracellular T. cruzi amastigotes. (A) Graphical overview of the protocol for γH2A detection in intracellular T. cruzi. Vero host cells were infected with T. cruzi tdTomato trypomastigotes at MOI 5 for 48 h. Cells were then treated with CTs or DMSO for 24 h before fixing with PFA and staining with anti-T. cruzi γH2A antibody. Samples were then imaged on an ImageXpress confocal microscope. (B) Representative images of γH2A response (green) in T. cruzi amastigotes (red) after treatment with DMSO control or CT5 with a 60× water objective (NA = 1.2). Scale bars = 5 µm. (C) Lower resolution representative images acquired with a 20× water objective (NA = 0.95) of γH2A response (green) in T. cruzi intracellular amastigote parasites (red) following treatment with DMSO or 20 µM CT1. Scale bars = 10 µm. (D) Quantitation of γH2A response in T. cruzi amastigotes. Infected cells were treated with DMSO or 20 µM of compounds (proteasome inhibitor is GNF6702, CT0-CT5 are cyanotriazoles). Quantitation was done by counting the total number of tdTomato-positive parasites and γH2A foci per image. Averages of at least three biological replicates, error bars represent standard deviation. Statistics calculated with unpaired parametric student’s t-test (ns = not significant, ****P < 0.0001).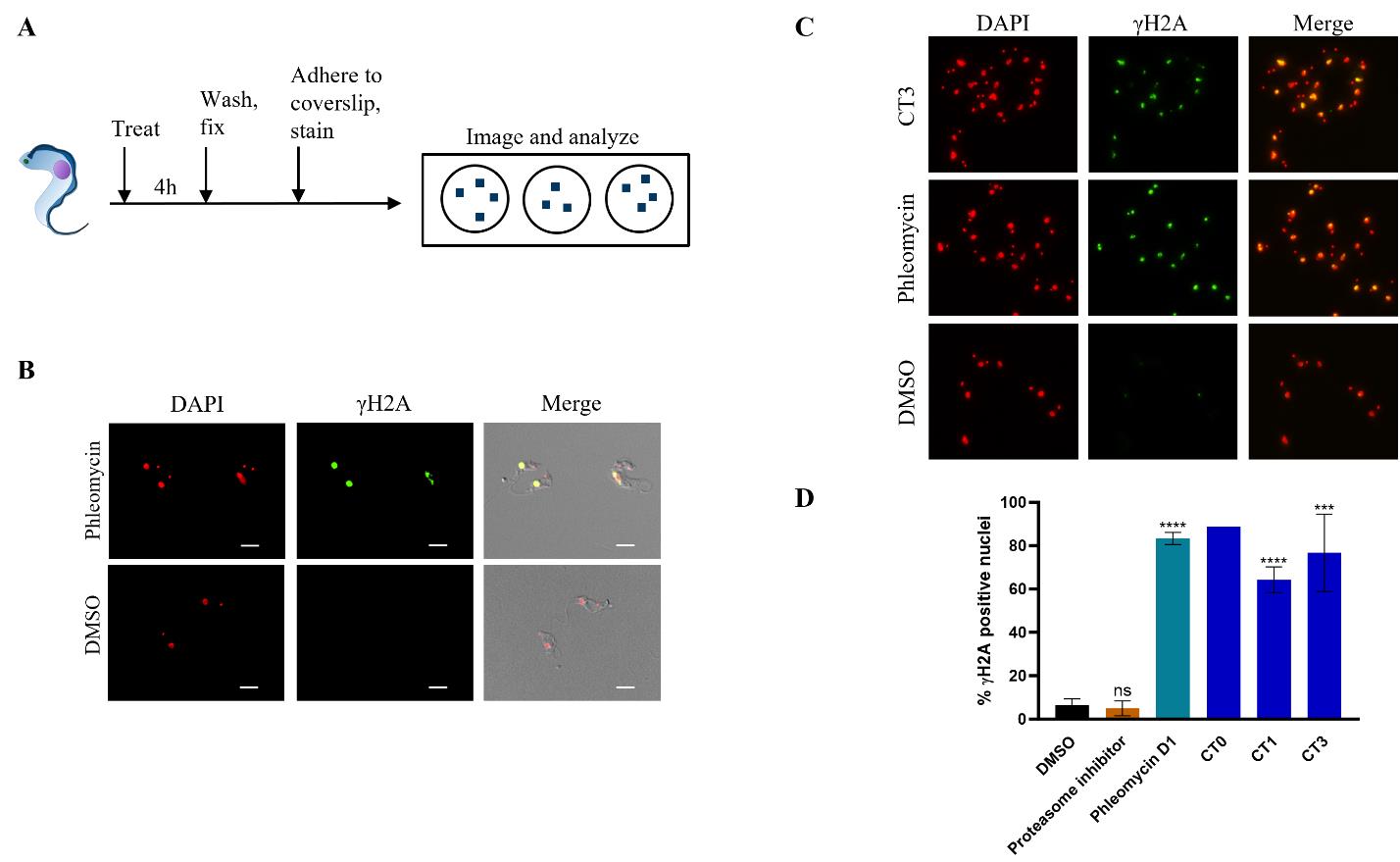
Figure 3. Detecting γH2A response in BSF T. brucei. (A) Graphical overview of the protocol for γH2A detection. T. brucei parasites were seeded in a 24-well plate and treated with CTs, phleomycin, or DMSO for 4 h. Parasites were then collected, fixed with PFA, stained with anti-Tb γH2A antibody, and mounted on slides with DAPI in mounting medium. Samples were imaged on a Nikon Ti2-E microscope. (B) Representative images of T. brucei parasites treated with phleomycin D1 or DMSO. Images were acquired using a 60× oil objective (NA = 1.4). Scale bars = 5 µm. (C) Representative images of T. brucei parasites after 4 h treatment with DMSO, phleomycin D1, or CT3. Images were acquired using a 40× air objective (NA = 0.6). (D) Quantitation of γH2A response in T. brucei treated with various compounds (proteasome inhibitor is GNF6702; CT0, CT1, and CT3 are cyanotriazoles) Quantitation was carried out by counting total nuclei and γH2A foci per image and calculating the percentage of nuclei positive for γH2A. Data are mean and standard deviation of at least two biological replicates (CT0, n = 1). Statistics calculated with unpaired, parametric student’s t-test (ns = not significant, ****P < 0.0001, ***P = 0.0002).
Figure 4. Chemical structures of compounds used in the study. GNF6702 is kinetoplastid proteasome inhibitor, CT0-CT5 are cyanotriazoles, Phleomycin is a known T. brucei DNA damaging agent, benznidazole is an approved drug for Chagas disease.
Validation of protocol
This protocol has been used and validated in the following research article:
Rao et al. [5]. Cyanotriazoles are selective topoisomerase II poisons that rapidly cure trypanosome infections. Science (Figure 3, panels B and C).
General notes and troubleshooting
Once microplates are stained and sealed, they can be stored at 4 °C for 1–2 months. It is critical that the wells do not dry during storage.
The protocol has been developed using T. cruzi CL Brener parasites. The protocol can be adapted to other parasite strains; however, the kinetics of γH2A induction could be different. We also note differences in the speed and duration of the γH2A response to different DNA-damaging agents, so some optimization for specific treatments may need to be made. Treatment times indicated in these protocols were optimized for parasite topoisomerase poisons and cyanotriazole (CT) compounds.
For T. cruzi, in place of 384-well microplates, 35 mm dishes (MatTek Corporation, catalog number: P35G-1.5-14-C), 96-well (or any other size well) plates, or similar can be used for the protocol using fibronectin coating and cell seeding methods as described in Jumani et al. [13]. For each vessel, cell numbers and volumes should be adjusted for surface area and height, respectively, based on the manufacturer’s recommendation.
Seeding of microplates can be done manually with single or multichannel pipettes, taking care to ensure the cell suspension is uniform during seeding, as this will enable equal distribution of cells between wells.
Washing steps for microplate-based assays can be automated with a BioTek EL406 or similar plate washer. For EL406, use slow mode, optimize protocol (for example, height, depth, and angle of the washer pins) to aspirate as much liquid as possible without disrupting monolayer, and dispense at the walls of the wells and not directly on the cells. Alternatively, washing can be done with a Multidrop or multichannel (or single channel) pipette, dispensing with a slow setting and dumping out liquid manually by inverting the plate and dabbing on absorbent laboratory wipe. Make sure to dispense liquid to the walls of the vessel and not directly onto the cells.
Image analysis can be performed using software like ImageJ/Fiji (can be adapted from Jumani et al. [13] altering the thresholding and size parameters), CellProfiler, Ilastik, or in-built microscope imaging software like NIS Elements or Molecular Devices.
For T. cruzi, it is critical to add PFA directly to cells before washing. Performing washing before fixing can cause Vero cell monolayers to disassociate.
The γH2A response in T. cruzi epimastigotes was validated with classical, non-CT DNA damaging agents hydroxyurea and phleomycin by western blot.
Acknowledgments
This work is part of the Rao et al. [5] manuscript published in Science, 2023. This research work was funded by Novartis BioMedical Research, and in part by the Wellcome Trust (Project Number: 219639/Z/19/Z). The authors thank Manuel Saldivia for coordinating production of T. brucei γH2A antibody, Debjani Patra for technical support, and to Manuel Saldivia, Ujjini H. Manjunatha, Jonathan E. Gable, Yen-Liang Chen, and the rest of the Novartis team for insightful discussions. We thank Gu Feng, Thomas Krucker, Jean Claude Poilevey, Emily Tongco-Wu and team for project management, alliance management and partnering, and legal and finance support.
Competing interests
All authors are Novartis employees, and some own shares in Novartis. CT compounds have been patented by Novartis with S.P.S.R. listed as an author (US patent application no. 17/253,737,2022).
Ethical considerations
No human or animal subjects were included in this study.
References
- de Sousa, A. S., Vermeij, D., Ramos, A. N. Jr, and Luquetti, A. O. (2024). Chagas disease. Lancet. 403(10422): 203–218.
- Büscher, P., Cecchi, G., Jamonneau, V. and Priotto, G. (2017). Human African trypanosomiasis. Lancet. 390(10110): 2397–2409.
- Mesu, V. K. B. K., Kalonji, W. M., Bardonneau, C., Mordt, O. V., Blesson, S., Simon, F., Delhomme, S., Bernhard, S., Kuziena, W., Lubaki, J. P., et al. (2018). Oral fexinidazole for late-stage African Trypanosoma brucei gambiense trypanosomiasis: a pivotal multicentre, randomised, non-inferiority trial. Lancet. 391(10116): 144–154.
- Betu Kumeso, V. K., Kalonji, W. M., Rembry, S., Valverde Mordt, O., Ngolo Tete, D., Prêtre, A., Delhomme, S., Ilunga Wa Kyhi, M., Camara, M., Catusse, J., et al. (2023). Efficacy and safety of acoziborole in patients with human African trypanosomiasis caused by Trypanosoma brucei gambiense: a multicentre, open-label, single-arm, phase 2/3 trial. Lancet Infect Dis. 23(4): 463–470.
- Rao, S. P. S., Gould, M. K., Noeske, J., Saldivia, M., Jumani, R. S., Ng, P. S., René, O., Chen, Y. L., Kaiser, M., Ritchie, R., et al. (2023). Cyanotriazoles are selective topoisomerase II poisons that rapidly cure trypanosome infections. Science. 380(6652): 1349–1356.
- Vann, K. R., Oviatt, A. A. and Osheroff, N. (2021). Topoisomerase II Poisons: Converting Essential Enzymes into Molecular Scissors. Biochemistry. 60(21): 1630–1641.
- Kinner, A., Wu, W., Staudt, C. and Iliakis, G. (2008). γ-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res. 36(17): 5678–5694.
- Glover, L. and Horn, D. (2012). Trypanosomal histone γH2A and the DNA damage response. Mol Biochem Parasitol. 183(1): 78–83.
- Gomes Passos Silva, D., da Silva Santos, S., Nardelli, S. C., Mendes, I. C., Freire, A. C. G., Repolês, B. M., Resende, B. C., Costa-Silva, H. M., da Silva, V. S., Oliveira, K. A. d., et al. (2018). The in vivo and in vitro roles of Trypanosoma cruzi Rad51 in the repair of DNA double strand breaks and oxidative lesions. PLoS NeglTrop Dis. 12(11): e0006875.
- UniProt, C. (2023). UniProt: the Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 51(D1): p. D523–D531.
- Amos, B., Aurrecoechea, C., Barba, M., Barreto, A., Basenko, E. Y., Bażant, W., Belnap, R., Blevins, A. S., Böhme, U., Brestelli, J., et al. (2021). VEuPathDB: the eukaryotic pathogen, vector and host bioinformatics resource center. Nucleic Acids Res. 50: D898–D911.
- Canavaci, A. M. C., Bustamante, J. M., Padilla, A. M., Perez Brandan, C. M., Simpson, L. J., Xu, D., Boehlke, C. L. and Tarleton, R. L. (2010). In Vitro and In Vivo High-Throughput Assays for the Testing of Anti-Trypanosoma cruzi Compounds. PLoS NeglTrop Dis. 4(7): e740.
- Jumani, R. S., Hasan, M. M., Stebbins, E. E., Donnelly, L., Miller, P., Klopfer, C., Bessoff, K., Teixeira, J. E., Love, M. S., McNamara, C. W., et al. (2019). A suite of phenotypic assays to ensure pipeline diversity when prioritizing drug-like Cryptosporidium growth inhibitors. Nat Commun. 10(1): 1862.
- Kumar, G., Thomas, B. and Mensa-Wilmot, K. (2022). Pseudokinase NRP1 facilitates endocytosis of transferrin in the African trypanosome. Sci Rep. 12(1): 18572.
Article Information
Publication history
Received: Feb 21, 2024
Accepted: May 30, 2024
Available online: Jun 24, 2024
Published: Jul 5, 2024
Copyright
© 2024 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Jumani, R. S., Thomas, B. and Rao, S. P. S. (2024). Imaging Assays to Detect DNA Damage in Trypanosome Parasites Using γH2A. Bio-protocol 14(13): e5026. DOI: 10.21769/BioProtoc.5026.
Rao, S. P. S., Gould, M. K., Noeske, J., Saldivia, M., Jumani, R. S., Ng, P. S., René, O., Chen, Y. L., Kaiser, M., Ritchie, R., et al. (2023). Cyanotriazoles are selective topoisomerase II poisons that rapidly cure trypanosome infections. Science. 380(6652): 1349–1356.
Category
Microbiology > Microbial cell biology > Cell staining
Cell Biology > Cell imaging > Confocal microscopy
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.