- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

A New Approach for Assessment of Neutrophil Extracellular Traps Through Immunofluorescence Staining in Whole Blood Smears

Published: Vol 14, Iss 11, Jun 5, 2024 DOI: 10.21769/BioProtoc.5010 Views: 3340
Reviewed by: Pilar Villacampa AlcubierreSaskia F. Erttmann
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Neutrophils, constituting 50%–70% of circulating leukocytes, play crucial roles in host defense and exhibit anti-tumorigenic properties. An elevated peripheral blood neutrophil-to-lymphocyte ratio is associated with decreased survival rates in cancer patients. In response to exposure to various antigens, neutrophils release neutrophil granular proteins, which combine to form web-like structures known as neutrophil extracellular traps (NETs). Previously, the relative percentage of NETs was found to be increased in resected tumor tissue samples from patients with gastrointestinal malignancies. The presence of NETs in peripheral blood is indicative of underlying pathological conditions. Hence, employing a non-invasive method to detect NETs in peripheral blood, along with other diagnostic tests, shows potential as a valuable tool not just for identifying different inflammatory disorders but also for assessing disease severity and determining patient suitability for surgical resection. While reliable methods exist for identifying NETs in tissue, accurately quantifying them in whole blood remains challenging. Many previous methods are time-consuming and rely on a limited set of markers that are inadequate for fully characterizing NETs. Therefore, we established a unique sensitive smear immunofluorescence assay based on blood smears to identify NETs in only as little as 2 μL of whole blood. To identify the NET complexes that have enhanced specificities, this combines the use of various antibodies against neutrophil-specific CD15, NET-specific myeloperoxidase (MPO), citrullinated histone H3 (Cit H3), and nuclear DNA. This protocol offers an easy, affordable, rapid, and non-invasive method for identifying NETs; thus, it can be utilized as a diagnostic marker and targeted through various therapeutic approaches for treating human malignancies.
Key features
• Characterization of neutrophil extracellular traps in whole blood smears through immunofluorescence staining.
• Affordable and quantitative approach to neutrophil extracellular trap detection.
Keywords: NETosisGraphical overview
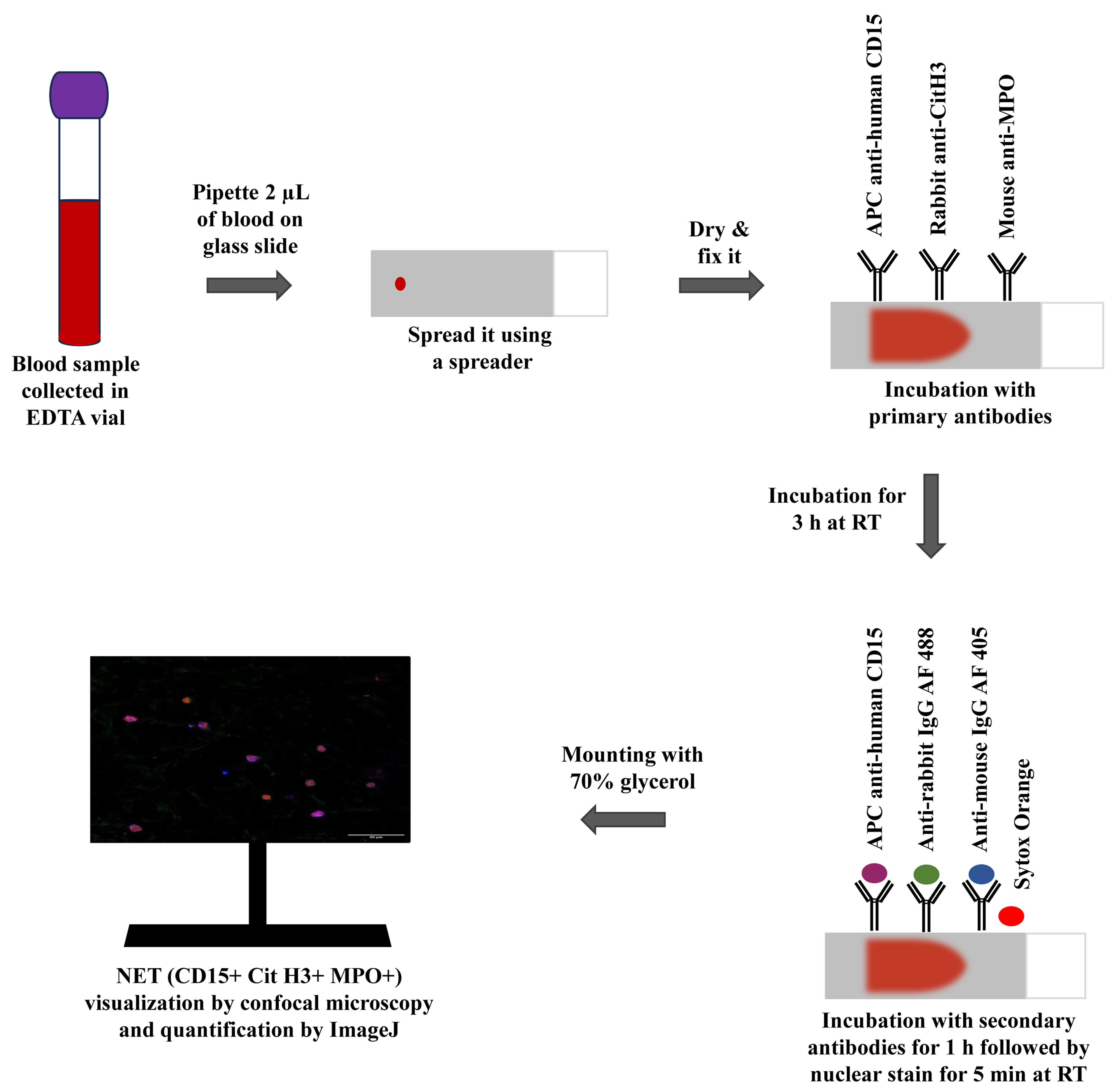
Graphical representation of the immunofluorescence-based blood smear assay to characterize neutrophil extracellular traps.
Background
Cancer is a serious global public health issue, and an upsurge in advanced-stage disease and mortality may result from delays in diagnosis and treatment [1]. Pancreatic ductal adenocarcinoma (PDAC), the most prevalent histologic variant of pancreatic cancer (PC), is considered one of the most aggressive solid tumors, having a high metastatic potential with a 5-year survival rate of less than 5% [2]. According to estimates, PDAC will overtake the principal cause of cancer mortality by 2030, ranking third globally in terms of cancer-associated fatalities [2,3]. It is currently widely acknowledged that immunological dysfunction and carcinogenesis are intimately related [4]. The most prevalent type of immune cell, neutrophils, comprising approximately 50%–70% of all circulating leukocytes, are essential for both the inflammatory and immune responses to solid tumors [5]. Neutrophils are responsible for maintaining the immune system of the host and provide not only the defense against foreign antigens but also anti-tumorigenic properties. The behavior of neutrophils can vary depending on the various stimuli: N1 neutrophils show pro-inflammatory and anti-tumorigenic properties, while N2 neutrophils show anti-inflammatory and pro-tumorigenic properties. According to recent research, a high peripheral blood neutrophil-to-lymphocyte ratio is associated with a poorer probability of survival for cancer patients [6]. Neutrophils, upon activation by cytokines or a variety of stimuli in the tumor microenvironment, undergo a cellular response called neutrophil extracellular trap (NET) formation (NETosis), in which DNA, along with histone and granule proteins, are extruded into the tissue surroundings and ultimately come into circulation [7]. A crucial enzyme in the production of NETs is peptidyl arginine deiminase 4 (PADI4), which catalyzes the deamination of arginine on histone H3 to induce chromatin decondensation and subsequent DNA extrusion [8]. The proteins that constitute NETs vary depending on the stimulus; nevertheless, the NET core signature proteins are histones, neutrophil elastase (NE), and myeloperoxidase (MPO), which are present regardless of the stimulus [9].
NETosis is a physiological phenomenon in which NETs are formed from neutrophils. Physiologically, NETs were found to be involved in host immune defense against fungi, parasites, and viruses [7]. However, studies on a variety of malignancies have revealed that NETs are crucial to the development of tumors [8]. Previously, the relative percentage of NETs was found to be increased in resected tumor tissue samples from patients with gastrointestinal malignancies [10,11]. Through the promotion of the epithelial-to-mesenchymal transition (EMT), NETs enhanced the ability of cancer cells to migrate and invade [12]. According to recent research, NETs trap circulating tumor cells, which promotes the growth and metastasis of the primary tumor, being directly correlated with the burden of metastatic disease [13,14]. Through the trapping of cancer cells and the reawakening of dormant cells through ECM remodeling, NETs may conspire to promote cancer recurrence [15]. NETs have also been observed to aid in the advancement of PC patients' tumor angiogenesis, venous thrombosis, and inhibition of apoptosis [16,17]. Overall, these findings indicate that NET detection could be significant for cancer diagnosis and prognosis.
Clinical evidence suggests that tumor-associated neutrophils (TANs) correlate with poor prognosis, and the tumor microenvironment plays a crucial role in controlling neutrophil recruitment. However, the extent of infiltration of neutrophils and their differentiation into NETs in the tumor microenvironment in a variety of cancers including PDAC remains unexplored [9]. Therefore, the need of the hour is to gain insight into the efficient characterization of NETs, so that their role in tumor progression can be deciphered. The purpose of this work was to identify NETs in the peripheral blood of PDAC patients. Detecting NETs in peripheral blood may indicate underlying pathological conditions. Thus, using a non-invasive approach to identify NETs, alongside other diagnostics, holds promise for identifying inflammatory disorders, assessing disease severity, and determining suitability for surgical resection. To that end, a unique smear immunofluorescence assay was established, which can identify NETs in only as little as 2 μL of blood. This study could aid in developing a diagnostic biomarker for various diseases and a therapeutic approach to boost immunotherapy following curative cancer excision.
Materials and reagents
APC anti-human CD15 (SSEA-1) (BioLegend, catalog number: 323008)
Mouse monoclonal antibody [2C7] to myeloperoxidase (MPO) (Abcam, catalog number: 25989)
Rabbit polyclonal antibody to histone H3 (anti-Cit H3 citrulline R2 + R8 + R17) (Abcam, catalog number: 5103)
Goat polyclonal antibody to rabbit IgG Alexa Fluor 488 (2 mg/mL) (Abcam, catalog number: ab150077)
Goat polyclonal antibody to mouse IgG Alexa Fluor 405 (2 mg/mL) (Abcam, catalog number: ab175660)
Sytox Orange nucleic acid stain, 5 mM solution in DMSO (Invitrogen, catalog number: S11368)
10× phosphate buffered saline (PBS), pH 7.2 (HiMedia, catalog number: TL1032-500mL)
Tween 20 (Sigma, catalog number: 9005-64-5)
Triton X-100 (HiMedia, catalog number: MB031-500mL)
Bovine serum albumin (BSA) (HiMedia, catalog number: MB083-100g)
Paraformaldehyde (PFA) (Nice Chemicals, catalog number: P64929)
Glycerol (≥ 99.5%) (HiMedia, catalog number: MB060)
Ultra-pure distilled water
Solutions
Wash buffer (see Recipes)
Fixing buffer (see Recipes)
Dilution buffer (see Recipes)
Permeabilization buffer (see Recipes)
Blocking buffer (see Recipes)
70% glycerol (see Recipes)
Recipes
Wash buffer (1× PBS)
Reagent Final concentration Quantity or Volume 10× PBS 1× PBS 5 mL Distilled water ~ 45 mL HCl 1 M Mix 5 mL of 10× PBS with 40 mL of distilled water. Adjust to pH 7.2 with 1 M HCl. Adjust the final volume up to 50 mL with distilled water. Store wash buffer at 4 °C.
Fixing buffer
Reagent Final concentration Quantity or Volume PFA 4% 10 g 1× PBS NA ~250 mL NaOH 1 M Dissolve 10 g of PFA in 200 mL of 1× PBS in a heating and stirring block (60 °C) and cool down upon complete dissolving of PFA. Add 1 M NaOH solution (4 g of NaOH pellets dissolved in 100 mL of distilled water) dropwise to adjust to pH 6.9. Adjust the final volume up to 250 mL with 1× PBS. Store aliquots at 4 °C (see General note 1).
Dilution buffer
Reagent Final concentration Quantity or Volume BSA 1% 0.5 g 1× PBS NA 50 mL Gently mix the solution by inverting the reaction tube. Sterile filter with 0.22 µm pore microfilter. Store at 4 °C.
Permeabilization buffer
Reagent Final concentration Quantity or Volume 1× PBS NA 100 mL Tween 20 0.1% 100 µL Triton X-100 0.5% 500 µL Prepare this solution in the dark and mix well. Store at 4 °C in air-tight amber bottles, as Tween 20 and Triton X-100 are light-sensitive.
Blocking buffer
Reagent Final concentration Quantity or Volume BSA 5% 0.5 g 1× PBS NA 10 mL Gently mix the solution by inverting the reaction tube. Sterile filter with 0.22 µm pore microfilter. Store at 4 °C.
70% glycerol
Reagent Final concentration Quantity or Volume Glycerol 70% 35 mL 1× PBS NA 15 mL Prepare 70% glycerol solution by mixing 35 mL of glycerol with 15 mL of 1× PBS. Use pH paper to ensure a pH of ~7.4. Acidic glycerol will cause rapid fading of fluorochromes. Store at 4 °C in air-tight amber bottles, as glycerol is light sensitive.
Laboratory supplies
15 mL conical centrifuge tubes (Tarsons, catalog number: 546021)
50 mL conical centrifuge tubes (Tarsons, catalog number: 546041)
BD Vacutainer K2EDTA vials (BD, catalog number: 454020)
BD EmeraldTM single-use syringe, 5 mL (BD, catalog number: 307725)
LifeLongTM 24 G needle, 0.55 mm × 25 mm (Lifelong, catalog number: 021824-H)
Microscope slides, 75 mm long × 25 mm wide, thickness 1.35 mm (Blue Star, catalog number: PIC-1)
Microscopic cover glass, rectangular (Special) 22 mm × 60 mm (Blue Star, catalog number: 5128789)
PierceTM microcentrifuge tubes, 1.5 mL (Thermo Scientific, catalog number: 69715)
PierceTM microcentrifuge tubes, 2.0 mL (Thermo Scientific, catalog number: 69720)
Syringe-driven filters, filter pore size 0.22 µm, diameter 30 mm (HiMedia, catalog number: SF137-100NO)
Advanced PAP Pen, 5 mm tip width (Sigma, catalog number: Z377821)
Borosilicate glass graduated round reagent bottles with screw caps (Borosil, catalog number: 1519)
Graduated cylinder (Borosil, catalog number: 3021)
Humidifier slide chamber (we used customized humidifier slide chamber of dimensions 35 cm × 30 cm × 6 cm from Seven Star Scientific Instruments. Researchers can also use Evergreen Scientific slide moisture chamber of dimensions 81/4×7×11/4" from VWR, catalog number: 76278-832) (see General note 4)
Qualigens Labolene Neutral pH 5 L (Thermo Fisher Scientific, catalog number: Q42218)
Sterile pipette tips 0.5–10 µL, 1–200 µL, 100–1,000 µL volume range (Tarsons)
Equipment
Micro-pipettes 0.5–10 µL, 10–100 µL, 20–200 µL, 100–1,000 µL (Eppendorf)
Class II biological safety cabinet (ESCO Lifesciences, model: AC2-4S8-NS)
SPINOTTM Digital Magnetic Stirrer Hot Plate (Tarsons, model: MC 02, Catalogue number: 6040)
Olympus Fluoview confocal laser scanning microscope (Olympus, model: FV3000)
RO-DI Ultra (Rions Labpure water solutions, model: ASTM Type I)
Software and datasets
ImageJ; Java-based program (1.5.4) (https://imagej.nih.gov/ij/download.html)
Microsoft Office Professional Plus 2019
Procedure
Collect pre-operative blood samples of PDAC patients and healthy controls in EDTA vials (all the patients/participants provided written, informed consent for their participation in this study).
With the help of a micro-pipette, put a sample of 2 μL of whole blood on a glass slide cleaned with 90% ethanol. Place the blood sample 1 cm away from one edge of the glass slide as illustrated in Figure 1.
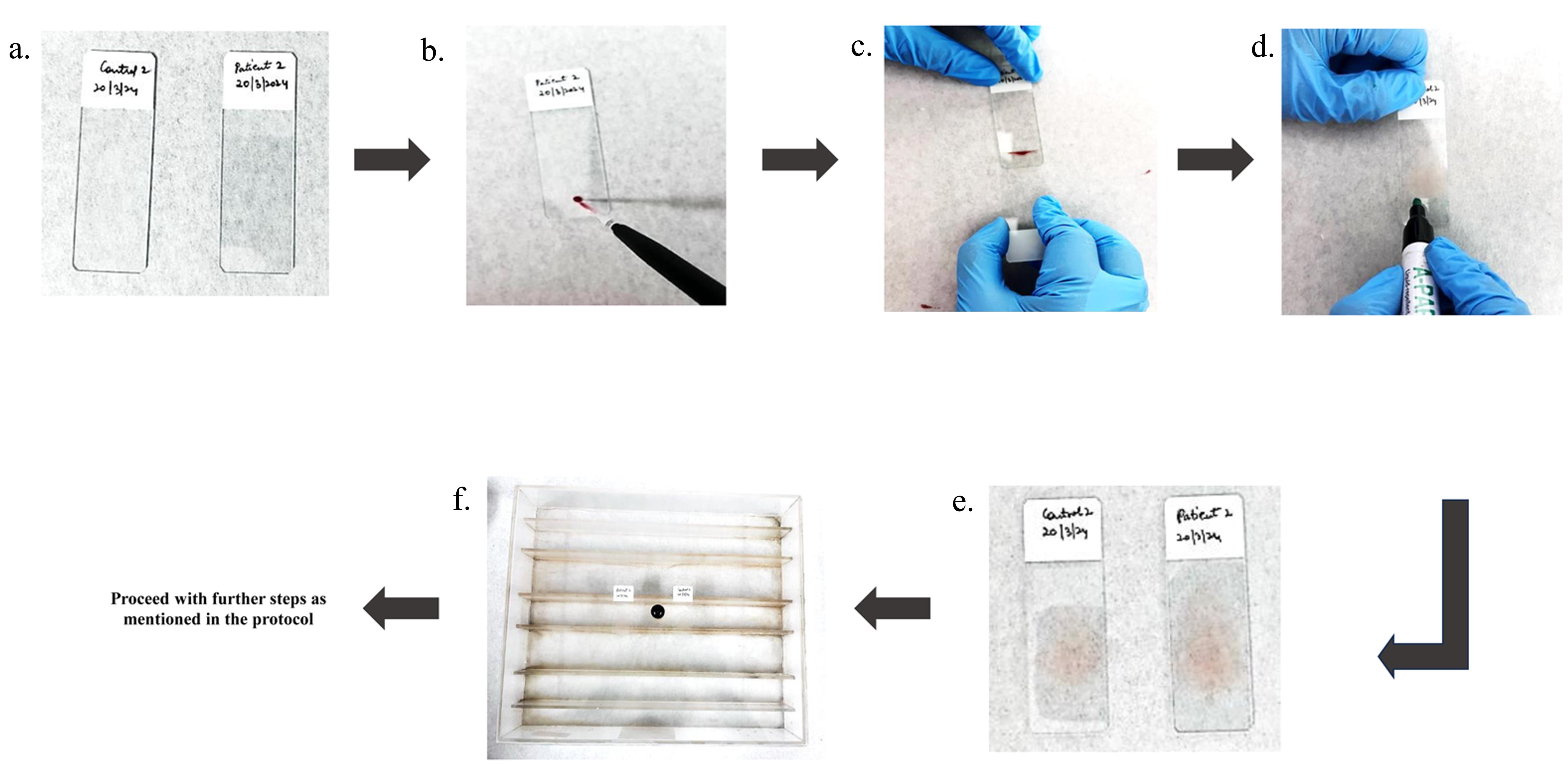
Figure 1. Stepwise protocol for whole-blood smear preparation. a. Clean the slides with 90% alcohol. b. Place 2 μL EDTA-treated blood on a slide with pipette. c. Position the spreader at a 45° angle on the blood drop and through a capillary mechanism, blood will spread along the edge in contact with the spreader. Move the spreader forward quickly, straight, and smoothly along the length of the slide. d. Make the boundary with advanced PAP pen around the smear to prevent the buffers spill out from the slide. e. Permit the smear to air-dry for 15 min. f. Position the slide above the racks of humidifier slide chamber.Position the spreader at a 45° angle in front of the blood drop. Push it back until it makes contact with the blood. Through a capillary mechanism, blood will spread along the edge in contact with the spreader. Move the spreader forward quickly, straight, and smoothly along the length of the slide (see General note 2).
Create a perimeter using the hydrophobic advanced PAP pen around the smear to prevent the spilling out of buffers from the slide. Allow the smear to air-dry naturally for 15 min at room temperature. Be cautious of excessive air-drying, as it can damage proteins and cellular structures, potentially affecting immunodetection outcomes.
Apply 200 μL of fixing buffer onto the smear using a micro-pipette to fix the samples and place them in a humidifier slide chamber for 10 min (see General note 3).
Tilt the slide gently to allow the excess PFA liquid to drain into the waste container. Following this, wash the slide three times with wash buffer (1 mL each time) and dispose of these washes in the same waste container to ensure proper disposal of toxic PFA residues.
Apply 200 μL of APC-labeled CD15 antibody diluted with dilution buffer and incubate in the dark for 30 min at room temperature in a humidifier slide chamber. Remove excess antibody by rinsing the slides three times with 1 mL of wash buffer and gently tilt the slides to discard the wash liquid into the waste container (see General note 4).
Apply 200 μL of permeabilization buffer to permeabilize the slides for 30 min at room temperature in the dark and wash the slides with 1 mL of wash buffer three times.
Apply 200 μL of blocking buffer on the smear for 30 min at room temperature in the dark to block the sections and then rinse the slides three times with 1 mL of wash buffer.
Apply 200 μL of diluted (as listed in Table 1) anti-MPO and anti-Cit H3 primary antibodies using the dilution buffer. Let the slides incubate in the dark for 3 h at room temperature in a humidifier slide chamber to avoid the tissue drying out and then wash the slides three times with 1 mL of wash buffer.
Table 1. Stock and working volume of antibodies
Antibody Stock solution Working solution APC-labeled CD15 1 mg/mL 1:200 Cit H3 1 mg/mL 1:500 MPO 1 mg/mL 1:500 Goat anti-rabbit Alexa Fluor 488 2 mg/mL 1:1,000 Goat anti-mouse Alexa Fluor 405 2 mg/mL 1:1,000 Sytox Orange 5 mM 1:1,500 Apply 200 μL of goat anti-rabbit Alexa Fluor 488 and goat anti-mouse Alexa Fluor 405 secondary antibodies diluted 1:1,000 in dilution buffer. Let the slides incubate in the dark for 1 h at room temperature in a humidifier slide chamber and wash the slides three times with 1 mL of wash buffer.
Stain slides with 5 mg/mL of Sytox Orange diluted 1:1500 in wash buffer for 5 min in the dark at room temperature. After staining, wash three times using 1 mL of wash buffer.
Apply a drop (20–30 μL) of 70% glycerol to the edge of the smear in the dark and then gently lower the coverslip onto the slide from that side, making sure there are no bubbles (see General note 5).
Let the slides dry for at least 15 min in a dark environment. Then, proceed with imaging of the slides using the Olympus Fluoview FV3000 confocal microscope.
Controls
Two types of negative controls can be utilized for quantifying positive staining of NETs:
1. Isotype control antibodies (mouse IgG and rabbit IgG) as a negative control in place of anti-MPO, and anti-citrullinated Histone 3 for primary staining at similar doses. As an alternative, incubate the slides in the host-specific serum in place of the primary antibodies (such as mouse and rabbit sera against MPO and citrullinated Histone 3 antibodies). The secondary staining is performed as described above.
2. Stain secondary antibodies in a manner akin to this, but without the addition of primary antibodies.
Data analysis
Image acquisition
Acquire images with a confocal microscope (Olympus Fluoview FV3000 in this case). Choose the suitable lasers; in this instance, 405 nm (MPO), 488 nm (Cit H3), 650 nm (CD15), and 555 nm (Sytox Orange). For identification of stained structures, adjust the voltages of the laser. Make sure staining is specific by comparing positive samples to control samples. After the lasers have been adjusted, utilize the same parameters for each slide you need to compare and examine. Extracellular structures that stain positively for CD15, MPO, and Cit H3 are referred to as NETs, illustrated in Figures 2 and 3.
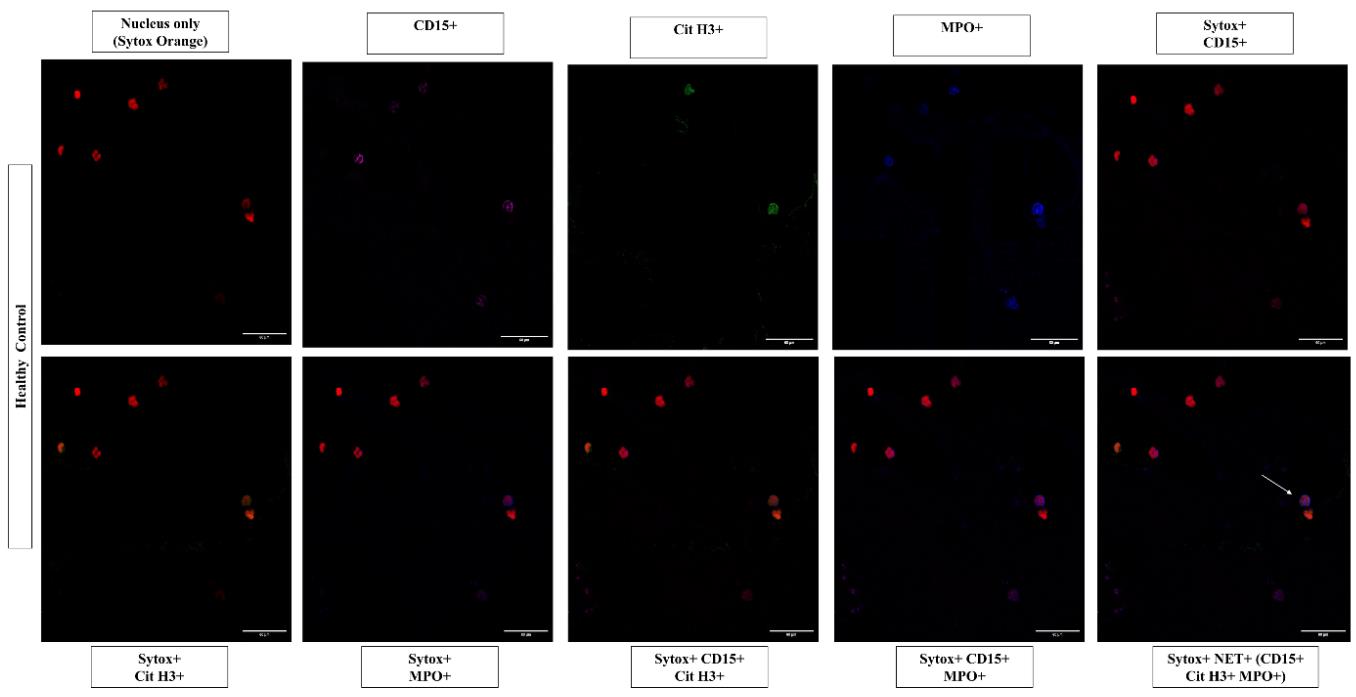
Figure 2. Representative images of neutrophil extracellular traps in peripheral blood smears of healthy controls. Individual images represent the positive staining for nuclear binding dye Sytox Orange (555 nm, red), CD15 (650 nm, magenta), MPO (405 nm, blue), Cit H3 (488 nm, green), and other combinations of CD15, Cit H3, and MPO, along with nuclear dye. Neutrophil extracellular traps are identified as extracellular structures where nuclear binding dye Sytox Orange (555 nm, red), colocalized with CD15 (650 nm, magenta), MPO (405 nm, blue), and Cit H3 (488 nm, green) in the merged image as indicated by the white arrows. Images acquired at 40× magnification, scale bars = 60 µm.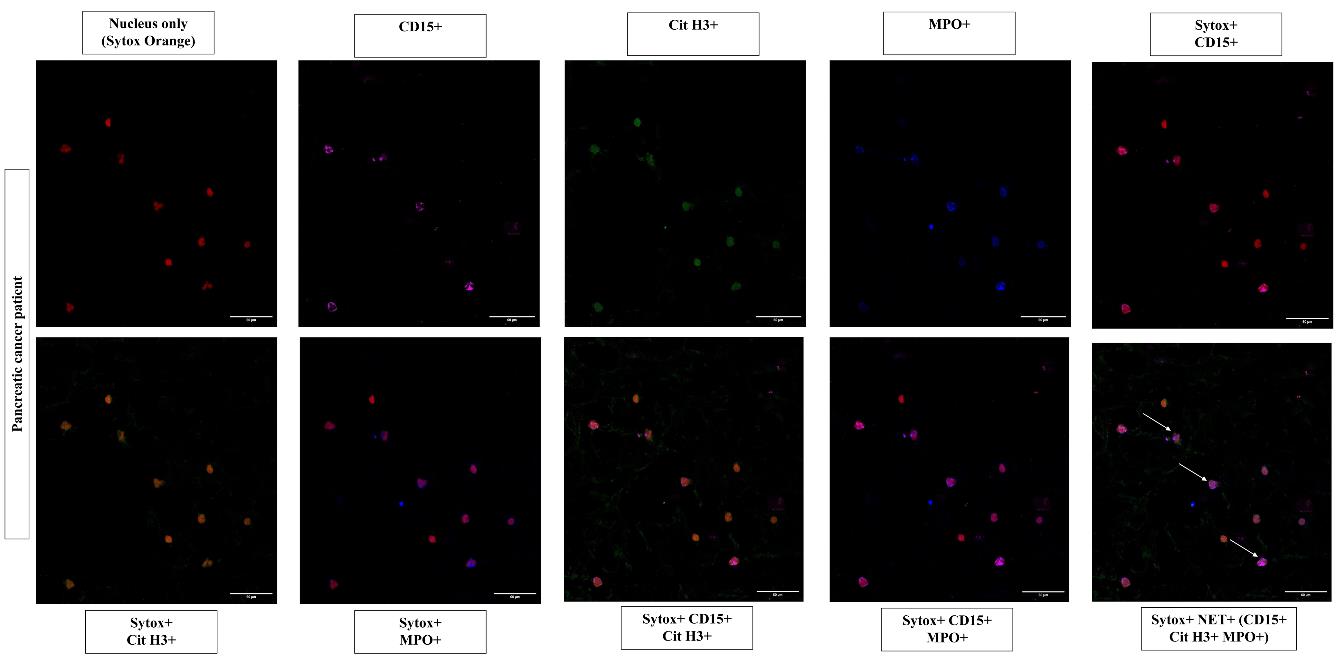
Figure 3. Representative images of neutrophil extracellular traps in peripheral blood smears of pancreatic cancer patients. Individual images represent the positive staining for nuclear binding dye Sytox Orange (555 nm, red), CD15 (650 nm, magenta), MPO (405 nm, blue), Cit H3 (488 nm, green), and other combinations of CD15, Cit H3, and MPO, along with nuclear dye. Neutrophil extracellular traps are identified as extracellular structures where nuclear binding dye Sytox Orange (555 nm, red), colocalized with CD15 (650 nm, magenta), MPO (405 nm, blue), and Cit H3 (488 nm, green) in the merged image as indicated by the white arrows. Images acquired at 40× magnification, scale bars = 60 µm.Quantitation of NETs
To further quantify the NETs, analyze the slides through confocal microscopy. Visualize the slides and take images of each slide at three high-power random fields, i.e., 40× magnification. Open the images in ImageJ and split the image into channels. Visualize the mentioned channels, i.e., blue (MPO), green (Cit H3), magenta (CD15), and red (Sytox Orange). The overlaid channels, which are positive for all markers (MPO, Cit H3, CD15, and Sytox Orange) represent the NETs. Count the NET-positive cells (CD15+, MPO+, and Cit H3+) and their subtypes (CD15+ MPO+ and CD15+ Cit H3+) in the peripheral blood smear images of both control and PC patients. Calculate the relative percentage of NETs and their subtypes as shown in Figure 4.
Note: Alternatively, researchers can use different immunofluorescence-based reporting methods available in literature such as fluorescence intensity to report the presence of markers.
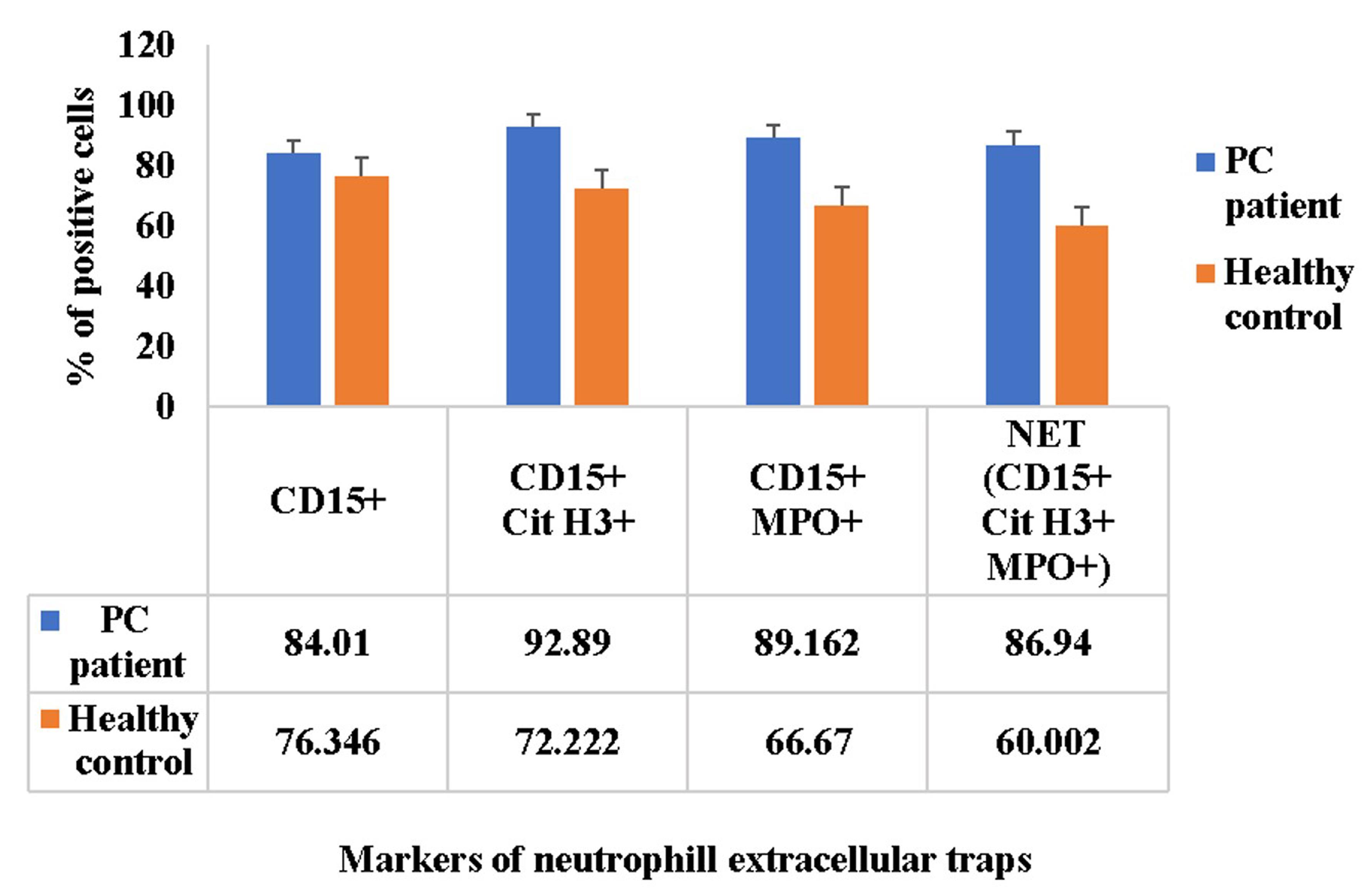
Figure 4. Comparison of neutrophil extracellular trap markers in peripheral blood smears. The bar graph indicates the relative percentage of neutrophils (CD15+), neutrophil extracellular traps (CD15+, MPO+, and Cit H3+), and their subtypes (CD15+ MPO+ and CD15+ Cit H3+) in relation with total Sytox positive cells in the peripheral blood smears of both control and pancreatic cancer patients. The x-axis represents the markers of neutrophil extracellular traps in the peripheral blood smears of both control and pancreatic cancer patients, whereas y-axis represents the relative percentage of various markers of neutrophil extracellular traps in relation with total Sytox positive cells. Data in bar graphs is represented in the form of mean ± standard error of mean, n = 5. PC, pancreatic cancer.
Validation of protocol
This protocol is a part of our ongoing research work on the role of neutrophil extracellular traps in promoting epithelial–mesenchymal transition and immunomodulation in pancreatic cancer. We have followed a standardized methodology to identify and characterize NETs in whole blood smears and provided every minute details regarding the same. The protocol will be cited accordingly in future publications. For related protocols, researchers can also refer to other immunofluorescence-based protocols for NETs detection performed in plasma samples (Matta, B., Battaglia, J. and Barnes, B. J., 2022. Detection of neutrophil extracellular traps in patient plasma: method development and validation in systemic lupus erythematosus and healthy donors that carry IRF5 genetic risk. Frontiers in immunology, 13, 951254. https://doi.org/10.3389/fimmu.2022.951254 [18]).
General notes and troubleshooting
General notes
Safety precautions must be taken when handling PFA, which is toxic and carcinogenic, and for NaOH pellets, which are highly corrosive. Prepare these solutions in a well-ventilated environment, wear a lab coat, gloves, safety glasses, and other protective gear, and avoid direct contact with these chemicals.
A spreader should be thinner than a glass slide when it comes to spreading blood on slides. The spreader's edge ought to be polished, smooth, and slim. The smear ought to extend halfway over the slide. It must be devoid of gaps, cracks, and bubbles. The smear should be smooth, leveled, and slightly curved in appearance. It must be sufficiently thin to produce a low power field of at least 10× in areas without the RBCs overlapping. Avoid applying too much pressure when spreading, and let the smear air-dry fully before staining in order to prevent RBC overlap. During staining, drying aids in preventing RBC overlapping.
To maintain high humidity, add 10 mL of water to the humidifier slide chamber. Slides should be placed atop the racks of the humidifier slide chamber. To preserve the moisture inside the container, close the lid of the humidifier slide chamber.
For washing, rinse the slides three times with 1 mL of wash buffer (each time gently without shaking) and gently tilt the slides to remove the liquid into the waste container.
Commercially available anti-fade mounting media can also be used instead of 70% glycerol. A refractive index (RI) of approximately 1.53 is suggested for the mounting medium. The brightness and clarity of the image increase with the proximity of the sample's and mounting medium's RI values to this value. With an RI of 1.47, glycerol boosts the sample's RI and helps in the emergence of a brighter, better-resolution image.
Troubleshooting
| Problem | Possible causes | Possible solutions |
|---|---|---|
| Sample signal | ||
| a. No signal | i. The fluorescence microscope might not be operating correctly. ii. Antigen detached from the sample during preparation. iii. PBS wash done too vigorously. iv. The antigen substrate will be degraded by microbial contamination of the specimen. v. Unclean glassware or not completely rinsed. vi. Loss of antigen adherence to slide may result from an excessively acidic or alkaline pH of PBS. vii. Improper storage of slides; prolonged exposure of fluorophores to light may cause the signal to fade. viii. Prolonged storage of slides. ix. Improper fixation. x. Improper dilution of the antibody (too diluted). xi. Not giving the specified incubation period. xii. Inappropriate utilization of the secondary antibody. xiii. Incorrect wavelength of excitation. | i. Use previously stained slides to examine under a microscope. If nothing is still visible, make sure the filters are set to the right wavelength, change the bulb, or realign it. ii. Avoid ever touching an antigen surface with your hands or a pipette. iii. Carefully follow the washing instructions. iv. Be cautious to prevent contamination when handling and collecting specimens. v. Properly clean glassware using Labolene cleaning agent. vi. Before using any buffer, always check the pH and adjust if needed. vii. Carry out incubations and keep slides in a dark place. For best outcomes, sections should be imaged right away after mounting. viii. To prevent antigenicity from being lost, use recently prepared slides. ix. Immediately incubate thoroughly in fixative. Use at least 4% formaldehyde to suppress endogenous phosphatases when using phospho-specific antibodies. x. Use low to high series dilutions or reference the antibody datasheet to determine the optimal antibody dilution. xi. Incubate slides with primary antibodies following a thoroughly standardized methodology to start from at least 3 h incubation to overnight incubation to obtain a consistent outcome. xii. Choose the suggested concentration and ensure that the secondary antibody matches the primary antibody's host species. xiii. Verify that the fluorophore(s)' excitation wavelength is matched by the illumination and detection (laser, excitation, emission filter). |
| b. Too low signal | Insufficient expression of the target protein. | i. Raise the antibody concentrations. ii. Adjust the method of detection by binding with a fluorophore that is more luminous. |
| c. Too high signal | i. Not enough blocking. ii. Improper dilution of the antibody (too concentrated primary or secondary antibody). | i. A normal serum that matches the same species as the secondary antibody should be utilized. ii. To find the suggested antibody dilution, go to the datasheet for the antibody product. |
| High background | i. Autofluorescence in samples. ii. Not enough blocking. iii. Improper dilution of the antibody (too concentrated primary or secondary antibody). iv. The samples became dry. v. Not enough washing. vi. Secondary cross-reactivity. | i. To ensure the levels of autofluorescence, use unstained samples as a control. Replace outdated formaldehyde stocks and make new solutions, because outdated fixatives have the potential to autofluoresce and may lead to high background staining (non-specific signals). For targets with low abundance, select channels with longer wavelengths. ii. Apply normal serum that belongs to the same species as the secondary antibody. iii. To find the suggested antibody dilution, go to the datasheet for the antibody product. iv. The sample must be kept immersed in liquid throughout the whole staining process. v. Wash to get rid of extra secondary antibodies, excess fixative, and weakly bound, non-specific antibody connections. vi. To find out if your secondary antibody is cross-reacting, employ isotype control antibodies. |
| Distorted morphology | i. Blood drop size. ii. Spreader sliding angle. iii. Spreading speed. iv. Viscosity of blood. | i. An extremely small drop might not allow for a lengthy enough smear for a sufficient diagnostic assessment; a large drop might cause the smear to stretch past the edge of the slide, losing cell clumping and preventing the development of the feathered edge. ii. A spreader sliding angle of more than 30° produces a thicker and smaller smear, while a smaller angle produces a thinner and lengthier smear. iii. While slower spreading can produce a lengthy or thicker smear, faster spreading can provide a narrower or thinner blood smear. iv. A patient with a higher hematocrit will have blood that is more viscous, which could lead to an excessively thick smear. In these situations, reducing the spreader slide's angle can aid in creating a thinner smear that is better suited for microscopic inspection. Anemia also causes a drop in blood viscosity, which leads to a thin smear preparation. In such scenarios, thickening the smear preparation will be achieved by angling the spreader slide more. |
Acknowledgments
We appreciate the engagement of every patient and volunteer. A Ph.D. scholarship from the Department of Biotechnology, Government of India, to Sakshi Bansal, helped with this work during the protocol's optimization (Award letter Number DBTHRDPMU/JRF/BET-21/I/2011-22/251). We express our gratitude to Ms. Meenakshi Sinha for her help with confocal microscopy, the Central Sophisticated Instrumentation Core (CSIC), and PGIMER, Chandigarh for their infrastructure support.
Competing interests
There are no competing interests to declare.
Ethical considerations
All described procedures for sample collection were consented to by the Institutional Ethics Committee of the Post Graduate Institute of Medical Education and Research, Chandigarh on February 27, 2023 (reference number IEC-INT/2023/PhD-885). All the patients/participants provided written, informed consent for their participation in this study.
References
- Yabroff, K. R., Wu, X. C., Negoita, S., Stevens, J., Coyle, L., Zhao, J., Mumphrey, B. J., Jemal, A. and Ward, K. C. (2021). Association of the COVID-19 Pandemic With Patterns of Statewide Cancer Services. J Natl Cancer Inst. 114(6): 907–909.
- Siegel, R. L., Miller, K. D., Fuchs, H. E. and Jemal, A. (2022). Cancer statistics, 2022. CA Cancer J Clin. 72(1): 7–33.
- Hu, J. X., Zhao, C. F., Chen, W. B., Liu, Q. C., Li, Q. W., Lin, Y. Y. and Gao, F. (2021). Pancreatic cancer: A review of epidemiology, trend, and risk factors. World J Gastroenterol. 27(27): 4298–4321.
- Gordon‐Weeks, A. N., Lim, S. Y., Yuzhalin, A. E., Jones, K., Markelc, B., Kim, K. J., Buzzelli, J. N., Fokas, E., Cao, Y., Smart, S., et al. (2017). Neutrophils promote hepatic metastasis growth through fibroblast growth factor 2–dependent angiogenesis in mice. Hepatology. 65(6): 1920–1935.
- Teramukai, S., Kitano, T., Kishida, Y., Kawahara, M., Kubota, K., Komuta, K., Minato, K., Mio, T., Fujita, Y., Yonei, T., et al. (2009). Pretreatment neutrophil count as an independent prognostic factor in advanced non-small-cell lung cancer: An analysis of Japan Multinational Trial Organisation LC00-03. Eur J Cancer. 45(11): 1950–1958.
- Schmidt, H., Suciu, S., Punt, C. J., Gore, M., Kruit, W., Patel, P., Lienard, D., von der Maase, H., Eggermont, A. M., Keilholz, U., et al. (2007). Pretreatment Levels of Peripheral Neutrophils and Leukocytes As Independent Predictors of Overall Survival in Patients With American Joint Committee on Cancer Stage IV Melanoma: Results of the EORTC 18951 Biochemotherapy Trial. J Clin Oncol. 25(12): 1562–1569.
- Brinkmann, V., Reichard, U., Goosmann, C., Fauler, B., Uhlemann, Y., Weiss, D. S., Weinrauch, Y. and Zychlinsky, A. (2004). Neutrophil Extracellular Traps Kill Bacteria. Science. 303(5663): 1532–1535.
- Mohanan, S., Cherrington, B. D., Horibata, S., McElwee, J. L., Thompson, P. R. and Coonrod, S. A. (2012). Potential Role of Peptidylarginine Deiminase Enzymes and Protein Citrullination in Cancer Pathogenesis. Biochem Res Int. 2012: 1–11.
- Jin, W., Yin, H., Li, H., Yu, X., Xu, H. and Liu, L. (2021). Neutrophil extracellular DNA traps promote pancreatic cancer cells migration and invasion by activating EGFR/ERK pathway. J Cell Mol Med. 25(12): 5443–5456.
- Demers, M., Wong, S. L., Martinod, K., Gallant, M., Cabral, J. E., Wang, Y. and Wagner, D. D. (2016). Priming of neutrophils toward NETosis promotes tumor growth. Oncoimmunology. 5(5): e1134073.
- Kanamaru, R., Ohzawa, H., Miyato, H., Matsumoto, S., Haruta, H., Kurashina, K., Saito, S., Hosoya, Y., Yamaguchi, H., Yamashita, H., et al. (2018). Low density neutrophils (LDN) in postoperative abdominal cavity assist the peritoneal recurrence through the production of neutrophil extracellular traps (NETs). Sci Rep. 8(1): 632.
- Demkow, U. (2021). Neutrophil Extracellular Traps (NETs) in Cancer Invasion, Evasion and Metastasis. Cancers (Basel). 13(17): 4495.
- Cools-Lartigue, J., Spicer, J., McDonald, B., Gowing, S., Chow, S., Giannias, B., Bourdeau, F., Kubes, P. and Ferri, L. (2013). Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. J Clin Invest. 123(8): 3446–3458.
- Martins-Cardoso, K., Almeida, V. H., Bagri, K. M., Rossi, M. I. D., Mermelstein, C. S., König, S. and Monteiro, R. Q. (2020). Neutrophil Extracellular Traps (NETs) Promote Pro-Metastatic Phenotype in Human Breast Cancer Cells through Epithelial–Mesenchymal Transition. Cancers (Basel). 12(6): 1542.
- Albrengues, J., Shields, M. A., Ng, D., Park, C. G., Ambrico, A., Poindexter, M. E., Upadhyay, P., Uyeminami, D. L., Pommier, A., Küttner, V., et al. (2018). Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science. 361(6409): eaao4227.
- Langiu, M., Palacios-Acedo, A. L., Crescence, L., Mege, D., Dubois, C. and Panicot-Dubois, L. (2022). Neutrophils, Cancer and Thrombosis: The New Bermuda Triangle in Cancer Research. Int J Mol Sci. 23(3): 1257.
- Boone, B. A., Murthy, P., Miller-Ocuin, J., Doerfler, W. R., Ellis, J. T., Liang, X., Ross, M. A., Wallace, C. T., Sperry, J. L., Lotze, M. T., et al. (2018). Chloroquine reduces hypercoagulability in pancreatic cancer through inhibition of neutrophil extracellular traps. BMC Cancer. 18(1): 678.
- Matta, B., Battaglia, J., Barnes, B. J. (2022). Detection of neutrophil extracellular traps in patient plasma: method development and validation in systemic lupus erythematosus and healthy donors that carry IRF5 genetic risk. Front Immunol. 13: 951254.
Article Information
Copyright
© 2024 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Bansal, S., Sharma, V., Gupta, R., Singh, H. and Aggarwal, A. (2024). A New Approach for Assessment of Neutrophil Extracellular Traps Through Immunofluorescence Staining in Whole Blood Smears. Bio-protocol 14(11): e5010. DOI: 10.21769/BioProtoc.5010.
Category
Cancer Biology > Tumor immunology > Immunological assays
Immunology > Immune cell staining > Immunodetection
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.