- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Electrophoretic Mobility Assay to Separate Supercoiled, Catenated, and Knotted DNA Molecules
Published: Vol 14, Iss 9, May 5, 2024 DOI: 10.21769/BioProtoc.4983 Views: 2386
Reviewed by: Pilar Villacampa AlcubierreAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Feb 2015
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Two-dimensional (2D) agarose gel electrophoresis is the method of choice to analyze DNA topology. The possibility to use E. coli strains with different genetic backgrounds in combination with nicking enzymes and different concentrations of norfloxacin improves the resolution of 2D gels to study the electrophoretic behavior of three different families of DNA topoisomers: supercoiled DNA molecules, post-replicative catenanes, and knotted DNA molecules. Here, we describe the materials and procedures required to optimize their separation by 2D gels. Understanding the differences in their electrophoretic behavior can help explain some important physical characteristics of these different types of DNA topoisomers.
Key features
• Preparative method to enrich DNA samples of supercoiled, catenated, and knotted families of topoisomers, later analyzed by 2D gels (or other techniques, e.g., microscopy).
• 2D gels facilitate the separation of the topoisomers of any given circular DNA molecule.
• Separation of DNA molecules with the same molecular masses but different shapes can be optimized by modifying the conditions of 2D gels.
• Evaluating the roles of electric field and agarose concentration on the electrophoretic mobility of DNA topoisomers sheds light on their physical characteristics.
Keywords: DNA topologyGraphical overview
Background
DNA molecules with different topology can be analyzed by the so-called single-molecule methods such as chromatin fiber autoradiography [1], dynamic molecular combing [2], transmission electron microscopy [3-5], atomic force microscopy [6], and magnetic tweezers [7,8]. DNA properties that are difficult to address experimentally can also be studied by computer simulations [9–13]. Two-dimensional (2D) agarose gel electrophoresis is the best experimental method currently available to allow the simultaneous identification of DNA molecules with different topology [e.g., supercoiled (Sc), catenated (Cats), and knotted (Kn) molecules]. This technique consists of two consecutive electrophoretic separations performed under different conditions and run at two orthogonal directions (4–8). The first dimension is resolved in a low-percentage (∼0.4%) agarose gel electrophoresis at a relatively low voltage (∼1 V/cm). The second dimension is run perpendicular to the first one so that an entire selected lane of the gel is used as replacement of the gel wells but in a higher-percentage (∼1%) agarose gel electrophoresis at higher electric field (∼5–6.6 V/cm). 2D gels were originally designed by Bell and Byers to separate branched and linear molecules [14], and it was early noticed that this method could also be successfully applied to study DNA topology. 2D gels were adapted to examine simultaneously thousands of molecules with different DNA topology, such as Sc forms, Kn forms, partially replicated forms (named pre-catenanes) with or without reversed forks, fully replicated catenanes (Cats), and replication intermediates (RIs) containing knotted bubbles [4,6,15–28]. 2D agarose gel electrophoresis has been extensively used to investigate the activity of topoisomerases in vitro and in vivo [29,30]. Additionally, 2D gels can also be used as a preparative method to enrich samples for specific DNA molecules that can be later examined by different techniques [4,6,18,19,31,32].
Plasmids are invaluable tools as a model to study DNA topology. The advantages of plasmids include their ease of isolation and the ability to quantitatively measure DNA supercoiling, knotting, and catenation in purified DNA samples [33]. Here, we present a protocol where 2D gels are used to analyze electrophoretic mobility of three families of topoisomers with the same molecular mass (8,766 bp): supercoiled dimers (ScDimers), monomeric-nicked catenanes (CatAs), and nicked-knotted dimers (KnDimers). We use monomeric (4,383 bp) or dimeric forms (8,766 bp) of pBR18, a derivative of pBR322 where the tetracycline resistance promoter was replaced with the poly-linker of pUC18 [34] to transform three different Escherichia coli strains. We describe the steps required to:
i. Obtain the three families of DNA samples for the analysis by 2D gels;
ii. Optimize their separation by 2D gels based on their extent of supercoiling, catenation, and knotting;
iii. Visualize them by non-radioactive detection.
Materials and reagents
Biological materials
Escherichia coli strains
DH5αF’: F’/gyrA96(Nalr) recA1 relA1 endA1 thi-1 hsdR17 (rk-mk+) glnV44 deoR Δ (lacZYA argF) U169[F80dΔ(lacZ)M15] (kindly gifted by Santiago Rodríguez de Córdoba) [35]
parE10: W3110 F- except [parE10 recA] (kindly gifted by Ian Grainge) [36]
LZ38 F-λ (P80 red114 xis-l cl857) zei-723::Tn10 parCK84::kanR (kindly gifted by Lynn Zechiedrich) [37,38]
Plasmid:
pBR18 (4,383 pb): a derivative of pBR322 where the tetracycline resistance promoter between EcoRI and HindIII was replaced with the poly-linker of pUC18 [32]
Luria-Broth (LB) (Invitrogen, catalog number: 12795084)
Ethanol absolute, molecular biology grade (Sigma, catalog number: 64-17-5)
Agarose (Seakem LE, Lonza, catalog number: 50004)
Norfloxacin (Abcam, catalog number: 70458-96-7)
Ethidium bromide (Sigma, catalog number: 1239-45-8)
Digoxigenin-High Prime kit (Roche, catalog number: 11585614910)
Anti-Digoxigenin-AP conjugate antibody (Roche, catalog number: 11093274910)
CDP-Star (Perkin Elmer, catalog number: NEL601001KT)
Brij® 58 (Sigma-Aldrich, catalog number: P-5884)
Sodium deoxycholate (Sigma-Aldrich, catalog number: D6750)
Tris-HCl (Sigma, catalog number: T2913)
Ethylenedinitrilotetraacetic acid, disodium salt (EDTA) (Sigma, catalog number: E7889)
Tris base (Sigma, catalog number: T1503)
Boric acid (Sigma, catalog number: PHR2664)
Hydrochloric acid (Sigma-Aldrich, catalog number: 258148)
Proteinase K, recombinant (Roche, catalog number: 3115879001)
Nb.BsmI 10,000 U/mL (New England Biolabs, catalog number: R0706S)
Calcium chloride (Flinn Scientific, catalog number: C0234)
Lambda DNA/HindIII Marker (Invitrogen, catalog number: SM0101)
BlueJuiceTM gel loading dye (10×) (Thermo Scientific, catalog number: 10816015)
Magnesium sulfate (MP Biomedicals, catalog number: 0219483394)
Sodium chloride (MP Biomedicals, catalog number: 0215257591)
Sucrose (Thermo Scientific Chemicals, catalog number: A15583.36)
Lysozyme (Thermo Scientific, catalog number: 10249843)
Polyethylene glycol (PEG) 6000 (Thermo Scientific, catalog number: J19972.A1)
Sodium hydroxide (Fisher Chemical, catalog number: 10675692)
Tri-sodium citrate (Fisher Chemical, catalog number: 10112880)
Disodium phosphate (MP Biomedicals, catalog number: 02191440.5)
Dextran sulfate (MP Biomedicals, catalog number: 11417880)
Sodium azide (Sigma, catalog number: S2002)
Sodium dodecyl sulfate (SDS) (Sigma, catalog number: 151-21-3)
Blotto (regular powdered milk at supermarket/pharmacy)
Sonicated and denatured salmon sperm DNA (Invitrogen, catalog number: 10605543)
Phenol:chloroform:isoamyl alcohol (25:24:1) equilibrated with 10 mM Tris–HCl, pH 8.0. (Sigma, catalog number: P3803)
0.5 M EDTA, pH 8.0
For bacterial transformation (Section A):
1 M magnesium sulfate
0.1 M calcium chloride
For isolation of plasmid DNA (Section B):
Tris-EDTA (TE) (see Recipes)
Sodium chloride-Tris-EDTA (STE) (see Recipes)
25% sucrose (see Recipes)
Lysozyme–RNase A solution (see Recipes)
Lysis buffer (see Recipes)
25% PEG 6000 in TE (see Recipes)
Proteinase K buffer (20 mg/mL) (see Recipes)
For 2D agarose gel electrophoresis (Section E):
10× Tris Borate EDTA (TBE) (see Recipes)
For Southern blotting (Section F):
Depurination solution (see Recipes)
Transfer solution (see Recipes)
20× saline sodium citrate (SSC) (see Recipes)
For nonradioactive hybridization (Section G):
20× saline sodium phosphate EDTA (SSPE) (see Recipes)
20% dextran sulfate (see Recipes)
20% SDS (see Recipes)
Prehybridization/hybridization solution (see Recipes)
Recipes
For isolation of plasmid DNA (Section B):
Tris-EDTA (TE)
10 mM Tris-HCl
1 mM EDTA, pH 8.0
Ultrapure water
Note: Sterilize and store at room temperature. Stable for one year.
Sodium chloride-Tris-EDTA (STE)
0.1 M sodium chloride
10 mM Tris-HCl, pH 8.0
1 mM EDTA, pH 8.0
Ultrapure water
Note: Sterilize and store at 4 °C. Stable for one year.
25% Sucrose
25% sucrose
0.25 M Tris-HCl, pH 8.0
Note: Sterilize and store at 4 °C. Stable for one month.
Lysozyme–RNase A solution
10 mg/mL lysozyme
0.2 mg/mL RNase A
0.25 M Tris-HCl, pH 8.0.
0.25 M EDTA, pH 8.0
Note: Prepare immediately prior to use; mix by inverting the tube and keep on ice.
Lysis buffer
50 mM Tris–HCl, pH 8.0
63 mM EDTA
1% Brij® 58
0.4% sodium deoxycholate
Ultrapure water
Note: Difficult to dissolve, use a magnetic stirrer. Stable for six months.
25% PEG 6000 in TE
25% PEG 6000
1.25 M sodium chloride
Note: Difficult to dissolve, use a magnetic stirrer. Store at 4 °C. Stable for two years.
Proteinase K buffer (20 mg/mL)
0.1 M sodium chloride
10 mM Tris-HCl, pH 8.0
1 mM EDTA, pH 8.0
0.1% SDS
Ultrapure water
Note: Preheat at 37 °C and add 100 μg/mL of Proteinase K before use. Proteinase K buffer should be stored at -20 °C for up to two years to maintain its activity.
For 2D agarose gel electrophoresis (Section E):
10× Tris Borate EDTA (TBE)
1.00 M Tris base
1.00 M boric acid
0.02 M EDTA
Ultrapure water
pH 8.2–8.4
Note: Prepare a 1:10 dilution with ultrapure water to make 1× TBE. Store at room temperature. Stable for one year.
For Southern blotting (Section F):
Depurination solution
0.25 M hydrochloric acid
Ultrapure water
Note: Prepare immediately prior to use.
Transfer solution
0.4 M sodium hydroxide
Ultrapure water
Note: Prepare immediately prior to use.
20× saline sodium citrate (SSC)
3 M sodium chloride
0.3 M Tri-sodium citrate
Ultrapure water
Note: Store at room temperature. Stable for two years.
For nonradioactive hybridization (Section G):
20× saline sodium phosphate EDTA (SSPE)
3.6 M sodium chloride
0.2 M disodium phosphate
20 mM EDTA
Ultrapure water
Note: Store at room temperature. Stable for two years.
20% dextran sulfate
20 g of dextran sulfate
0.2 g of sodium azide
Ultrapure water
Note: Prepare aliquots and freeze (-20 °C). Stock solutions are stable for up to six months.
20% SDS
3 M sodium chloride
0.3 M sodium citrate
Ultrapure water
pH 7.0
Note: Store at room temperature. Stable for one year.
Prehybridization/hybridization solution
2× SSPE
1% SDS
0.5% Blotto
10% dextran sulfate
0.5 mg/mL sonicated and denatured salmon sperm DNA
Ultrapure water
Note: Prepare aliquots and freeze (-20 °C). Stock solutions are stable for up to 2 years.
Laboratory supplies
500 mL centrifuge bottles (Thermo Scientific, catalog number: 010-1493)
50 mL high-speed centrifuge tubes (Thermo Scientific, catalog number: 3118-0085)
50 mL Falcon tubes (Fisherbrand, catalog number: 11512303)
15 mL Falcon tubes (Fisherbrand, catalog number: 11765075)
1.5 mL microcentrifuge tubes (Thermo Scientific, PierceTM, catalog number: 10177443)
Whatman filter paper (GE Healthcare, catalog number: 3030917)
Zeta-Probe membrane (Bio-Rad, catalog number: 1620159)
Glass or plastic pipette
Film (Fuji, catalog number: 4741019236)
Cell spreader (Sigma-Aldrich, model: Cell Spreader silver stainless steel, bar L 33 mm, catalog number: HS86655)
Equipment
Superspeed floor centrifuge (Thermo Scientific, catalog number: 75006580)
Fixed-angle rotor for 500 mL bottles (Thermo Scientific, catalog number: 096-062375)
Swinging-bucket rotor for 50 mL round or conical tubes (Thermo Scientific, catalog number: 75003010)
Horizontal electrophoresis system with inter-electrode distances of at least 30 cm that can hold a 15 × 20 cm tray (Bio-Rad, catalog number: 1704403)
Basic power supply (Bio-Rad, catalog number: 11645050)
UV transilluminator (Biorad, model: Gel Doc XR+ System, catalog number: 1708195)
Microbiological shaking incubator (VWR, catalog number: 76628-472)
Thermoblock (Labnet, model: AccuBlock, catalog number: D1301)
Vacuum pump (Millipore, model: Millivac-Mini Vacuum Pump XF5423050)
Hybridization oven (Techne®, Hybrigene, catalog number: 445-0024)
X-ray film cassette or ChemiDocTM Imaging System (Bio-Rad, catalog number: 12003153)
Microcentrifuge (Eppendorf, model: 5418R, catalog number: EPP5401000013)
Software and datasets
ImageJ (https://imagej.nih.gov/ij/download.html) (Access date, 02/01/2024)
Procedure
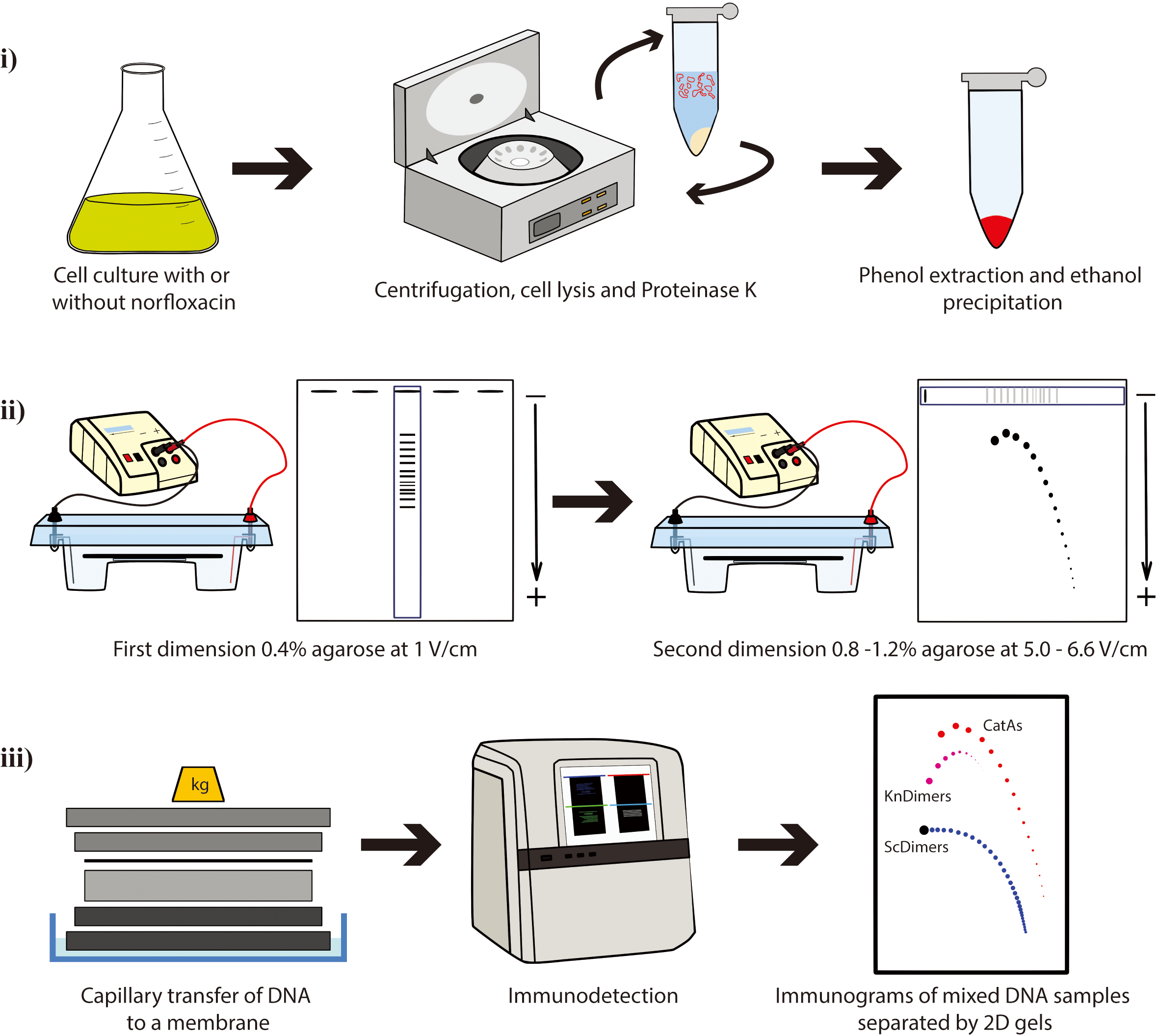
We describe below the step-by-step procedure where 2D gels are used to analyze electrophoretic mobility of DNA molecules with the same mass (8,766 pb) but different topology: dimeric forms of a supercoiled molecule (ScDimers), nicked-catenated monomers (CatAs), and nicked-knotted dimers (KnDimers) (Figure 1). We use monomeric (4,383 bp) or dimeric forms (8,766 bp) of pBR18 to transform three different Escherichia coli strains (See Section A):
- E. coli LZ38 cells are recA+ and accumulate multimeric forms of the plasmid. In addition, they bear a mutation in the parC gene that turns Topo IV resistant to norfloxacin. Therefore, in these cells, norfloxacin inhibits only DNA gyrase, leading to the accumulation of poorly supercoiled molecules, and both monomeric (ScMonomers) and dimeric (ScDimers) forms become clearly distinguished in the same 2D gel.
- E. coli DH5∝F’ cells bear a mutation in the gyrA gene that turns DNA gyrase resistant to norfloxacin. Therefore, in these cells, norfloxacin inhibits only Topo IV, leading to the accumulation of catenated molecules (Cats).
- E. coli parE10 cells bear a thermosensitive mutation in the parE gene. Therefore, in these cells, norfloxacin inhibits only DNA gyrase, leading to the accumulation of relaxed molecules. In this way, supercoiled (Sc Dimers) and knotted molecules (KnDimers) become clearly distinguished in the same 2D gel.
Cell treatment with norfloxacin, an inhibitor of wild-type DNA gyrase and topoisomerase IV (Topo IV) [39], and DNA digestion with nicking enzyme Nb.BsmI are used to visualize Sc, CatAs, and Kn family of topoisomers in 2D agarose gels (Figure 1). The data generated using this protocol can be used to study the different reactions of Sc, CatAs, and Kn families of topoisomers to changes in agarose concentration and voltage during electrophoresis or it can be used as a preparative method to enrich samples for specific DNA molecules [6,18,31].
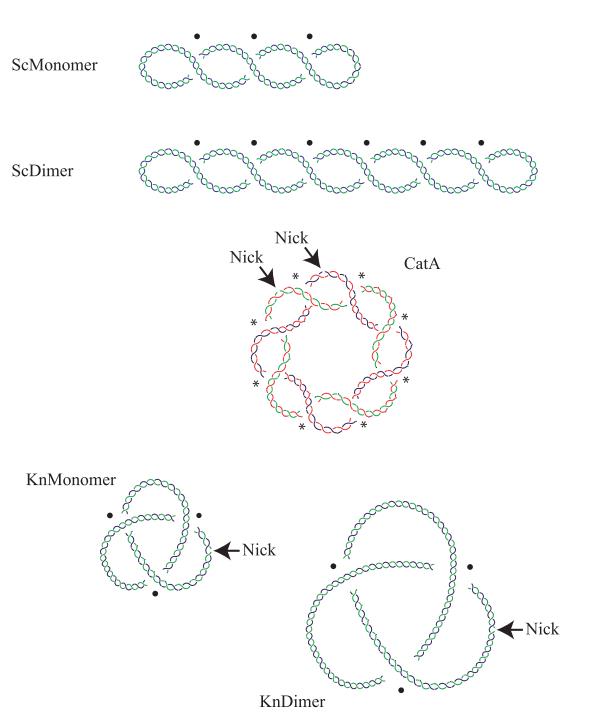
Figure 1. Cartoons of DNA molecules with different topology. Supercoiled monomer (ScMonomer), supercoiled dimer (ScDimer), nicked-catenated monomers (CatA), nicked-knotted monomer (KnMonomer), and nicked-knotted dimer (KnDimer) are represented. ScDimers, CatAs, and KnDimers were chosen in our study because all have the same mass and similar density of crossings, which allows us to study their electrophoretic mobility based only on their topology. Intramolecular crossings are marked with a black spot, while intermolecular ones are pointed with an asterisk. The catenated rings in CatA are drawn in blue and red and in green and red to distinguish each.
Bacterial transformation
Inoculate the selected bacterial strain in 30 mL of LB broth and grow culture in a shaking incubator at 250 rpm overnight.
Incubate DH5αF’ and LZ38 cells at 37 °C.
Incubate parE10 cells at 30 °C.
Make a 100-fold dilution in 30 mL of LB plus 10 mM magnesium sulfate.
Grow the culture up to an OD600 of 0.3–0.4 for approximately 3–5 h.
Incubate DH5αF’ and LZ38 cells at 37 °C.
Incubate parE10 cells at 30 °C.
Centrifuge 1 mL of the culture in a 1.5 mL Eppendorf tube at 9,300× g for 4 min at room temperature.
Carefully resuspend the pellet in 100 μL of 0.1 M cold calcium chloride.
Incubate the tube for 20 min on ice.
Add 10 ng of plasmid DNA, either monomeric (4,383bp) or dimeric forms (8,766 bp) of pBR18, in a 10 μL volume and incubate for another 10 min on ice:
To obtain ScDimers of pBR18, transform E. coli LZ38 cells with pBR18 monomers.
To obtain CatAs, transform E. coli DH5∝F’ cells with pBR18 monomers.
To obtain KnDimers, transform E. coli parE10 cells with pBR18 dimers.
Heat-shock the cells by placing the tube at 37 °C for 5 min or at 42 °C for 2 min.
Add 1 mL of LB and incubate at 37 °C for 1 h with moderate agitation.
Spread the cells on an agar plate containing 75 μg/mL ampicillin to select for cells with the plasmid.
Isolation of plasmid DNA (Note 1)
Inoculate the selected transformed bacterial strain (step A10) in 25 mL of LB broth and grow culture in a shaking incubator at 250 rpm overnight.
Dilute E. coli cells from the overnight culture 40-fold into 1 L of fresh LB medium and incubate in a shaking incubator at 250 rpm at 37 °C.
Grow the cells to exponential phase (OD600 0.4–0.6, approximately 3–5 h) and chill the culture quickly on ice. Transfer to 500 mL bottles and centrifuge at 3,500× g for 15 min at 4 °C in a floor centrifuge using a fixed-angle rotor.
Remove the media, resuspend the pellet in 20 mL of STE, transfer to a 50 mL Falcon tube, and centrifuge at 2,100× g for 15 min at 4 °C.
Harvest the cells by centrifugation and resuspend them in 50 mL high-speed centrifuge tubes with 5 mL of 25% sucrose (see Recipes). Keep the tubes on ice.
Add 1 mL of lysozyme–RNase A solution (see Recipes) and incubate on ice for 5 min.
Add 2 mL of 0.25 M EDTA, pH 8.0, mix by inverting the tube, and incubate on ice for 5 min.
Lyse the cells by adding 8 mL of lysis buffer (see Recipes) and inverting the tubes 3–4 times.
Incubate the samples for 1 h on ice.
Centrifuge at 30,000× g for 60 min at 4 °C to pellet the chromosomal DNA and other bacterial debris.
Transfer the supernatant with plasmid DNA (~10 mL) to a 50 mL high-speed centrifuge tube and precipitate it by adding 2/3 volumes of 25% PEG 6000 in TE (see Recipes).
Incubate overnight at 4 °C.
Pellet the precipitated DNA by centrifugation at 12,500× g for 15 min at 4 °C. Carefully remove the supernatant with a glass or plastic pipette.
Resuspend the pellet in 5 mL of a preheated proteinase K digestion buffer supplied with 25 μL of proteinase K (20 mg/mL) (see Recipes) and incubate for 30 min at 37 °C.
Under a fume hood, add v/v of phenol:chloroform:isoamyl alcohol (25:24:1) to your sample and incubate in tube rotator for 15 min at room temperature.
Centrifuge at 1,600× g for 5 min at room temperature. Carefully remove the upper aqueous phase and transfer the layer to a fresh tube. Be sure not to carry over any phenol during pipetting.
Repeat steps B15 and B16 at least once more.
Add one volume of chloroform:isoamyl alcohol (24:1) to your sample and incubate in a tube rotator for 15 min at room temperature.
Centrifuge at 1,600× g for 5 min at room temperature. Carefully remove the upper aqueous phase and transfer the layer to a 50 mL high-speed centrifuge tube.
Precipitate the DNA overnight at -20 °C with 2.5 volumes of cold absolute ethanol.
Centrifuge at 12,800× g for 60 min at 4 °C. Carefully remove the upper aqueous phase and wash the pellet with cold 70% ethanol.
Centrifuge at 12,800× g for 15 min at 4 °C. Aspirate the supernatant, air dry the pellet for 2 h, and resuspend the pellet in 150–200 μL of ultrapure water or 1× TE.
Inhibition of DNA Gyrase and Topo IV in vivo
Start growing cells as described above (Steps B1–B3) until they reach the exponential phase.
To accomplish inhibition of topoisomerases in vivo, add norfloxacin to a final concentration of 15 or 150 μM and incubate for 15–30 min with orbital shaking (see Note 2):
To partially relax supercoiled monomeric (ScMonomers) and dimeric (ScDimers) topoisomers, expose transformed E. coli LZ38 to 150 μM Norfloxacin.
To obtain catenanes monomers (Cats), expose transformed E. coli DH5∝F’ cells to 15 µM Norfloxacin.
To separate knotted dimers (KnDimers) and supercoiled molecules, expose transformed E. coli parE10 cells to 15 μM Norfloxacin.
To inhibit Topo IV in parE10 strain, start growing the culture at the permissive temperature (30 °C) until reaching the exponential phase and then incubate it at the restrictive temperature (43 °C) in shaking incubator at 250 rpm for 1 h.
Harvest the cells by centrifugation and proceed to plasmid DNA isolation as described before.
DNA treatments
To obtain nicked catenated monomers (CatAs) and nicked-knotted dimers (KnDimer): digest DNA from transformed E. coli DH5∝F’ and parE10 cells with Nb.BsmI (8 U/μg of DNA) for 1 h at 50 °C (see Note 3).
DNA (0.1 μg/μL) 10 μL
Buffer 10× 10 μL
RNase 1 mg/mL 10 μL
Ultrapure water 62 μL
Nb.BsmI (10,000 U/mL) 8 μL
Precipitate the DNA by adding 2.5 volumes (250 μL) of cold absolute ethanol.
Centrifuge at 16,800× g for 30 min at 4 °C. Carefully remove the aqueous phase and wash the pellet with cold 70% ethanol.
Centrifuge at 16,800× g for 15 min at 4 °C. Aspirate the supernatant, air dry the pellet for 15–30 min, and resuspend the pellet in 10 μL of ultrapure water or 1× TE.
2D agarose gel electrophoresis
We use a mix of plasmid DNAs isolated from the different bacterial strains to analyze the electrophoretic mobility of ScDimers and CatAs on one hand (Mix A) and ScDimers and KnDimers on the other hand (Mix B) (Figure 2).

Figure 2. Representative immunodetections of mixed DNA samples enriched for supercoiled dimers (ScDimers), nicked-catenated monomers (CatAs), and nicked-knotted dimers (KnDimers) analyzed by 2D agarose gel electrophoresis. A. Mixed DNA sample of ScDimers and CatAs. B. Mixed DNA sample of ScDimers and Kn Dimers. The first dimension was run at 1 V/cm (i.e., 30 V in our gel electrophoresis chamber) for 30 h in a 0.4% agarose gel; the second dimension was run at 5 V/cm (i.e., 150 V in our gel electrophoresis chamber) for 10 h in a 1% agarose gel. Interpretative diagrams are shown to the right. OC Dimers refers to the mobility of dimeric open circles used to align the different immunodetections.Assemble a gel casting set with a 20-teeth (0.5 × 0.15 cm each) comb and pour a 0.4% agarose gel in 1× TBE. Let the gel solidify (15 × 20 cm).
Place the gel into a gel tank and pour 1× TBE buffer covering the gel. Carefully remove the comb.
Prepare the DNA samples in a final volume of 10 μL:
Mix A (ScDimers and CatAs):
1–3 μL of each DNA (200 ng of plasmid DNA).
1 μL of gel loading dye (10×) of 200 ng of plasmid DNA.
Fill with ultrapure water until a final volume of 10 μL (3–7 μL).
Mix B (ScDimers and KnDimers):
1–3 μL of each DNA (200 ng of plasmid DNA).
1 μL of gel loading dye (10×) of 200 ng of plasmid DNA.
Fill with ultrapure water until a final volume of 10 μL (3–7 μL).
Load the samples (Mix A or Mix B) in duplicates in two different lanes of the gel electrophoresis (e.g., lane 1 and lane 4). Lane 1 will be used only as a control to verify that the first dimension was carried out correctly (Note 4). Lane 4 (your target lane) will be used for the second dimension.
Run the first dimension at 1.0 V/cm (i.e., 30 V in our gel electrophoresis chamber) for 25–30 h at room temperature.
To prepare the second dimension, take the gel casting tray with the gel out of the tank and place it on a clean surface. Cut out the target lane from the first dimension (lane 4 in this protocol). Rotate the slice by 90º counterclockwise and place it on top of another gel casting (Figure 3).
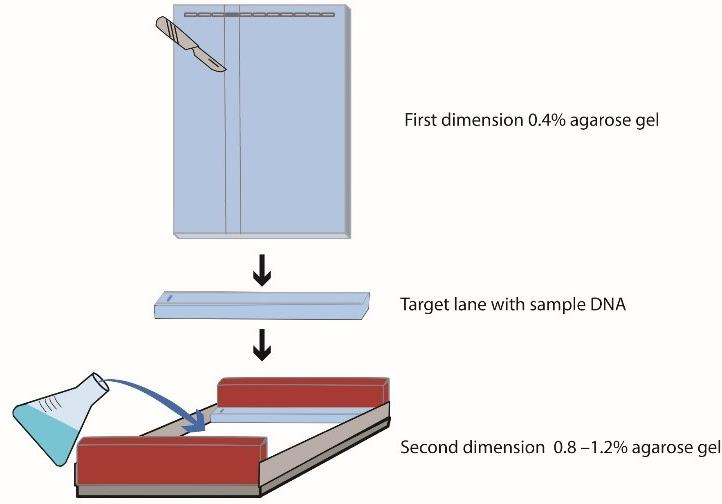
Figure 3. Preparation of the second dimension. Cut out the target lane from the first dimension. Rotate the slice 90° and place it on top of another gel casting. Pour the dissolved agarose (0.8%–1.2%) around the excised agarose lane.Melt 0.8%–1.2% agarose gel in 1× TBE, cool it down to 50–55 °C, and pour the dissolved agarose around the excised agarose lane from the first dimension. Let the gel polymerize at room temperature.
Pre-cool the 1× TBE buffer and the gel tray at 4 °C.
Place the gel into a gel tank and pour the cooled 1× TBE buffer covering the gel.
Run the second dimension at 5.0–6.6 V/cm (i.e., 150–200 V in our gel electrophoresis chamber) for 8–22 h in cold chamber at 4 °C.
After electrophoresis, transfer the DNA from the gels to an appropriate membrane by Southern blotting.
Southern blotting
Rinse the gel briefly with sterile distilled water.
To depurinate the DNA prior to transfer, submerge the gels for 10–15 min in depurination solution (see Recipes), with moderate shaking at room temperature. Transfer the gel to a clean recipient with sterile distilled water.
Set up the blot transfer as follows (see Figure 4): place three sheets of Whatman 3 MM paper that has been soaked with 0.4 N sodium hydroxide on top of a “bridge” that rests in a shallow reservoir of transfer solution. Place the gel on top of the three soaked sheets of Whatman 3 MM paper. Roll a clean pipette over the sandwich to remove all trapped air bubbles from between the gel and the paper.
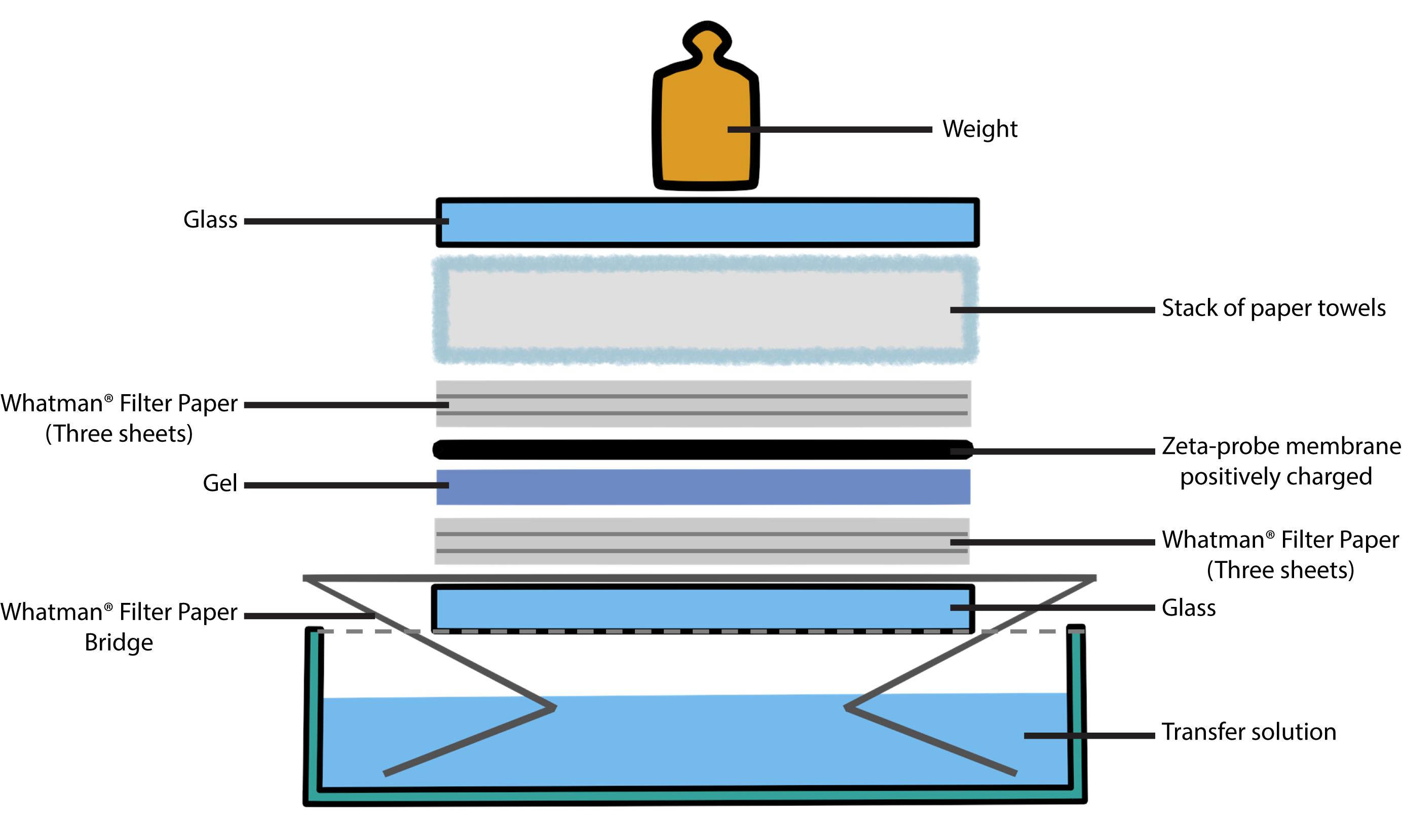
Figure 4. Transferring DNA to a membrane. How to set up a Southern blotting of agarose gels by capillary transfer. Capillary transfer of the denatured fragments to a membrane uses the flow of the 0.4 N sodium hydroxide solution (transfer solution) to deposit the DNA fragments on the membrane. A wet sheet of blotting paper acts as a wick for the transfer solution (bridge), which is drawn up by a stack of dry paper through the gel/membrane sandwich. We use the alkaline transfer protocol described by López-Estraño et al. [40].Cut a piece of positively charged Zeta-probe blotting membrane to the size of the gel. Pre-soak the membrane in sterile distilled water and place it on top of the gel. Use a pipette to eliminate air bubbles as above.
Add three sheets of Whatman 3 MM paper soaked in sterile distilled water, a stack of paper towels, a glass plate, and a 200–500 g weight (see Note 5).
Leave the blot transfer in transfer solution (see Recipes) overnight.
After the transfer, peel the membrane from the gel; rinse it twice in 2× SSC (see Recipes) for 5 min.
Nonradioactive hybridization
Label the DNA probes with digoxigenin using the Digoxigenin-High Prime kit following manufacturer’s recommendations.
In the meantime, prehybridize the membrane in a 20 mL prehybridization/hybridization solution for 4–6 h at 65 °C in hybridization bottles on a rotisserie inside a hybridization oven set at 65 °C.
Denature the labeled DNA probe by heating it at 95–100 °C for 5 min and chill it quickly in an ice bath.
Add the probe to the hybridization bottles to a final concentration of 20 ng/mL, place the bottles back into the oven, and hybridize overnight at 65 °C.
Wash the hybridized membranes sequentially with 2× SSC and 0.1% SDS for 5 min at room temperature twice, and then twice with 0.1× SSC and 0.1 % SDS for 15 min at 68 °C.
Perform the detection with an Anti-Digoxigenin-AP conjugate antibody and CDP-Star, according to the instructions provided by the manufacturer.
Data analysis
Scan the immunodetections and quantify the signals of interest by densitometry using Image J. The immunodetections are prepared according to the program manual, and the area and degree of saturation of each topoisomer are determined. The electrophoretic mobility values are obtained by measuring the distance of each topoisomer with respect to a signal common to all the immunodetections. We use the trivial knot, without supercoiling, called OC (Open Circle) as a reference. The electrophoretic mobility of individual spots of each family of topoisomers identified in immunodetections, with their corresponding increase in topological complexity (ΔC), expressed in mm/h, was compared for the different electrophoretic conditions (% agarose and voltage) used during the second dimension. (See Table 1 and Figure 5 for the electrophoretic mobility of topoisomers of Sc Dimers and CatAs under three different voltages applied during the second dimension.)
Table 1. Electrophoretic mobility of individual spots of ScDimers and CatAs identified in the immunodetections
Second dimension |
| Second dimension |
| |||||
| ΔC | 150V | 175V | 200V | ΔC | 150V | 175V | 200V | |
| 1 | 5.5 | 10.55 | 14.19 | 1 | 2.65 | 4.91 | 5.50 | |
| 2 | 5.5 | 10.59 | 14.25 | 2 | 2.35 | 4.32 | 4.50 | |
| 3 | 5.5 | 10.64 | 14.38 | 3 | 2.5 | 4.55 | 4.56 | |
| 4 | 5.5 | 10.64 | 14.44 | 4 | 2.8 | 5.14 | 4.81 | |
| 5 | 5.6 | 10.64 | 14.50 | 5 | 3.25 | 5.82 | 5.38 | |
| 6 | 5.65 | 10.68 | 14.56 | 6 | 3.8 | 6.64 | 6.19 | |
| 7 | 5.8 | 10.68 | 14.69 | 7 | 4.45 | 7.45 | ||
| 8 | 5.9 | 10.73 | 14.75 | 8 | 5.05 | 8.32 | ||
| 9 | 6.15 | 10.77 | 14.81 | 9 | 5.75 | 9.09 | ||
| 10 | 6.35 | 10.91 | 14.88 | |||||
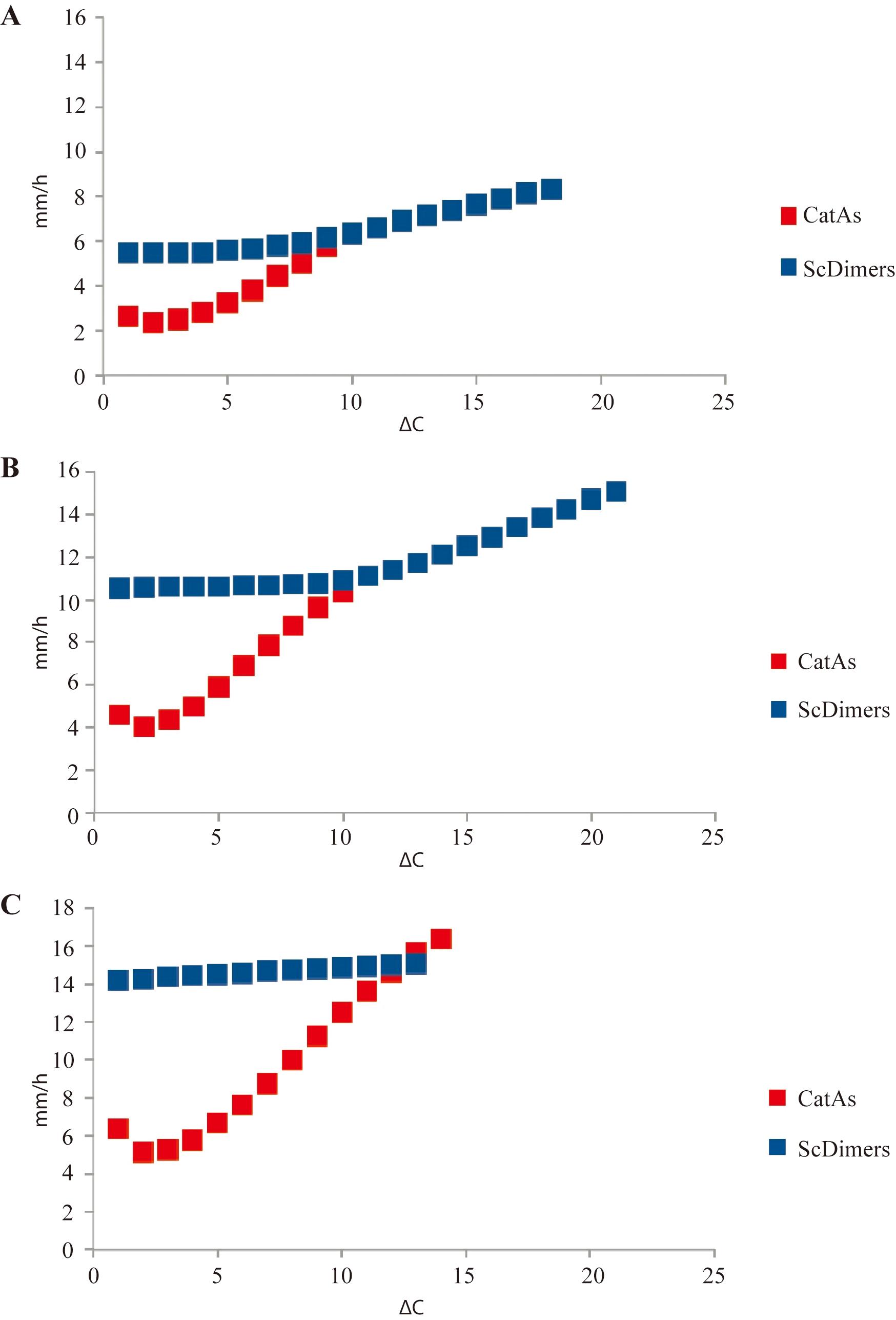
Figure 5. Comparison of electrophoretic mobility of supercoiled dimers (ScDimers) and monomeric-nicked catenanes (CatAs) under three different voltages during the second dimension of 2D gels. Electrophoretic mobility as a function of topological complexity (ΔC) is expressed in mm/h for the ScDimers and CatAs under three different voltages during the second dimension. A. 1% agarose gel run at 5 V/cm (i.e., 150 V in our electrophoretic chambers) for 10 h. B. 1% agarose gel run at 5.8 V/cm (i.e., 175 V) for 10 h. C. 1% agarose gel run at 6.6 V/cm (i.e., 200 V) for 10 h.
Validation of protocol
This protocol has been used and validated in the following research article:
Cebrián et al. (2015). Electrophoretic mobility of supercoiled, catenated and knotted DNA molecules. Nucleic Acids Res. Doi: 10.1093/nar/gku1255 (Figures 1, 2 and 5; Table 1).
General notes and troubleshooting
General notes
Commercial miniprep kits produce random DNA breaks in the plasmids molecules during their isolation and are not suitable for obtaining enriched samples of supercoiled, catenated, and knotted forms of the plasmids [34].
Treatment with suboptimal concentrations of the drug turns plasmids poorly supercoiled and, in this way, monomeric as well as dimeric topoisomers become clearly distinguished. In this way, ScDimers and KnDimers can be visualized in the same 2D gel. A similar result could be obtained using chloroquine 2D gels. However, chloroquine is not useful to resolve and identify nicked DNA (KnDimers and CatAs) as in these molecules the DNA is not under a torsional constraint (supercoiling relaxation), hence chloroquine does not have any topological effect.
DNA digestion with nicking enzymes releases torsional constrain (supercoiling relaxation) of the plasmids. In this protocol, DNA digestion with nicking enzyme Nb.BsmI is performed to remove DNA supercoiling present in our molecules; therefore, we convert all type of catenanes into CatAs and knotted DNA molecules are revealed.
A duplicate sample is loaded in the first lane of the gel only to check that first dimension of electrophoresis has been carried out correctly. Once the first dimension has finished, cut out the lane, place it carefully in ethidium bromide, and verify the correct migration of DNA samples with a UV transilluminator before proceeding to the second dimension of electrophoresis. This procedure is performed only after the first dimension. Once migration is verified, discard the gel and cut out your target lane (Figure 3). The ethidium bromide step must be performed with an extra sample (in a different lane) because ethidium bromide is a planar molecule that intercalates between the two strands of the DNA double helix altering DNA topology. Your target lane should not be exposed to ethidium bromide at any time during the electrophoresis to prevent DNA topology modification.
The capillary method uses paper towels to draw the transfer buffer through the gel to the membrane, placed on top of your gel. Make sure there is no shortcut between the wick and the paper towels; otherwise, if your paper towel layer touches the wick, the capillary action will bypass your gel and the DNA will not migrate from the gel onto the membrane. Parafilm or other wrap is often placed around the gel to frame its edges and prevent shortcuts.
Acknowledgments
The authors acknowledge the critics and continuous support of Andrzej Stasiak and all the current and former members of their groups. This work was sustained by grant BFU2011- 22489 to J.B.S. from the Spanish Ministerio de Economía y Competitividad. MJFN and VM were partially supported by an award granted by the National Council of Science and Technology (CONACYT), as Active Researchers of the National Incentive Program for Researchers (PRONII). We dedicate this work to the memory of our friend, mentor, and colleague, Jorge B. Schvartzman.
Competing interests
Authors declare no competing interests.
References
- Huberman, J. A. and Riggs, A. D. (1966). Autoradiography of chromosomal DNA fibers from Chinese hamster cells. Proc. Natl. Acad. Sci. U S A 55(3): 599–606. https://doi.org/10.1073/pnas.55.3.599.
- Michalet, X., Ekong, R., Fougerousse, F., Rousseaux, S., Schurra, C., Hornigold, N., van Slegtenhorst, M., Wolfe, J., Povey, S., Beckmann, J. S., et al. (1997). Dynamic molecular combing: stretching the whole human genome for high-resolution studies. Science 277(5331): 1518–1523. https://doi.org/10.1126/science.277.5331.1518.
- Sogo, J. M., Stasiak, A., Martinez-Robles, M. L., Krimer, D. B., Hernandez, P. and Schvartzman, J. B. (1999). Formation of knots in partially replicated DNA molecules. J. Mol. Biol. 286(3): 637–643. https://doi.org/10.1006/jmbi.1998.2510.
- Viguera, E., Hernandez, P., Krimer, D. B., Lurz, R. and Schvartzman, J. B. (2000). Visualisation of plasmid replication intermediates containing reversed forks. Nucleic Acids Res. 28(2): 498–503. https://doi.org/10.1093/nar/28.2.498.
- Sogo, J. M., Stahl, H., Koller, T. and Knippers, R. (1986). Structure of replicating simian virus 40 minichromosomes. The replication fork, core histone segregation and terminal structures. J. Mol. Biol. 189(1): 189–204. https://doi.org/10.1016/0022-2836(86)90390-6.
- Lopez, V., Martinez-Robles, M. L., Hernandez, P., Krimer, D. B. and Schvartzman, J. B. (2012). Topo IV is the topoisomerase that knots and unknots sister duplexes during DNA replication. Nucleic Acids Res. 40(8): 3563–3573. https://doi.org/10.1093/nar/gkr1237.
- Charvin, G., Bensimon, D. and Croquette, V. (2003). Single-molecule study of DNA unlinking by eukaryotic and prokaryotic type-II topoisomerases. Proc. Natl. Acad. Sci. U S A 100(17): 9820–9825. https://doi.org/10.1073/pnas.1631550100.
- Neuman, K. C., Charvin, G., Bensimon, D. and Croquette, V. (2009). Mechanisms of chiral discrimination by topoisomerase IV. Proc. Natl. Acad. Sci. U S A 106(17): 6986–6991. https://doi.org/10.1073/pnas.0900574106.
- Witz, G. and Stasiak, A. (2010). DNA supercoiling and its role in DNA decatenation and unknotting. Nucleic Acids Res. 38(7): 2119–2133. https://doi.org/10.1093/nar/gkp1161.
- Weber, C., Stasiak, A., De Los Rios, P. and Dietler, G. (2006). Numerical simulation of gel electrophoresis of DNA knots in weak and strong electric fields. Biophys. J. 90(9): 3100–3105. https://doi.org/10.1529/biophysj.105.070128.
- Martinez, V., Schaerer, C., Hernandez, P., Krimer, D. B., Schvartzman, J. B. and Fernandez-Nestosa, M. J. (2021). Distribution of torsional stress between the un-replicated and replicated regions in partially replicated molecules. J. Biomol. Struct. Dyn. 39(6): 2266–2277. https://doi.org/10.1080/07391102.2020.1751294.
- O'Donnol, D., Stasiak, A. and Buck, D. (2018). Two convergent pathways of DNA knotting in replicating DNA molecules as revealed by theta-curve analysis. Nucleic Acids Res. 46(17): 9181–9188. https://doi.org/10.1093/nar/gky559.
- Rawdon, E. J., Dorier, J., Racko, D., Millett, K. C. and Stasiak, A. (2016). How topoisomerase IV can efficiently unknot and decatenate negatively supercoiled DNA molecules without causing their torsional relaxation. Nucleic Acids Res. 44(10): 4528–4538. https://doi.org/10.1093/nar/gkw311.
- Bell, L. and Byers, B. (1983). Separation of branched from linear DNA by two-dimensional gel electrophoresis. Anal. Biochem. 130(2): 527–535. https://doi.org/10.1016/0003-2697(83)90628-0.
- Adams, D. E., Shekhtman, E. M., Zechiedrich, E. L., Schmid, M. B. and Cozzarelli, N. R. (1992). The role of topoisomerase IV in partitioning bacterial replicons and the structure of catenated intermediates in DNA replication. Cell 71(2): 277–288. https://doi.org/10.1016/0092-8674(92)90356-h.
- Brewer, B. J. and Fangman, W. L. (1987). The localization of replication origins on ARS plasmids in S. cerevisiae. Cell 51(3): 463–471. https://doi.org/10.1016/0092-8674(87)90642-8.
- Brewer, B. J., Sena, E. P. and Fangman, W. L. (1988). Analysis of replication intermediates by two-dimensional agarose gel electrophoresis. Cancer Cells 6: 229–234.
- Fierro-Fernandez, M., Hernandez, P., Krimer, D. B. and Schvartzman, J. B. (2007). Replication fork reversal occurs spontaneously after digestion but is constrained in supercoiled domains. J. Biol. Chem. 282(25): 18190–18196. https://doi.org/10.1074/jbc.M701559200.
- Fierro-Fernandez, M., Hernandez, P., Krimer, D. B., Stasiak, A. and Schvartzman, J. B. (2007). Topological locking restrains replication fork reversal. Proc. Natl. Acad. Sci. U S A 104(5): 1500–1505. https://doi.org/10.1073/pnas.0609204104.
- Keller, W. (1975). Determination of the number of superhelical turns in simian virus 40 DNA by gel electrophoresis. Proc. Natl. Acad. Sci. U S A 72(12): 4876–4880. https://doi.org/10.1073/pnas.72.12.4876.
- Martinez-Robles, M. L., Witz, G., Hernandez, P., Schvartzman, J. B., Stasiak, A. and Krimer, D. B. (2009). Interplay of DNA supercoiling and catenation during the segregation of sister duplexes. Nucleic Acids Res. 37(15): 5126–5137. https://doi.org/10.1093/nar/gkp530.
- Schvartzman, J. B. and Stasiak, A. (2004). A topological view of the replicon. EMBO Rep. 5(3): 256–261. https://doi.org/10.1038/sj.embor.7400101.
- Schvartzman, J. B., Hernandez, P., Krimer, D. B., Dorier, J. and Stasiak, A. (2019). Closing the DNA replication cycle: from simple circular molecules to supercoiled and knotted DNA catenanes. Nucleic Acids Res. 47(14): 7182–7198. https://doi.org/10.1093/nar/gkz586.
- Stellwagen, N. C. (2009). Electrophoresis of DNA in agarose gels, polyacrylamide gels and in free solution. Electrophoresis 30 Suppl 1(Suppl 1): S188–195. https://doi.org/10.1002/elps.200900052.
- Sundin, O. and Varshavsky, A. (1980). Terminal stages of SV40 DNA replication proceed via multiply intertwined catenated dimers. Cell 21(1): 103–114. https://doi.org/10.1016/0092-8674(80)90118-x.
- Thorne, H. V. (1966). Electrophoretic separation of polyoma virus DNA from host cell DNA. Virology 29(2): 234–239. https://doi.org/10.1016/0042-6822(66)90029-8.
- Neelsen, K. J. and Lopes, M. (2015). Replication fork reversal in eukaryotes: from dead end to dynamic response. Nat. Rev. Mol. Cell Biol. 16(4): 207–220. https://doi.org/10.1038/nrm3935.
- Cebrian, J., Castan, A., Martinez, V., Kadomatsu-Hermosa, M. J., Parra, C., Fernandez-Nestosa, M. J., Schaerer, C., Hernandez, P., Krimer, D. B. and Schvartzman, J. B. (2015). Direct Evidence for the Formation of Precatenanes during DNA Replication. J. Biol. Chem. 290(22): 13725–13735. https://doi.org/10.1074/jbc.M115.642272.
- Cebrian, J., Martinez, V., Hernandez, P., Krimer, D. B., Fernandez-Nestosa, M. J. and Schvartzman, J. B. (2021). Two-Dimensional Gel Electrophoresis to Study the Activity of Type IIA Topoisomerases on Plasmid Replication Intermediates. Biology (Basel) 10(11). https://doi.org/10.3390/biology10111195.
- Cebrian, J., Monturus, E., Martinez-Robles, M. L., Hernandez, P., Krimer, D. B. and Schvartzman, J. B. (2014). Topoisomerase 2 is dispensable for the replication and segregation of small yeast artificial chromosomes (YACs). PLoS One 9(8): e104995. https://doi.org/10.1371/journal.pone.0104995.
- Sogo, J. M., Lopes, M. and Foiani, M. (2002). Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science 297(5581): 599–602. https://doi.org/10.1126/science.1074023.
- Santamaria, D., Hernandez, P., Martinez-Robles, M. L., Krimer, D. B. and Schvartzman, J. B. (2000). Premature termination of DNA replication in plasmids carrying two inversely oriented ColE1 origins. J. Mol. Biol. 300(1): 75–82. https://doi.org/10.1006/jmbi.2000.3843.
- Cebrian, J., Kadomatsu-Hermosa, M. J., Castan, A., Martinez, V., Parra, C., Fernandez-Nestosa, M. J., Schaerer, C., Martinez-Robles, M. L., Hernandez, P., Krimer, D. B., et al. (2015). Electrophoretic mobility of supercoiled, catenated and knotted DNA molecules. Nucleic Acids Res. 43(4): e24. https://doi.org/10.1093/nar/gku1255.
- Martin-Parras, L., Lucas, I., Martinez-Robles, M. L., Hernandez, P., Krimer, D. B., Hyrien, O. and Schvartzman, J. B. (1998). Topological complexity of different populations of pBR322 as visualized by two-dimensional agarose gel electrophoresis. Nucleic Acids Res. 26(14): 3424–3432. https://doi.org/10.1093/nar/26.14.3424.
- Hanahan, D. (1983). Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol. 166(4): 557–580. https://doi.org/10.1016/s0022-2836(83)80284-8.
- Kato, J., Nishimura, Y., Imamura, R., Niki, H., Hiraga, S. and Suzuki, H. (1990). New topoisomerase essential for chromosome segregation in E. coli. Cell 63(2): 393–404. https://doi.org/10.1016/0092-8674(90)90172-b.
- Zechiedrich, E. L., Khodursky, A. B. and Cozzarelli, N. R. (1997). Topoisomerase IV, not gyrase, decatenates products of site-specific recombination in Escherichia coli. Genes Dev. 11(19): 2580–2592. https://doi.org/10.1101/gad.11.19.2580.
- Zechiedrich, E. L., Khodursky, A. B., Bachellier, S., Schneider, R., Chen, D., Lilley, D. M. and Cozzarelli, N. R. (2000). Roles of topoisomerases in maintaining steady-state DNA supercoiling in Escherichia coli. J. Biol. Chem. 275(11): 8103–8113. https://doi.org/10.1074/jbc.275.11.8103.
- Drlica, K. (1999). Mechanism of fluoroquinolone action. Curr. Opin. Microbiol. 2(5): 504-508. https://doi.org/10.1016/s1369-5274(99)00008-9.
- Lopez-estrano, C., Schvartzman, J. B., Krimer, D. B. and Hernandez, P. (1998). Co-localization of polar replication fork barriers and rRNA transcription terminators in mouse rDNA. J. Mol. Biol. 277(2): 249–256. https://doi.org/10.1006/jmbi.1997.1607.
Article Information
Copyright
© 2024 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Cebrián, J., Martínez, V., Hernández, P., Krimer, D. B., Martínez-Robles, M. L., Schvartzman, J. B. and Fernández-Nestosa, M. J. (2024). Electrophoretic Mobility Assay to Separate Supercoiled, Catenated, and Knotted DNA Molecules. Bio-protocol 14(9): e4983. DOI: 10.21769/BioProtoc.4983.
Category
Molecular Biology > DNA > DNA structure
Molecular Biology > DNA > Electrophoresis
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.