- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

CRISPR/Cas9 Ribonucleoprotein-Mediated Mutagenesis in Sporisorium reilianum

Published: Vol 14, Iss 8, Apr 20, 2024 DOI: 10.21769/BioProtoc.4978 Views: 2767
Reviewed by: Shweta PanchalEugenio LlorensXiaofei Liang
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9 (CRISPR/Cas9) has become the state of the art for mutagenesis in filamentous fungi. Here, we describe a ribonucleoprotein complex (RNP)-mediated CRISPR/Cas9 for mutagenesis in Sporisorium reilianum. The efficiency of the method was tested in vitro with a cleavage assay as well as in vivo with a GFP-expressing S. reilianum strain. We applied this method to generate frameshift- and knock-out mutants in S. reilianum without a resistance marker by using an auto-replicating plasmid for selection. The RNP-mediated CRISPR/Cas9 increased the mutagenesis efficiency, can be applied for all kinds of mutations, and enables a marker-free genome editing in S. reilianum.
Key features
• First CRISPR/Cas9 application in S. reilianum.
• Generation of S. reilianum mutants without genomic integration of resistance marker.
• Allows the generation of multiple gene knockouts as well as deletion of large genomic regions.
Keywords: CRISPR/Cas9Background
The smut fungi consist of more than 1,500 species, being highly economically important due to their infection of relevant crops such as barley, sorghum, wheat, and maize [1]. The majority of smut fungi infect their host systemically through the roots and replace the inflorescences with teliospores without causing symptoms during early infection stages [2,3]. One example of this systemic infection is Sporisorium reilianum f. sp. zeae, which is the causal agent of maize head smut. S. reilianum is closely related to the intensively investigated model organism Ustilago maydis. However, they differ in their mode of infection as well as in the site of symptom development [4,5]. In 2010, a genome sequence of S. reilianum f. sp. zeae was published, which, together with the U. maydis genome, provided the foundation for systematic identification and genetic manipulation of effector genes contributing to virulence [6,7]. Genome comparison of U. maydis and S. reilianum revealed conserved effector genes even though they differ drastically in their pathogenesis on the same host, Zea mays. To characterize effector genes and their contribution to virulence, knock-out mutants are generated and compared to the wild type. In the past, U. maydis knock-out mutants were generated using PCR-amplified donor templates with resistance markers for gene replacements [8]. Importantly, it was shown that not only the genomic locus but also the integration of resistance markers can negatively influence the expression of reintegrated genes [9]. Recently, the mutagenesis of U. maydis was drastically improved with a marker-free approach using clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9 (CRISPR/Cas9) [10,11] and further developed for a seamless gene conversion approach [12]. In contrast to U. maydis, the generation of knockout mutants in S. reilianum is still dependent on resistance markers, and multiple gene knockouts are hampered by the limited number (i.e., carboxine, hygromycin, nourseothricin, and phleomycin) of available resistance markers [8]. However, the plasmidbased CRISPR/Cas9 transformation as used in U. maydis has not been successful for S. reilianum.
CRISPR/Cas9, originating from the adaptive immune system of Streptococcus pyogenes, has been broadly adapted to many eukaryotic systems. It is a versatile tool for mutagenesis in various filamentous fungi [13]. The delivery strategies of CRISPR/Cas9 differ between fungal species: (i) stable genomic integration of cas9, (ii) transient delivery of Cas9 where the expression of Cas9 is dependent on selection pressure of a self-replicating plasmid or a telomere vector [10,14], or (iii) ribonucleoprotein complex (RNP)mediated transformation [15,14]. Here, we describe CRISPR/Cas9 applications in S. reilianum using an RNPmediated transformation approach. We demonstrate the generation of frameshifts as well as knock-out mutants mediated by RNPs, thereby generally improving the mutagenesis, and, for the first time, enable a marker-free editing in S. reilianum.
Materials and reagents
Biological materials
S. reilianum strains were stored at -80 °C in 30% glycerol. For transformation, S. reilianum wildtype strains SRZ1 and SRZ2 [7] were used.
Reagents
Single-guide RNA (sgRNA) synthesis
T4 DNA polymerase (New England Biolabs, catalog number: M0203S), storage: -20 °C
NEBufferTM r2.1 (50 mM NaCl, 10 mM Tris-HCl, 10 mM MgCl2, 100 μg/mL BSA, pH 7.9), storage: -20 °C (New England Biolabs, catalog number: B7202S)
dNTPs (DNA) (Carl Roth, catalog number: K039.1), storage: -20 °C
NucleoSpin® Gel and PCR clean up (Machery and Nagel, catalog number: 740.609.250), storage: RT
HiScribe® T7 High Yield RNA Synthesis kit (New England Biolabs, catalog number: E2040S), storage: -20 °C
DNase I (Thermo Fisher, catalog number: EN0521), storage: -20 °C
DNase I buffer (Thermo Fisher, catalog number: EN0521), storage: -20 °C
RNA Clean & Concentrator 25 kit (Zymo Research, catalog numbers: R1017 and R1018), storage: RT
Purple loading dye (New England Biolabs, catalog number: B7024S); ingredients: 2.5% Ficoll®-400, 10 mM EDTA, 0.08% SDS, 0.02% Dye 1, 0.02% Dye 2, pH 8; storage: RT
Nuclease-free water, storage: RT
Formation of RNP and in vitro cleavage assay
EnGen® Spy Cas9 NLS + NEB buffer r3.1 (New England Biolabs, catalog number: M0667)
500 mM Ethylenediaminetetraacetic acid (EDTA) (Carl Roth, catalog number: 8043.2)
Proteinase K (Thermo Fisher, catalog number: EO0491)
100 bp ladder (New England Biolabs, catalog number: N3231S)
Universal agarose (Bio-Budget, catalog number: 10-35-1020)
1% Ethidium bromide solution (Carl Roth, catalog number: 2218.2)
Potato dextrose agar (PDA) plates (39 g/L) (BD, DifcoTM, catalog number: 213400)
Tris base (Sigma, catalog number: 102262896)
Acetic acid (VWR, catalog number: 20103.330)
EDTA 0.5 M pH 8.0 (Carl Roth, catalog number: 8043.2)
Protoplasting and transformation of S. reilianum
Novozym 234 [Novo Nordisc; Denmark, not available anymore; alternative: lysing enzyme from Trichoderma harzianum (Sigma, catalog number: SLBJ0553V)]
Sodium citrate (Carl Roth, catalog number. 3580.1)
Sorbitol (Sigma, catalog number: 102466217)
Citrate acid (Carl Roth, catalog number: X863.2)
Sorbitol (Sigma, catalog number: 102466217)
Tris-HCl (Carl Roth, catalog number: 9090.3)
CaCl2 (Sigma, catalog number: 1002825086)
Poly(ethylene glycol) PEG MW3350 (Sigma, P4338, catalog number: 102604683)
BactoTM-Yeast-Extract (Thermo Fisher, Gibco, catalog number: 212720)
BactoTMPeptone (BD, Difco, catalog number: 211820)
Sucrose (Carl Roth, catalog number: 4621.2)
Sorbitol (Sigma, catalog number: 102466217)
BactoTM-Agar (BD, catalog number: 214030)
Potato dextrose agar (PDA) plates (BD, DifcoTM, catalog number: 213400)
Carboxine (5 mg/mL) (Sigma, catalog number: 102085144)
Heparin sodium salt from porcine intestinal mucosa (15 mg/mL) (Sigma, catalog number: 1001937695)
Solutions
50× TAE buffer (see Recipes)
1× TAE buffer (see Recipes)
SCS buffer (see Recipes)
STC buffer (see Recipes)
STC/40% PEG (see Recipes)
Regeneration agar light (see Recipes)
Recipes
50× TAE buffer
Reagent Final concentration Quantity or Volume Tris base 2 M (v/v) 242.0 g Acetic acid 2 M (v/v) 57.1 mL EDTA 0.5 M pH 8.0 10% (v/v) 100.0 mL 1× TAE buffer
Reagent Final concentration Quantity or Volume 50× TAE buffer 2% (v/v) 20.0 mL Deionized water 98% (v/v) 980.0 mL SCS buffer
Reagent Final concentration Quantity or Volume Solution 1: Sodium citrate, pH 5 0.6% (w/v) 5.9 ml Sorbitol 18.2% (w/v) 182.0 g Solution 2: Citrate acid 0.4% (w/v) 4.2 g Sorbitol 18.2% (w/v) 182.0 g Solution 1 and 2 are mixed until pH 5.8 is reached (ratio ~5:1) and autoclaved.
STC buffer
Reagent Final concentration Quantity or Volume Sorbitol 50% (v/v) 500.0 mL Tris-HCl, 1 M pH 7.5 1% (v/v) 5.0 mL CaCl2, 1 M, sterile-filtrated (100 mL total volume is enough) 10% (v/v) 50.0 mL STC/40% PEG
Reagent Final concentration Quantity or Volume STC buffer 60% (v/v) 600.0 mL Poly(ethylene glycol) PEG, MW3350; sterilefiltrated, (50 mL total volume is enough) 40% (w/v) 400.0 g Regeneration agar light
Reagent Final concentration Quantity or Volume BactoTM yeast extract 1% (w/v) 10.0 g BactoTM peptone 0.4% (w/v) 20.0 g Sucrose 0.4% (w/v) 20.0 g Sorbitol 18.2% (w/v) 182.2 g BactoTM agar 1.5% (w/v) 15.0 g
Laboratory supplies
PCR machine (Bio-Rad, model: T100TM Thermal Cycler)
Microfuge for PCR tubes (VWR, model: Ministar)
Tabletop centrifuge (VWR, model: Microstar 17)
37 °C incubator (Memmert, model: UN110)
28 °C incubator/room
Optional: Polyacrylamide gel electrophoresis (SDS-PAGE) equipment (Bio-Rad, model: PowerPacTM Basic, Mini-Protean® Tetra System)
Agarose gel electrophoresis equipment
Nanodrop (Thermo Scientific, model: Nanodrop 2000c)
ChemiDocTM MP imaging system (or equivalent imaging system), with GFP filter (Bio-Rad, model: Universal Hood III)
Geldoc: visualization of DNA by UV radiation using a gel documentation unit (Peqlab/VWR, model: EBOX VX5)
Equipment
PCR tubes and 1.5 ml Eppendorf tubes
Sterile cut tips (1,000 µL and 20 µL)
Procedure
In vitro transcription of sgRNA
Design protospacer in CHOPCHOP sgRNA designer (https://chopchop.cbu.uib.no/) using S. reilianum as target organism. Choose the protospacer sequence starting with a G, which is needed for initiating the transcription by T7 RNA polymerase. If there is no desired protospacer starting with G, add an additional G upstream of the chosen protospacer sequence (21 nt).
Add T7 RNA polymerase promoter sequence and overlapping scaffold sequence upstream and downstream of the chosen protospacer sequence, respectively, and order the gene-specific oligonucleotide (Table 1). In addition, a reverse complementary constant oligonucleotide is needed, which harbors the scaffold and terminator sequence and a 20 nt overlap to the scaffold sequence of the gene-specific oligonucleotide (Table 1).
Table 1. Sequences of oligonucleotides for sgRNA synthesis
Oligo Sequence Gene-specific CAAAATTCCATTCTACAAC-GNNNNNNNNNNNNNNNNNNN-GTTTTAGAGCTAGAAATAGCAAG Constant AAAAGCACCGACTCGGTGCCACTTTTTCAAGTTGATAACGGACTAGCCTTATTTTAACTTGCTATTTCTAGCTCTAAAAC Note: The underlined sequence of the constant oligo depicts the overlapping part with the gene-specific oligo.
Mix both oligonucleotides in a 1:1 ratio as follows:
1 µL of protospacer oligo 100 µM stock
1 µL of constant oligo 100 µM stock
8 µL of H2O
10 µL total
Anneal the oligos using the following program in PCR machine:
95 °C for 5 min
95 °C to 85 °C at -2 °C/s
85 °C to 25 °C at -0.1 °C/s
4 °C pause
Add T4 DNA polymerase to fill in the overhangs:
2.5 µL of dNTPs (10 mM)
2 µL of NEBufferTM r2.1(10×)
5 µL of H2O
0.5 µL of T4 DNA polymerase
10 µL total
Incubate at 12 °C for 20 min in a PCR machine.
Purify the product with a PCR clean-up kit, measure the concentration with Nanodrop, and verify the PCR product on a 2%–3% TAE agarose gel.
Use 2 µg of the resulting DNA from above as template and the HiScribe T7 High Yield RNA Synthesis kit for the following reaction (NEB, protocol for small RNAs):
6 µL of NTPs (25 mM each in stock)
2 µL of 10× T7 buffer
1.5 µL of T7 RNA polymerase mix
X µL of Template (2 µg DNA template, step 6)
Y µL of nuclease-free H2O (add to 20 µL)
20 µL total
Flip the tube, vortex shortly, and incubate at 37 °C overnight.
The next day, add 14 µL of nuclease-free H2O, 4 µL of DNase I buffer (10×), and 2 µL of DNase I and incubate at 37 °C for 15 min.
Caution: Small RNA is easy degradable; work in a RNase-free space and use gloves and a lab coat for all following steps!
Purify the resulting sgRNA with the RNA Clean & Concentrator 25 kit and use the manufacturer's protocol (manual, page 5).
Optional: Check the quality of the RNA on 10% denaturing PAA gel using TBE buffer (89 mM Tris base, 89 mM boric acid, and 2 mM sodium EDTA) and 8 M urea and TBE as running buffer [14].
Measure the concentration by Nanodrop and proceed with in vitro cleavage assay (section B).
Pause point: You can freeze the sgRNA at -80 °C and continue the next day; long-term storage of sgRNA is also possible at -80 °C. See Troubleshooting 1.
In vitro cleavage assay
To test the in vitro efficiency of the designed sgRNA, mix 1.5 µL of Cas9 (NEB) and ~1.5 µg of the sgRNA (1:1 molar ratio) and incubate it for 10 min at RT (Figure 1B).
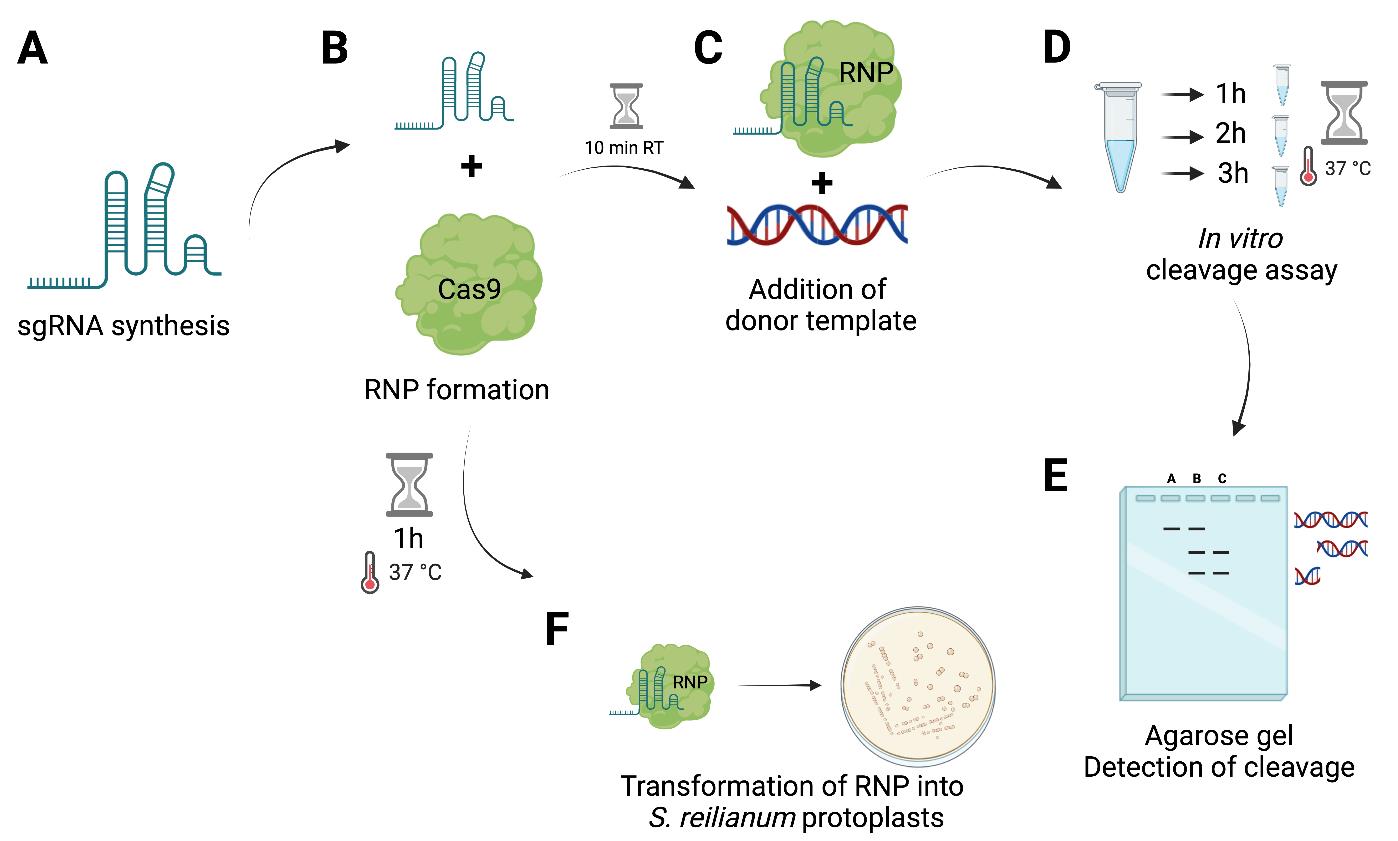
Figure 1. Graphical overview of the workflow of ribonucleoprotein complex (RNP)-mediated transformation in S. reilianum. (A) In vitro synthesis of sgRNA using T7 HiScribe kit. (B) RNP formation of in vitro–transcribed sgRNA with SpCas9. (C) Alternatively: To perform an in vitro cleavage assay, incubation at room temperature (RT) for 10 min and subsequent addition of a donor template (amplification of the gene of interest region) and incubation at 37 °C for 3 h is conducted. (D) Sampling of 10 µL of reaction mix after 1, 2, and 3 h (or alternatively overnight). (E) Visualization of in vitro cleavage on a 1.5% agarose gel using 100 bp ladder. (F) RNP incubation for 1 h at 37 °C prior to transformation into S. reilianum protoplasts. Figure was created with biorender.com.Afterwards, add 333 ng of a DNA cleavage template (PCR product of the region of interest) (Figure 1C).
After 1, 2, and 3 h take 10 µL samples (Figure 1D) and stop the reaction by adding 1 µL of 500 mM EDTA, pH 8.
Subsequently, add 1 µL of proteinase K to the reaction and incubate the reaction mix for 30 min at 50 °C for degradation of Cas9.
Stop the reaction by the addition of 1× purple loading dye.
After the collection of all samples, check cleavage on an 1.5% agarose gel with 100 bp ladder (stained with ethidium bromide solution) visualized using a Gel-Doc (Figure 1E, see Troubleshooting 2).
Assembly of RNP for transformation into S. reilianum
Use 2 µg of the in vitro–transcribed sgRNA targeting the gene of interest and mix it with 6 µg of SpCas9.
Subsequently, add 1× NEBufferTM 3.1 and water in a minimum volume (Figure 1B).
After mixing and centrifugation, incubate the reaction for 1 h at 37 °C prior to transformation (Figure 1B).
Transformation of S. reilianum
Prepare S. reilianum protoplasts using Novozym 234 as described previously [8].
For RNP transformation (Figure 1E), thaw the protoplasts for 5 min on ice.
Add a self-replicating plasmid with antibiotic resistance cassette [e.g., pNEBUC—Carboxine (Cbx); Brachmann et al. [8], replicating in S. reilianum], the RNP (formed in section C), 1 µL of 15 mg/mL heparin, and, optionally, 1.5 µg of a donor template to the protoplasts (Figure 2).
Note: The self-replicating plasmid is lost when the selection for Cbx resistance is stopped. So far, we could not report an integration into the genome of S. reilianum.
Incubate the protoplasts for 10 min on ice.
Add 500 µL of STC/40% PEG and resuspend the protoplasts carefully with a tip-cut blue tip until the liquid looks homogenous without clumps (5–8 times pipetting up and down).
Incubate the protoplasts for another 15 min on ice.
Spread the protoplasts on a regeneration agar light plate with two layers [bottom layer: corresponding selective antibiotic (for pNEBUC—carboxin: 2.5 µg/mL), top layer: without antibiotic resistance].
The next day, flip the transformation plate upside down.
After four days, use a blue tip to single out transformants from regeneration agar to PDA + Carboxin (2.5 µg/mL) for 2–3 days.
Afterwards, transfer a single colony for two days to PDA plates to lose the resistance.
Subsequently, DNA is isolated [16] and used for further confirmation (see section E).
RNP-assisted homologous recombination to generate a knockout in S. reilianum
For the generation of an antibiotic-resistance-free knock-out mutant in S. reilianum, a CRISPR/Cas9-mediated homology-directed repair was exploited. To do this, a donor template is generated by cloning the 1 kb homology flanking regions of the target gene into a MOCLO vector TK#1_pAGM1311 by Gibson assembly (Figure 2).
For the transformation of S. reilianum protoplasts (see section D), add the donor template together with a self-replicating plasmid (pNEBUC), the RNP (with a sgRNA against the target region), and 1 µL of heparin.
Note: The transformation efficiency is high > 100 colonies; if your efficiency is lower, repeat protoplasting and transformation.
Transfer obtained transformants as described above (see section C).
Isolate DNA of the transformants and the wild type.
Conduct a PCR using the forward primer of the left flank and the reverse primer of the right flank (Figure 2C) and compare the band sizes to the wild type (Figure 2D).
Putative positive mutants from PCR are selected for further verification via Southern blot [17,18] using the deletion construct (left flank + right flank), previously used as a donor template, as probe for hybridization.
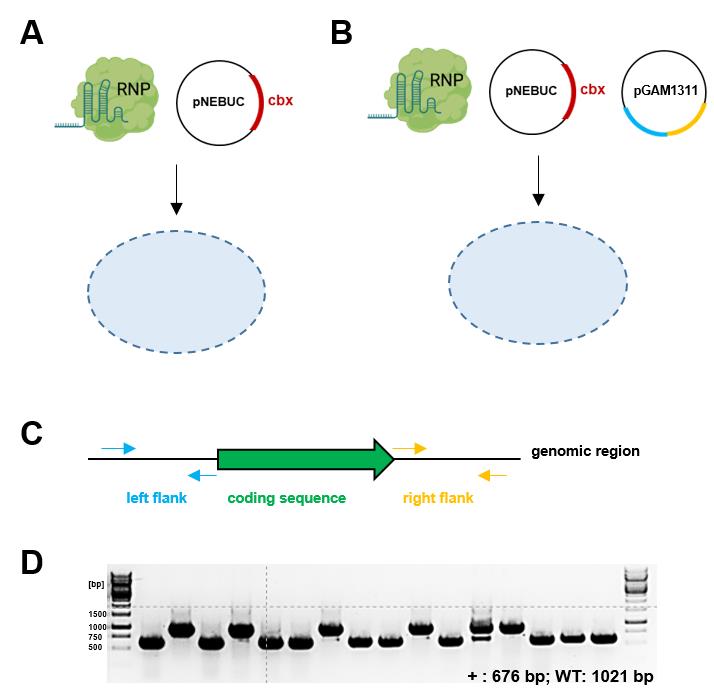
Figure 2. Transformation of S. reilianum using ribonucleoprotein complex (RNP) with and without a donor template. (A) Generation of frameshift mutants using RNP-mediated transformation. Generated frameshift mutants are screened by sequencing or as described in Figure 3 by the loss of fluorescence. (B) Generation of knock-out mutants using RNP-assisted homologous recombination with a donor template. (C) Donor template design for the generation of a knockout. 1 kb flanking regions of the coding sequence are amplified by PCR with overhangs for Gibson assembly into backbone TK#1_pAGM1311 (MOCLO backbone, Level -1). (D) Example of deletion mutant verification PCR using Left Flank (LF) forward primer and Right Flank (RF) reverse primer [expected sizes: WT, 1021 bp; mutant (+), 676 bp; efficiency: 62.5% (10/16)]. The efficiency can vary between different genomic loci. Figure 2A and B were created with biorender.com.
Validation of protocol
The efficiency of the RNP CRISPR/Cas9 can, for instance, be tested with GFP fluorescence as a readout (Figure 3). To test the efficiency in S. reilianum, a strain harboring a single integration of GFP controlled by pOTEF (constitutive promoter) was generated in the ip locus of SRZ2 strain and confirmed via Southern blot (Figure S1). For the transformation of S. reilianum protoplasts, a sgRNA against GFP together with the Cas9 in a RNP (see section C) and an auto-replicating plasmid (pNEBUC) for selection on regeneration agar was used. Transformants were singled out after four days of incubation at 28 °C on PDA + Cbx (2.5 mg/mL) and, after two days, were transferred to PDA plates and checked for their fluorescence using a Chemi-Doc.
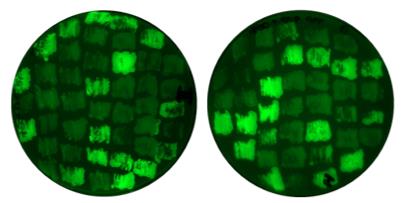
Figure 3. GFP as target for ribonucleoprotein complex (RNP)-mediated transformation in S. reilianum. An example shows the efficiency of RNPmediated CRISPR/Cas9 transformation in S. reilianum. A S. reilianum SRZ2 strain expressing GFP under pOTEF promoter was generated. sgRNA+Cas9 targeting GFP coding sequence was transformed, and mutants with frameshift lose the GFP signal. Efficiency for GFP sgRNA: ~41% (34/83).
General notes and troubleshooting
Troubleshooting
| No. | Step | Problem | Suggestion/solution |
| 1 | sgRNA synthesis | Low concentration (<500 ng/µL) | Do not proceed with transformation, repeat synthesis; high concentration in minimum volume is needed |
| 2 | In vitro cleavage assay | No bands after cleavage | 1) Test functionality of Cas9 enzyme (use a control) 2) Design of new sgRNAs |
Acknowledgments
This project has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (grant agreement No. 771035), as well as funding by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy, EXC-2048/1- Project ID: 390686111. This protocol was adapted from previous work [14].
References
- Begerow, D., Göker, M., Lutz, M. and Stoll, M. (2004). On the evolution of smut fungi on their hosts. Front. Basidiomycote Mycol. 81–98.
- Martinez, C., Roux, C., Jauneau, A. and Dargent, R. (2002). The biological cycle ofSporisorium reilianumf. sp. zeae: an overview using microscopy. Mycologia 94(3): 505–514.
- Laurie, J. D., Ali, S., Linning, R., Mannhaupt, G., Wong, P., Güldener, U., Münsterkötter, M., Moore, R., Kahmann, R., Bakkeren, G., et al. (2012). Genome Comparison of Barley and Maize Smut Fungi Reveals Targeted Loss of RNA Silencing Components and Species-Specific Presence of Transposable Elements. Plant Cell 24(5): 1733–1745.
- Begerow, D., Stoll, M. and Bauer, R. (2006). A phylogenetic hypothesis of Ustilaginomycotina based on multiple gene analyses and morphological data. Mycologia 98(6): 906–916.
- Steinberg, G. and Perez-Martin, J. (2008). Ustilago maydis, a new fungal model system for cell biology. Trends Cell Biol. 18(2): 61–67.
- Kämper, J., Kahmann, R., Bölker, M., Ma, L. J., Brefort, T., Saville, B. J., Banuett, F., Kronstad, J. W., Gold, S. E., Müller, O., et al. (2006). Insights from the genome of the biotrophic fungal plant pathogen Ustilago maydis. Nature 444(7115): 97–101.
- Schirawski, J., Mannhaupt, G., Münch, K., Brefort, T., Schipper, K., Doehlemann, G., Di Stasio, M., Rössel, N., Mendoza-Mendoza, A., Pester, D., et al. (2010). Pathogenicity Determinants in Smut Fungi Revealed by Genome Comparison. Science 330(6010): 1546–1548.
- Brachmann, A., König, J., Julius, C. and Feldbrügge, M. (2004). A reverse genetic approach for generating gene replacement mutants in Ustilago maydis. Mol. Genet. Genomics 272(2): 216–226.
- Schmitz, L., Kronstad, J. W. and Heimel, K. (2019). Conditional gene expression reveals stage‐specific functions of the unfolded protein response in the Ustilago maydis–maize pathosystem. Mol. Plant Pathol. 21(2): 258–271.
- Schuster, M., Schweizer, G., Reissmann, S. and Kahmann, R. (2016). Genome editing in Ustilago maydis using the CRISPR–Cas system. Fungal Genet. Biol. 89: 3–9.
- Zuo, W., Depotter, J. R. and Doehlemann, G. (2020). Cas9HF1 enhanced specificity in Ustilago maydis. Fungal Biol. 124: 228–234.
- Zuo, W., Depotter, J. R. L., Gupta, D. K., Thines, M. and Doehlemann, G. (2021). Cross‐species analysis between the maize smut fungi Ustilago maydis and Sporisorium reilianum highlights the role of transcriptional change of effector orthologs for virulence and disease. New Phytol. 232(2): 719–733.
- Schuster, M. and Kahmann, R. (2019). CRISPR-Cas9 genome editing approaches in filamentous fungi and oomycetes. Fungal Genet. Biol. 130: 43–53.
- Leisen, T., Bietz, F., Werner, J., Wegner, A., Schaffrath, U., Scheuring, D., Willmund, F., Mosbach, A., Scalliet, G., Hahn, M., et al. (2020). CRISPR/Cas with ribonucleoprotein complexes and transiently selected telomere vectors allows highly efficient marker-free and multiple genome editing in Botrytis cinerea. PLoS Pathog. 16(8): e1008326.
- Foster, A. J., Martin-Urdiroz, M., Yan, X., Wright, H. S., Soanes, D. M. and Talbot, N. J. (2018). CRISPR-Cas9 ribonucleoprotein-mediated co-editing and counterselection in the rice blast fungus. Sci. Rep. 8(1): 14355.
- Hoffman, C. S. and Winston, F. (1987). A ten-minute DNA preparation from yeast efficiently releases autonomous plasmids for transformaion of Escherichia coli. Gene 57: 267–272.
- Southern, E. (1975). Detection of specific sequences among DNA fragments separated by gel electrophoresis. J. Mol. Biol. 98(3): 503–517.
- Sambrook, J., Fritsch, E. F. and Maniatis, T. (1989). Molecular cloning: a laboratory manual (No. Ed. 2). Cold Spring Harbor Laboratory Press.
Supplementary information
The following supporting information can be downloaded here:
- Figure S1. Southern blot of single integrated GFP into SRZ2 strain.
Article Information
Copyright
© 2024 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Werner, J., Zuo, W. and Doehlemann, G. (2024). CRISPR/Cas9 Ribonucleoprotein-Mediated Mutagenesis in Sporisorium reilianum. Bio-protocol 14(8): e4978. DOI: 10.21769/BioProtoc.4978.
Category
Microbiology > Microbial genetics > CRISPR-Cas9
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.