- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

CRISPR/dCas9-Tet1-Mediated DNA Methylation Editing
Published: Vol 14, Iss 8, Apr 20, 2024 DOI: 10.21769/BioProtoc.4976 Views: 4710
Reviewed by: Clara Morral MartinezPhilipp WörsdörferAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jan 2023

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
DNA methylation is a key epigenetic mechanism underlying many biological processes, and its aberrant regulation has been tightly associated with various human diseases. Precise manipulation of DNA methylation holds the promise to advance our understanding of this critical mechanism and to develop novel therapeutic methods. Previously, we were only able to alter genome-wide DNA methylation by treating with small molecules (e.g., 5-Aza-2-deoxycytidine) or perturbing relevant genes (e.g., DNA methyltransferase) targetlessly, which makes it challenging to investigate the functional significance of this epigenetic mark at specific genomic loci. By fusing the catalytic domain of a key enzyme in the DNA demethylation process (Ten-eleven translocation dioxygenases 1, Tet1) with a reprogrammable sequence-specific DNA-targeting molecular protein, dCas9, we developed a DNA methylation editing tool (dCas9-Tet1) to demethylate specific genomic loci in a targeted manner. This dCas9-Tet1 system allows us to study the role of DNA methylation at almost any given loci with only the replacement of a single-guide RNA. Here, we describe a protocol that enables modular and scalable manipulation of DNA methylation at specific genomic loci in various cell cultures with high efficiency and specificity using the dCas9-Tet1 system.
Key features
• Precisely editing the DNA methylation of specific genomic loci in a targeted manner.
• Fine-tuning gene expression without changing DNA sequence.
• Applicable to many types of cell cultures and with the potential for ex vitro and in vivo applications.
Keywords: CRISPRGraphical overview
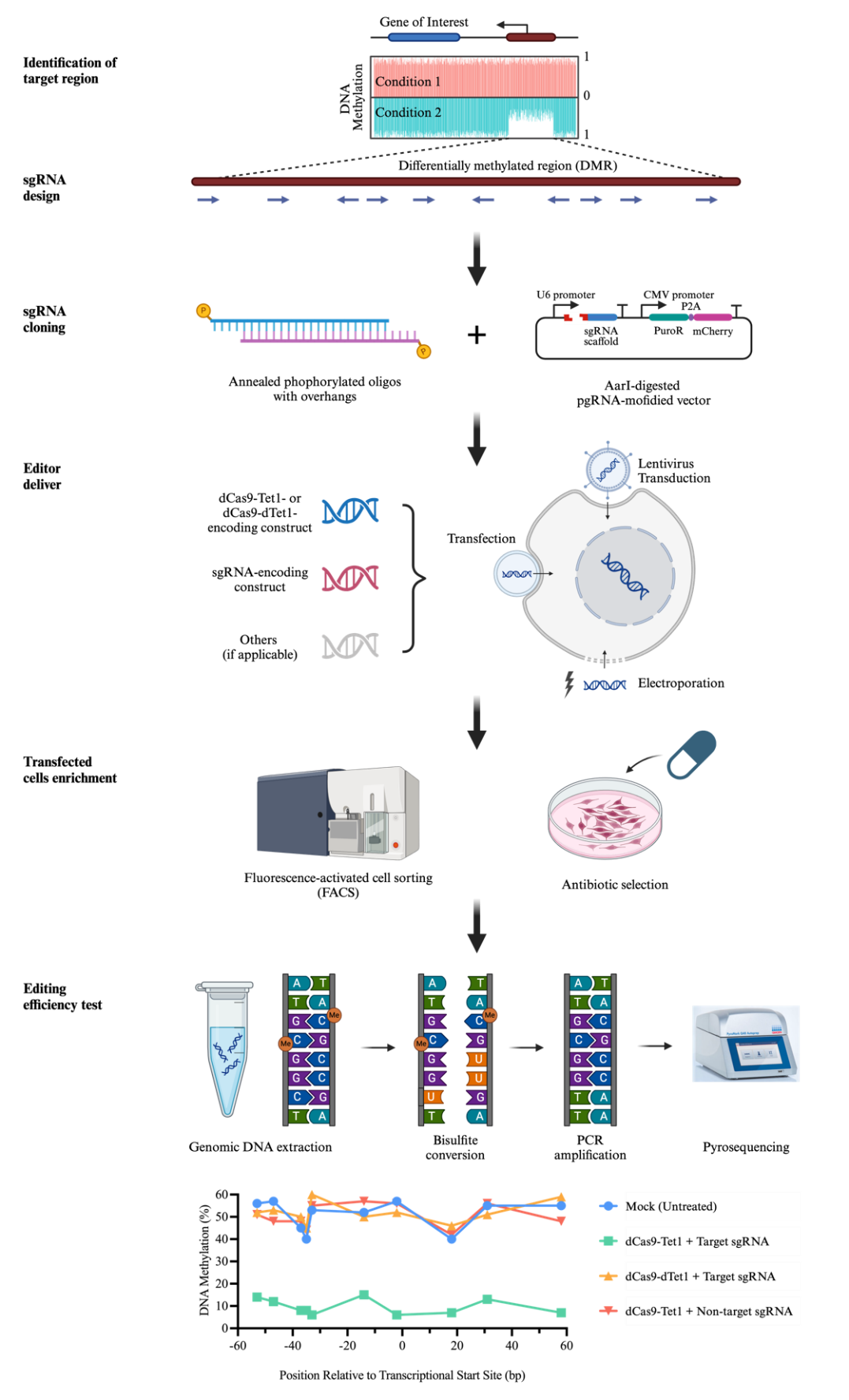
Overview of dCas9-Tet1-mediated DNA methylation editing
Background
DNA methylation is one of the most important epigenetic modifications, which refers to the covalent modification of the cytosine of DNA by a methyl group. In mammals, DNA methylation often occurs in the context of cytosine-phosphate-guanosine (CpG) residues [1]. It plays a critical role in various biological processes including gene regulation, genomic imprinting, and X chromosome inactivation, as well as the preservation of chromosome stability [2,3]. The proper regulation of DNA methylation is pivotal in maintaining normal cellular activities, and its dysregulation has been tightly associated with a myriad of human diseases [4].
Given its significance, the ability to manipulate DNA methylation with precision could revolutionize both basic research and therapeutic fields. Over the years, several epigenetic editing tools have been developed to manipulate DNA methylation at specific sites by tethering DNA methylation-associated effectors to sequence-specific DNA-binding domains, comprising mainly zinc finger proteins (ZFPs), transcription activator-like effectors (TALEs), and the clustered regularly interspaced short palindromic repeats (CRISPR) system [5–7]. Our lab pioneered a CRISPR-based DNA-methylation editing system using the fusion of a catalytically dead Cas9 (dCas9) with Ten-eleven translocation 1 (Tet1) hydroxylase catalytic domain (dCas9-Tet1), allowing for the erasure of DNA methylation in the mammalian genome with high specificity [8]. It has been proved as a powerful technology in many aspects, including mechanistically dissecting the functional significance of DNA methylation as well as treating incurable diseases by rewriting the DNA methylation of disease-causing genes. For instance, we demonstrated that targeted demethylation of the MyoD distal enhancer by dCas9-Tet1 along with 5-Aza (5-Aza-2-deoxycytidine, a DNA methylation inhibitor) treatment facilitated myogenic reprogramming of fibroblasts [8]. With the application of dCas9-Tet1, we also studied the hypermethylation of the CGG repeat expansion mutation at the 5’ UTR of fragile X mental retardation 1 (FMR1) gene. We demonstrated that demethylation of the CGG repeats unlocked the epigenetic silencing of FMR1 and restored FMRP expression in Fragile X syndrome cells [9]. More recently, we reported that dCas9-Tet1-mediated DNA demethylation of the promoter of methyl CpG-binding protein 2 (MECP2) gene can reactivate the expression of MECP2 from the inactive X chromosome in Rett syndrome (RTT)-like human embryonic stem cells (hESCs) and in hESCs-derived neurons, which could be a potential therapeutic approach for Rett syndrome [10].
Compared to ZFPs- or TALEs-based approaches, our methods displayed higher efficacy, specificity, and resolution of DNA methylation editing [8]. Importantly, unlike ZFPs and TALEs, which require design and optimization of corresponding distinct proteins to target different DNA sequences, only a replacement of single-guide RNA (sgRNA) is needed for CRISPR/dCas9 system to target different genomic loci. The versatility to flexibly target almost any given genomic loci enable researchers to precisely manipulate DNA methylation with ease. Nevertheless, the size and structural complexity of dCas9-Tet1 system might limit its application in vivo.
Herein, we describe a step-by-step protocol for editing DNA methylation in cell cultures using the dCas9-Tet1 system. This protocol is divided into three subsections to outline 1) the identification of sgRNA target sequences and clone sequences into sgRNA scaffold construct, 2) the delivery of the constructs encoding dCas9-Tet1 and sgRNA into the cells of interest, and 3) the examination of editing results by pyrosequencing.
Materials and reagents
Biological materials
Human embryonic kidney (HEK) 293T cells (ATCC, catalog number: CRL-11268)
Human embryonic stem cells (hESCs) [National Institutes of Health (NIH) registration number: WIBR-2, #29]
Mouse embryonic fibroblasts (MEFs) (ATCC NIH3T3, catalog number: CRL-1658 or derived from other labs)
Stbl3 chemical competent cells (Invitrogen, catalog number: C737303)
pgRNA-modified (Addgene, catalog number: 84477)
Fuw-dCas9-Tet1-P2A-BFP (Addgene, catalog number: 108245)
Fuw-dCas9-dTet1-P2A-BFP (Addgene, catalog number: 108246)
PiggyBac transposase (Lab stock, available upon request)
dCas9-Tet1 on PiggyBac transposon vector (Lab stock, available upon request)
pCMV-dR8.74 (Addgene, catalog number: 22036)
pCMV-VSVG (Addgene, catalog number: 8454)
Reagents
DNA oligos (Eton Bioscience, customized order)
T4 ligase and its buffer (New England Biolabs, catalog number: M0202)
T4 polynucleotide kinase (PNK) (New England Biolabs, catalog number: M0201)
AarI enzyme (Thermo Scientific, catalog number: ER1581)
Agarose (Fisher Scientific, catalog number: BP1356-500)
Zymoclean Gel DNA Recovery kit (Zymo Research, catalog number: D4007)
S.O.C medium (Invitrogen, catalog number: 15544034)
mTeSR1 medium (Stemcell, catalog number: 85850)
LB Broth, Miller (Fisher Scientific, catalog number: BP1426-500)
Agar (Fisher Scientific, catalog number: BP1423-500)
Carbenicillin disodium salt (Sigma Aldrich, catalog number: C1389)
E.Z.N.A. plasmid mini kit I (Omega Biotek Inc, catalog number: D6943)
E.Z.N.A. plasmid mini kit II (Omega Biotek Inc, catalog number: D6945)
ZymoPURE II plasmid purification kit, Maxiprep (Zymo Research, catalog number: D4203)
X-tremeGENE DNA transfection reagent (Sigma Aldrich, catalog number: 06365787001)
Opti-MEM I reduced serum medium (Gibco, catalog number: 11058021)
DMEM, high glucose (Gibco, catalog number: 11095092)
Fetal bovine serum (FBS) (Gibco, catalog number: 10082-147)
200 mM L-glutamine (Gibco, catalog number: 25030081)
MEM non-essential amino acids solution, 100× (Gibco, catalog number: 11140076)
Penicillin-Streptomycin (10,000 U/mL) (Gibco, catalog number: 15140122)
Trypsin-EDTA (0.25%), phenol red (Gibco, catalog number: 25200056)
DMEM/F12 medium (Invitrogen, catalog number: 11320-033)
Knockout serum replacement (Invitrogen, catalog number: 10828028)
β-mercaptoethanol (Life Tech, catalog number: 21985023)
Fibroblast growth factor 2 (FGF2) (Gibco, catalog number: PHG0263)
Calcium- and magnesium-free phosphate-buffered saline (PBS) (Gibco, catalog number: 10010023)
Bovine serum albumin (BSA) Fraction V solution (7.5%) (Gibco, catalog number: 15260037)
0.5 M EDTA, pH 8.0 (Invitrogen, catalog number: 15575038)
1 M HEPES (Hyclone, catalog number: SH30237.01)
DNeasy blood & tissue kit (Qiagen, catalog number: 69504)
QIAamp DNA micro kit (Qiagen, catalog number: 56304)
EZ DNA Methylation-Gold kit (Zymo Research, catalog number: D5006)
PyroMark PCR Master Mix kit (Qiagen, catalog number: 978703)
PyroMark Q48 Advanced CpG Reagents (4 × 48) (Qiagen, catalog number: 974002)
Rock inhibitor (Sigma-Aldrich, catalog number: S4317)
Doxycycline (Sigma-Aldrich, catalog number: D9891)
Solutions
HEK293T cell culture medium (see Recipes)
hESCs culture medium (see Recipes)
FACS buffer (see Recipes)
Recipes
HEK293T cell culture medium
Reagent Final concentration Volume DMEM medium n/a 435 mL FBS 10% 50 mL 200 mM L-glutamine 2 mM 5 mL Non-essential amino acids 1% 5 mL Penicillin-streptomycin 1% (100 U/mL) 5 mL Total n/a 500 mL hESCs culture medium
Reagent Final concentration Quantity or Volume DMEM/F12 medium n/a 385 mL FBS 15% 75 mL Knockout serum replacement 5% 25 mL 200 mM L-glutamine 2 mM 5 mL Non-essential amino acids 1% 5 mL Penicillin-streptomycin 1% (100 U/mL) 5 mL β-mercaptoethanol 0.1 mM 0.5 mL FGF2 4 ng/mL 2 μg Total n/a 500 mL FACS buffer
Reagent Final concentration Volume PBS n/a 142.62 mL 7.5% BSA 0.5% 5 mL 0.5 M EDTA 0.5 mM 150 μL 1 M HEPES 15 mM 2.25 mL Total n/a 150 mL
Laboratory supplies
1.5 mL microcentrifuge tube (Axygen, catalog number: MCT-175-C)
PCR tube (Axygen, catalog number: PCR-0208-CP-C)
12-well plate (Corning, catalog number: 3513)
96-well plate (Corning, catalog number: 3596)
T-175 flask (Corning, catalog number: 431080)
0.22 μm filter (Corning, catalog number: 430513)
0.45 μm low protein binding filter (Thermo Scientific, catalog number: 121-0045)
Ultracentrifuge tube (Beckman Coulter, catalog number: 344058)
0.4 cm electroporation cuvette (Bio-Rad, catalog number: 1652081)
Equipment
Thermocycler (Applied Biosystem, ProFlex)
PyroMark Q48 Autoprep sequencer (Qiagen, catalog number: 9002471)
Bio-Rad gene pulser Xcell electroporation systems (Bio-Rad, catalog number: 1652660)
FACSAria cell sorter (BD, model: BD FACSAria™ II)
Fluorescence microscope (Nikon, model: Eclipse TS2R)
Centrifuge (Qiagen, model: 5810 with rotor A-2-DWP-AT)
Ultracentrifuge and rotor (Beckman, model: SW32Ti)
Software and datasets
PyroMark Assay Design (Version 2.0.2, 1/10/2024)
PyroMark Q48 Autoprep (Version 4.3.3, 1/10/2024)
Procedure
Identification of sgRNA target sequences and clone into sgRNA scaffold construct
A targeting region is often selected within the CpG island (CGI) of a given gene or a differentially methylated region (DMR) of interest between different cell types or conditions identified by publicly available databases or determined by experiments.
Design the protospacer sequence of sgRNA for targeting dCas9-Tet1 under the following guidelines:
The sequence should be immediately adjacent to a protospacer adjacent motif (PAM), which is 5'-NGG-3' for dSpCas9 used in our system. Do not include PAM sequence as part of target sequence.
The length is typically 20 nt, but can be as short as 17 nt.
Avoid overlapping with CpG sites, as it will protect them from being demethylated.
The average effective range of dCas9-Tet1 with a single sgRNA is 150–200 bp downstream PAM sequence in most cases. Multiple protospacer sequences may be designed to target as many CpG sites within the targeting region as possible.
We design all the possible sgRNAs based on NGG PAM availability within the target region and then run these sgRNAs via online design programs such as CRISPick (https://portals.broadinstitute.org/gppx/crispick/public) and Off-Spotter (https://cm.jefferson.edu/Off-Spotter/) to pick up the one with high on-target score by CRISPRa mode and lower off-target prediction.
For each sgRNA, synthesize two oligos with overhangs to be compatible with sgRNA backbone construct. The overhang sequences are specific to the sgRNA scaffold construct used. Herein, we will use the one commonly used in our lab as an example (pgRNA modified).
sgRNA-sense: 5'-TTGG(G)-N1N2N3 N4N5N6N7N8N 9N10N11N12N13N 14N15N16N17N18N 19N20-3'
sgRNA-antisense:5'-AAAC-N*20N*19N*18 N*17N*16N*15N*14N* 13N*12N*11N*10N*9 N*8N*7N*6N*5N*4 N*3N*2N*1-(C)-3' (Nx and N*x are complemented)
Clone protospacer sequences into sgRNA scaffold construct.
Oligo phosphorylation and annealing. Set up the following reaction in a PCR tube on ice (Table 1):
Table 1. Reaction for oligo phosphorylation and annealing
Reagent Quantity sgRNA-sense oligo (100 μM) 1 μL sgRNA-antisense oligo (100 μM) 1 μL 10× T4 ligation buffer 1 μL Nuclease-free water 6.5 μL T4 PNK 0.5 μL Total 10 μL Incubate in a thermocycler using the following program (Table 2 ):
Table 2.Thermocycling conditions for the oligo phosphorylation and annealing reaction
Temperature (°C) Duration 37 30 min 95 5 min Ramp down to 25 °C at 1 °C/min 4 ∞ Dilute the phosphorylated and annealed oligo duplex 1:50 in nuclease-free water. Annealed oligos can be stored at -20 °C and are stable through at least 2–3 freeze-thaw cycles.
Linearize the sgRNA scaffold vector by restriction endonuclease AarI digestion. Set up the following reaction in a PCR tube in a microcentrifuge tube on ice (Table 3):
Table 3. Reaction for the linearization of sgRNA scaffold vector by AarI digestion
Reagent Quantity pgRNA-modified (Plasmid DNA) 1 μg 10× AarI buffer 2 μL 50× oligonucleotide (0.025 mM) 0.4 μL Nuclease-free water Up to 30 μL AarI 1 μL Total 30 μL Incubate at 37 °C for 1 h. Gel purify the digested plasmid using Zymoclean Gel DNA Recovery kit and determine the concentration.
Ligate the annealed oligo duplex with protospacer sequence to the digested sgRNA scaffold construct with a standard T4 ligase reaction. Set up the following reaction in a PCR tube or in a microcentrifuge tube on ice (Table 4):
Table 4. Reaction for ligating annealed phosphorylated oligos into sgRNA scaffold vector
Reagent Quantity Diluted phosphorylated and annealed oligo duplex 1 μL AarI-digested pgRNA-modified DNA 50 ng 10× T4 ligation buffer 1 μL Nuclease-free water Up to 10 μL T4 ligase 1 μL Total 10 μL Incubate at room temperature for 30 min or at 16 °C overnight followed by heat inactivation at 65 °C for 10 min. Chill the ligation reaction on ice.
Transform the ligation product to Stbl3 E. coli cells.
Thaw Stbl3 competent cells on ice.
Aseptically add 5 μL of ligation reaction to 20 μL of competent cells.
Incubate on ice for 30 min.
Heat shock at 42 °C for 45 s.
Immediately place the cells on ice for 2–3 min.
Add 250 μL of S.O.C medium. Shake at 37 °C for 1 h.
Spread 100 μL on a LB agar plate with carbenicillin. Invert the plate and incubate at 37 °C overnight.
Construct validation and amplification.
Pick 2–3 clones for each sgRNA construct and propagate them in 2 mL of LB medium with carbenicillin at 37 °C with shaking overnight.
Extract the plasmid DNA using Omega E.Z.N.A. Plasmid Mini kit I or II and following manufacturer’s instructions. Submit samples for Sanger sequencing. The following oligonucleotide can be used as the sequencing primer to read out the final protospacer sequence for each clone: 5'-GAAACTCACCCTAACTG-3'.
Delivery of the constructs encoding dCas9-Tet1 and sgRNA into the cells of interest
Considering the different ability to accept exogenous genes of various cell types, we will describe three most used gene delivery methods: 1) chemically transient transfection (HEK293T cells as an example); 2) electroporation (hESCs as an example); and 3) lentivirus transduction (hESCs and hESCs-derived neurons as examples). These three approaches could be generalized to any other tissue cultures, though optimizations of reaction conditions may be needed to achieve the best performance.
Chemically transient transfection for HEK293T cells
HEK293T cells are maintained in HEK293T cell culture medium (see Recipe 1) at 37 °C with 5% CO2. Chemically transient transfection experiment in HEK293T cells is performed in 12-well plates using X-tremeGENE 9 DNA transfection reagent.
Plate approximately from 1 × 105 to 2 × 105 cells/well in a sterile 12-well plate 24 h before transfection. Make sure cells are at optimal concentration (50%–80% confluency) at the time of transfection.
Prior to transfection, bring transfection reagent, reduced serum medium (Opti-MEM I reduced serum medium), and plasmid DNA to room temperature.
Add 700 ng of plasmids DNA encoding dCas9-Tet1 (Fuw-dCas9-Tet1-P2A-BFP) and 300 ng of plasmids DNA encoding target sgRNAs to a sterile microcentrifuge tube and vortex to mix thoroughly. Additionally, prepare dCas9-catalytically dead Tet1 (Fuw-dCas9-dTet1-P2A-BFP) + target sgRNA, and dCas9-Tet1 + scramble sgRNA mixture as experimental control groups. Blue fluorescence protein (BFP) gene is co-expressed with dCas9-Tet1 or dCas9-dTet1, and the puromycin resistance gene and the gene encoding a red fluorescence protein, mCherry, are co-expressed with sgRNAs.
For each experimental group, add 3 μL of transfection reagent (transfection reagent:DNA ratio = 1:3) to 100 μL of Opti-MEM I reduced serum medium. Incubate at room temperature for 5 min.
Add plasmids DNA mixture. Pipette to mix thoroughly. Incubate at room temperature for 15 min.
Add transfection complex to the cells in a dropwise manner. Gently shake or swirl the plate to ensure even distribution over the entire well.
Forty-eight hours post transfection, isolate cells that are successfully transfected.
Dissociating cells with trypsin/EDTA: remove and discard medium. Wash cells with PBS once. Add 300 μL of prewarmed trypsin/EDTA solution to each well and incubate at 37 °C for 5 min or until cells detach.
Add 600 μL of prewarmed growth medium to inactivate trypsin. Gently disperse the medium by pipetting and transfer the cell suspension to a tube.
Spin at 250× g for 5 min at room temperature. Remove and discard the supernatant.
Prepare single-cell suspension by resuspending cell pellet in FACS buffer.
Perform FACS to isolate live BFP- and mCherry-double positive cells.
Plate sorted cells into plate and culture at the same condition for 3–5 more days.
Five to seven days post transfection, harvest cells for further analysis.
Electroporation for hESCs
hESCs are maintained either with mTeSR1 medium on Matrigel-coated plates or on irradiated MEFs with standard hESCs medium (see Recipe 2). The electroporation is performed with Bio-Rad Gene Pulser Xcell electroporator. We generally use PiggyBac transposon system to create a stable cell line for the ease of experiment.
The day before electroporation, for each experimental group, feed one 6-well plate of hESCs at confluency with medium containing Rock inhibitor (10 μM).
Prepare single-cell suspension:
Remove medium and wash with PBS once.
Add 1 mL of Accutase to each well. Incubate at 37 °C for 5 min.
Harvest cells by pipetting gently to dissociate cells and collect with 1 mL of hESCs culture medium supplemented with Rock inhibitor.
Spin down the cells at 250× g for 5 min.
Resuspend the cell pellet with 450 μL of PBS pre-cooled on ice to make the final volume 500 μL.
In a sterile microcentrifuge tube, add 11 μg of plasmid DNA encoding PiggyBac transposase and 33 μg of plasmid DNA encoding dCas9-Tet1 and targeting sgRNA. Add PBS to 300 μL and vortex to mix thoroughly. Additionally, prepare dCas9-dTet1 + target sgRNA, and dCas9-Tet1 + scramble sgRNA mixture as experimental control groups. dCas9-Tet1/dTet1 is under control by a TRE3G promoter, whose expression can be induced by the addition of doxycycline (Dox) at the presence of reverse tetracycline-controlled transactivator (rtTA). The plasmid also contains constitutively expressed genes encoding rtTA and a selection maker (e.g., PuroR for puromycin selection).
Perform electroporation:
For each experimental group, pre-cool an electroporation cuvette on ice.
Add DNA mixture to single-cell suspension and gently pipette to mix thoroughly.
Transfer the total of 800 μL mixture into a pre-cooled 0.4 cm electroporation cuvette.
Place the cuvette on ice for 5 min.
Put the cuvette into the chamber in the required orientation. Electroporate with the following settings:
Exponential setting
Voltage = 250 V
Capacitance = 500 μF
Resistance = infinity
Cuvette = 4 mm
One pulse with expected time 12–14 μs
Place cuvette on ice for 5 min.
Plate the treated cells from the cuvette into one 6-well plate with medium containing Rock inhibitor.
Keep cells in medium with Rock inhibitor.
Three days post electroporation, switch to standard medium without Rock inhibitor.
The next day, add selection chemical (e.g., puromycin for cells receiving construct with PuroR genes).
Keep selection for extended days until visible colonies are formed. Change the medium every day during the process.
Add Dox to induce the expression of dCas9-Tet1/dTet1 for 5–7 days and harvest cells for further analysis.
Lentivirus transduction for hESCs and neurons
Lentivirus packaging:
HEK293T cells are maintained in HEK293T cell culture medium (See Recipe 1) at 37 °C with 5% CO2. Early passage is preferred.
The day before transfection, plate approximately 8 million cells per T-175 flask and plate two flasks per construct. Make sure cells are at optimal concentration (70%–80% confluency) at the time of transfection.
Prior to transfection, bring the transfection reagent, reduced serum medium, and plasmid DNA to room temperature.
Add 28 μg of pCMV-dR8.74 plasmid DNA, 4.7 μg of pCMV-VSVG plasmid DNA, and 37.3 μg of dCas9-Tet1/dTet1 or 19 μg of sgRNA plasmid DNA to a sterile microcentrifuge tube and vortex to mix thoroughly.
Note: All plasmids DNA should be prepared by endotoxin-free Maxi-prep kit (e.g., ZymoPURE II plasmid purification kit, Maxiprep).
For each construct, add 210 μL of transfection reagent to 5 mL of Opti-MEM I reduced serum medium. Incubate at room temperature for 5 min.
Add premixed plasmid DNA. Mix thoroughly. Incubate at room temperature for 15 min.
Add transfection complex to the cells in a dropwise manner (2.5 mL/flask). Gently shake or swirl the plate to ensure even distribution over the entire flask.
The next day, change to fresh medium. From this point forward, consider that everything that comes in contact with cells contains infectious lentivirus particles. Use BSL2 safety protocols with respect to PPE, decontamination of plasticware and media, etc.
Two days after medium change, harvest the medium and replace with fresh medium. Store the collected medium at 4 °C.
Two days after the first medium collection, harvest the medium. Store the collected medium at 4 °C.
Lentivirus concentration
Filter the collected medium with a 0.45 μm filter with a low protein binding membrane.
Spin down the lentivirus at 90,000× g for 1 h and 45 min at 4 °C.
Discard the supernatant. Resuspend the virus pellet in 200 μL of PBS per centrifuge tube.
Rotate the combined viruses in PBS at 4 °C overnight. Aliquot to the volume needed for single-use to avoid freeze-thaw cycles.
Lentivirus titration
Seed 1 × 104 HEK293T cells/well in a 96-well plate one day in advance.
Perform gradient dilution of the lentiviral particles to 1:10, 1:102, 1:103, 1:10 4, 1:105, and 1:106 in 100 μL of final volume in culture medium.
A total 100 μL of viral particle mixture should be added to each well with at least three replicates per virus.
Two days post infection, count the fluorescent positive cells using fluorescent microscopy and select the dilution factor with a proper fluorescent positive proportion (10%–30% positive cells/well). Count the triplicates and average the number of positive cells.
Estimate the functional lentivirus titer using the following formulation:
Virus titer (T) was calculated based on the infection efficiency for HEK293T cells, where T = (P*N)/(V), T = titer (TU/μL), P = % of infection positive cells according to the fluorescence marker, N = number of cells at the time of transduction, V = total volume of virus used.
Note: TU stands for transduction unit.
Lentivirus transduction:
Depending on the cell type, add virus to each well with a multiplicity of infection (MOI) of 10–100. For post-mitotic neurons, a high MOI of 50–100 is recommended to achieve high transduction efficiency and editing performance, while a MOI of 10 works well for dividing cells like HEK293T cells.
Incubate cells with viruses at 37 °C for 15 min.
Spin cells with viruses at 800× g for 1 h at room temperature (Qiagen 5810 with rotor A-2-DWP-AT).
Incubate for 24 h and then perform half media change.
Twenty-four hours later, do a full medium change.
Isolate infected cells by FACS or eliminate non-infected cells by chemical selection, depending on the selection marker on the used viral vector.
Harvest cells for further analysis.
Editing results examination by pyrosequencing
Extract genomic DNA of edited cells using Qiagen DNeasy blood & tissue kit or Qiagen QIAamp DNA micro kit following manufacturer’s instructions.
Perform bisulfite conversion of genomic DNA using EZ DNA Methylation-Gold kit following manufacturer’s instructions. Unmethylated cytosine (C) is converted to uracil during bisulfite conversion, which will be read as thymine (T) during PCR amplification. Methylated cytosine (mC) remains unchanged.
Pyrosequencing assay design.
The assay is designed using Qiagen PyroMark Assay Design software following software guidance.
Pick the high-scored primer set (forward and reverse PCR primer and sequencing primer) with no unspecific binding.
PCR to amplify the genomic region of interest from bisulfite-converted genomic DNA using PyroMark PCR kit. Set up the reaction on ice (Table 5):
Table 5. Reaction for pyro-PCR
Reagent Quantity 2× PyroMark PCR master mix 12.5 μL 10× CoralLoad Concentrate 2.5 μL Forward primer (5 μM) 0.5 μL Reverse primer (5 μM) 0.5 Nuclease-free water Up to 25 Bisulfite converted genomic DNA (10–20 ng) Total 25 μL Run the following program in a thermocycler (Table 6):
Table 6. Thermocycling conditions for the PCR reaction
Step Temp. (°C) Duration No. of cycles Initial denaturation 98 1 Denaturation 98 10 s 45 Annealing 56 30 s Extension 72 Final extension 72 1 Hold 4 ∞ - Take a small aliquot of PCR product (10 μL) to run in an agarose gel. Make sure there is only one single and intense band with size matched as predicted.
Perform pyrosequencing to quantify the T:C ratio at each CpG site within assayed regions and convert into a percentage of DNA methylation.
Program the sequencing run using PyroMark Q48 Autoprep software following manufacturer’s instructions.
Add the reagents supplied by PyroMark Q48 Advanced CpG Reagents (4 × 48) kit (nucleotides, sequencing primer, buffer, substrate, and enzyme mix) into the appropriate wells of the cartridge according to the volume calculated by the sequencer software. Load PCR products into the corresponding wells of the pyrosequencing disc and place the disc into the sequencer.
Start the run.
Once the sequencing is done, analyze the results using PyroMark Q48 Autoprep software.
Data analysis
The analysis of DNA methylation by pyrosequencing has been reported previously [11,12]. We used the PyroMark Q48 Autoprep software by Qiagen. The result file generated by the sequencer was directly opened and analyzed by PyroMark Q48 Autoprep software with default settings. Figure 1 shows a typical program analyzing 11 CpGs in the promoter region of mouse Snrpn genes. The methylation percentage is displayed above bars highlighted in blue, which stands for every cytosine in the context of CpG within the assayed region. The background color of the percentage shows the quality assessment by the software: blue means passed QC, yellow for check, and red for failed. We normally only use the ones with high sequencing quality (blue and yellow). The bar with an orange background is the bisulfite treatment controls to assess successful bisulfite treatment; it is normally a cytosine site in non-CpG context and known to be unmethylated. As a result of successful bisulfite conversion reaction, this unmethylated cytosine will be fully converted to T after PCR and sequenced as T in pyrosequencing. Make sure the examined DNA is efficiently bisulfite converted.
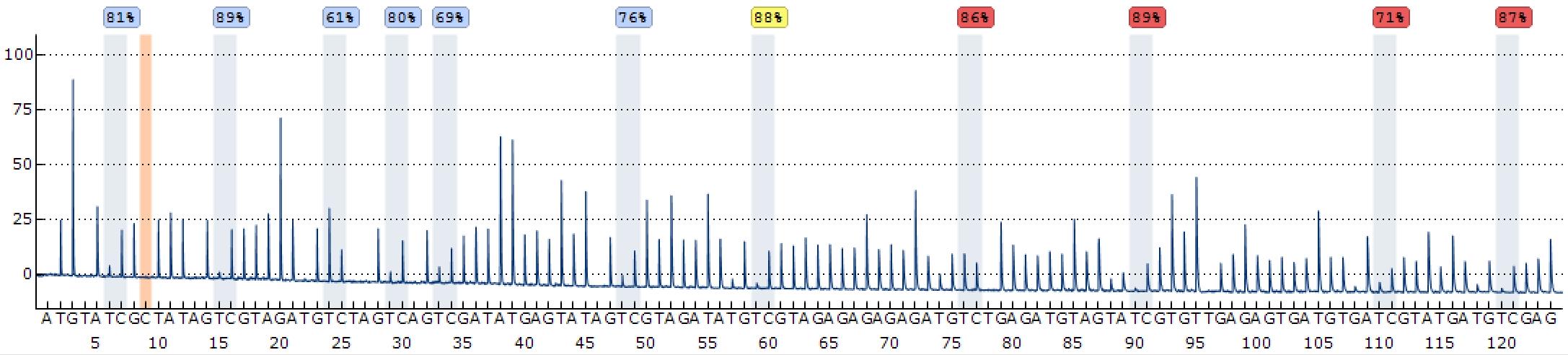
Figure 1. Example program analyzing 11 cytosine-phosphate-guanosines (CpGs) in the promoter region of mouse Snrpn promoter. The y-axis shows the signal intensity in arbitrary units, while the x-axis represents the dispensation order. The dispensations corresponding to the cytosines of assayed CpGs are highlighted in blue. The methylation percentages at individual CpG site are displayed above the respective bars. The background color of percentage shows the quality assessment by the software: blue for passed, yellow for check, and red for failed.
To examine the editing outcome, compare the DNA methylation percentage of CpGs with high sequencing quality between samples with different treatment. If the targeted genomic region is successfully edited, the DNA methylation level of assayed CpGs from dCas9-Tet1 + target sgRNA-treated cells should be lower than the ones from untreated cells or dCas9-dTet1 + target sgRNA- or dCas9-dTet1 + scramble sgRNA-treated cells. For further examination of the direct outcome of DNA methylation change, RT-qPCR and western blotting may be needed to quantify the change in RNA and protein level of edited genes, respectively.
Validation of protocol
This protocol or parts of it has been used and validated in the following research article:
Qian et al. [10]. Multiplex epigenome editing of MECP2 to rescue Rett syndrome neurons. Science Translational Medicine (Figure 1, panel c).
General notes and troubleshooting
General notes
We design all the possible sgRNAs based on NGG pam availability within the target region and then run these sgRNAs via online design programs such as CRISPick (https://portals.broadinstitute.org/gppx/crispick/public) and Off-Spotter (https://cm.jefferson.edu/Off-Spotter/) to pick up the ones with high on-target score by CRISPRa mode and lower off-target prediction. Two critical points to consider are: 1) the editing window for dCas9-Tet1 described here is within 150–200 bp downstream of PAM site and 2) the CpG sites within the sgRNA target sequence are often protected against methylation editing.
Compared to classical CRISPR-Cas9 knockout systems where sgRNAs are always designed within coding regions to disrupt gene expression, the sgRNAs for demethylation editing are recommended to be designed upstream of the transcription start site to avoid dCas9-mediated steric hinderance and transcription interference.
In order to achieve the highest editing efficiency, a small screening may be conducted to test multiple sgRNAs and their combinations to identify the most effective sgRNA(s).
The expression level of dCas9-Tet1 is critical to achieving high editing efficiency. The period of dCas9-Tet1 expression is critical to control off-target effects. Therefore, choosing the proper expression system (inducible or constitutive) and controlling the expression time for editors (dCas9-Tet1 and sgRNA) are key to specifically edit DNA methylation in a variety of targeted cells.
Demethylation in neurons is slower than in dividing cells. A high titer of lentiviral editors expressing dCas9-Tet1 and sgRNA are required to edit methylation in neurons. To avoid neuronal cell death upon lentivirus transduction, a high density of neuronal culture is preferred. Alternatively, a Dox-inducible dCas9-Tet1 expression cassette can be engineered in hESCs, and editing DNA methylation in neurons can be achieved after neuronal differentiation.
The dynamics of dCas9-Tet1-mediated demethylation largely depends on cell types. In general, the editing effect reaches its peak after 5–7 days of editing. However, it could be slower in non-dividing cells such as post-mitotic neurons, as both passive and active demethylation can operate in dividing cells, but only active demethylation occurs in cells that are not dividing.
The duration and reversibility are locus-specific and influenced by local chromatin environment as well as cell type. In general, edited DNA demethylation can be maintained in dividing cells but is not stable in post-mitotic cells such as neurons. For instance, the demethylation status of FMR1 on the active X chromosome can be maintained in vitro for weeks [9], whereas the demethylation status of MECP2 on the inactive X chromosome is not stable in post-mitotic neurons [10].
When doing lentivirus transduction, the MOIs will depend on the purpose of each experiment and the type of cell lines. We used higher MOIs (50–100) for post-mitotic neurons and lower MOIs (approximately 10) for dividing cells such as HEK293. Please note that this MOI is based on the virus titer (T) that is calculated based on the infection efficiency for HEK293T cells, where T = (P*N)/(V), T = titer (TU/µL), p = % of infection positive cells according to the fluorescence marker, N = number of cells at the time of transduction, V = total volume of virus used. Note that TU stands for transduction unit.
Troubleshooting
Problem 1: The efficiency of methylation editing is low as measured by pyrosequencing.
Possible cause: The titer of lentiviral dCas9-Tet1 is too low to induce methylation editing.
Solution: Increase the titer of lentiviral dCas9-Tet1 by performing ultracentrifugation of at least 38 mL of viral supernatant harvested from transfected HEK293T cells and then reconstituting in 100 μL of PBS for effective infection to deliver dCas9-Tet1 into targeted cells.
Acknowledgments
Funding was obtained from the NIH grant R00MH113813 (X.S.L.), Rett Syndrome Research Trust grant (X.S.L.), and Columbia University Startup grant UR011118 (J.Q. and X.S.L.). This protocol is previously described and validated in Qian et al. [10]. Multiplex epigenome editing of MECP2 to rescue Rett syndrome neurons. Science Translational Medicine. The graphical abstract was created with BioRender.com.
Competing interests
X.S.L. is cofounder of Epitor Therapeutics.
References
- Jaenisch, R. and Bird, A. (2003). Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat. Genet. 33: 245–254. https://doi.org/10.1038/ng1089.
- Smith, Z. D. and Meissner, A. (2013). DNA methylation: roles in mammalian development. Nat. Rev. Genet. 14(3): 204–220. https://doi.org/10.1038/nrg3354.
- Mattei, A. L., Bailly, N. and Meissner, A. (2022). DNA methylation: a historical perspective. Trends Genet. 38(7):676–707. https://doi.org/10.1016/j.tig.2022.03.010.
- Greenberg, M. V. C. and Bourc'his, D. (2019). The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 20(10): 590–607. https://doi.org/10.1038/s41580-019-0159-6.
- Holtzman, L. and Gersbach, C. A. (2018). Editing the Epigenome: Reshaping the Genomic Landscape. Annu. Rev. Genomics Hum. Genet. 19: 43–71. https://doi.org/10.1146/annurev-genom-083117-021632.
- Liu, X. S. and Jaenisch, R. (2019). Editing the Epigenome to Tackle Brain Disorders. Trends Neurosci. 42(12): 861–870. https://doi.org/10.1016/j.tins.2019.10.003.
- Nakamura, M., Gao, Y., Dominguez, A. A. and Qi, L. S. (2021). CRISPR technologies for precise epigenome editing. Nat. Cell Biol. 23(1): 11–22. https://doi.org/10.1038/s41556-020-00620-7.
- Liu, X. S., Wu, H., Ji, X., Stelzer, Y., Wu, X., Czauderna, S., Shu, J., Dadon, D., Young, R. A. and Jaenisch, R. (2016). Editing DNA Methylation in the Mammalian Genome. Cell 167(1): 233–247.e217. https://doi.org/10.1016/j.cell.2016.08.056.
- Liu, X. S., Wu, H., Krzisch, M., Wu, X., Graef, J., Muffat, J., Hnisz, D., Li, C. H., Yuan, B., Xu, C., et al. (2018). Rescue of Fragile X Syndrome Neurons by DNA Methylation Editing of the FMR1 Gene. Cell 172(5): 979–992.e976. https://doi.org/10.1016/j.cell.2018.01.012.
- Qian, J., Guan, X., Xie, B., Xu, C., Niu, J., Tang, X., Li, C. H., Colecraft, H. M., Jaenisch, R. and Liu, X. S. (2023). Multiplex epigenome editing of MECP2 to rescue Rett syndrome neurons. Sci. Transl. Med. 15(679): eadd4666. https://doi.org/10.1126/scitranslmed.add4666.
- Tost, J. and Gut, I. G. (2007). DNA methylation analysis by pyrosequencing. Nat. Protoc. 2(9): 2265–2275. https://doi.org/10.1038/nprot.2007.314.
- Delaney, C., Garg, S. K. and Yung, R. (2015). Analysis of DNA Methylation by Pyrosequencing. Methods Mol. Biol. 1343: 249–264. https://doi.org/10.1007/978-1-4939-2963-4_19.
Article Information
Copyright
© 2024 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Qian, J. and Liu, S. X. (2024). CRISPR/dCas9-Tet1-Mediated DNA Methylation Editing. Bio-protocol 14(8): e4976. DOI: 10.21769/BioProtoc.4976.
- Qian, J., Guan, X., Xie, B., Xu, C., Niu, J., Tang, X., Li, C. H., Colecraft, H. M., Jaenisch, R. and Liu, X. S. (2023). Multiplex epigenome editing of MECP2 to rescue Rett syndrome neurons. Sci. Transl. Med. 15(679): eadd4666. https://doi.org/10.1126/scitranslmed.add4666.
Category
Molecular Biology > DNA > DNA modification
Biological Sciences > Biological techniques > CRISPR/Cas9
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.