- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Preparation and Purification of β-1,3-glucan-Linked Candida glabrata Cell Wall Proteases by Ion-Exchange Chromatography, Gel Filtration, and MDPF-Gelatin-Zymography Assay
(*contributed equally to this work) Published: Vol 14, Iss 6, Mar 20, 2024 DOI: 10.21769/BioProtoc.4958 Views: 1730
Reviewed by: Alba BlesaSuresh PantheeSascha Brunke
Original research article
The authors used this protocol in:

Dec 2020
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Candida glabrata is an opportunistic pathogen that may cause serious infections in an immunocompromised host. C. glabrata cell wall proteases directly interact with host cells and affect yeast virulence and host immune responses. This protocol describes methods to purify β-1,3-glucan-bonded cell wall proteases from C. glabrata. These cell wall proteases are detached from the cell wall glucan network by lyticase treatment, which hydrolyzes β-1,3-glucan bonds specifically without rupturing cells. The cell wall supernatant is further fractioned by centrifugal devices with cut-offs of 10 and 50 kDa, ion-exchange filtration(charge), and gel filtration (size exclusion). The enzymatic activity of C. glabrata proteases is verified with MDPF-gelatin zymography and the degradation of gelatin is visualized by loss of gelatin fluorescence. With this procedure, the enzymatic activities of the fractions are kept intact, differing from methods used in previous studies with trypsin digestion of the yeast cell wall. The protein bands may be eventually located from a parallel silver-stained gel and identified with LC–MS/MS spectrometry. The advantage of this methodology is that it allows further host protein degradation assays; the protocol is also suitable for studying other Candida yeast species.
Key features
• Uses basic materials and laboratory equipment, enabling low-cost studies.
• Facilitates the selection and identification of proteases with certain molecular weights.
• Enables further functional studies with host proteins, such as structural or immune response–related, or enzymes and candidate protease inhibitors(e.g., from natural substances).
• This protocol has been optimized for C. glabrata but may be applied with modifications to other Candida species.
Graphical overview
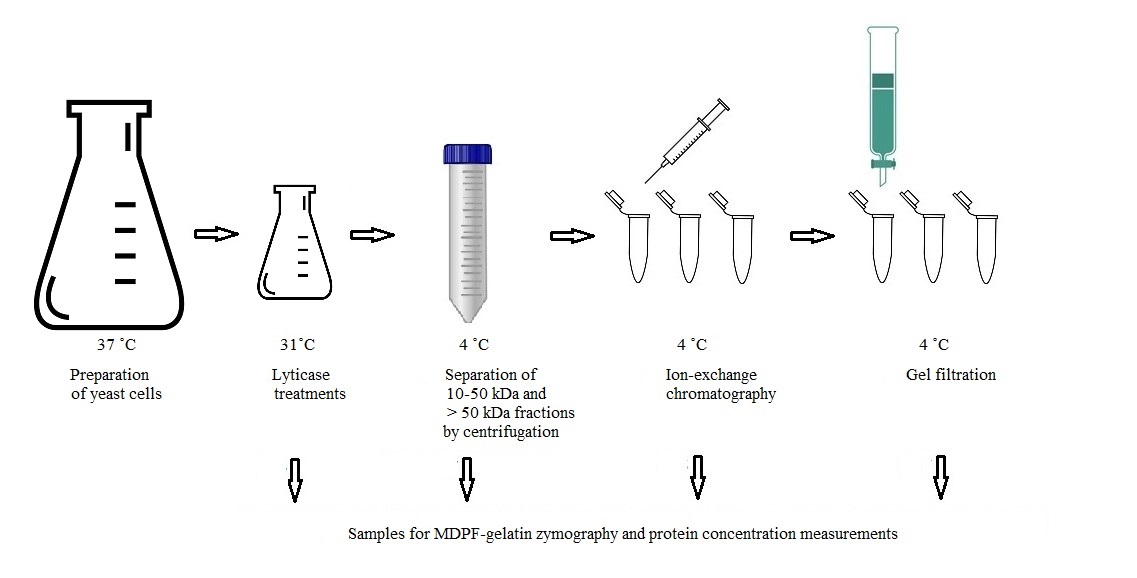
Background
Candida glabrata is an opportunistic pathogen causing infections in vulnerable hosts. As C. glabrata is often resistant to the most used azole antimycotics, the search for novel and safe antimycotics is ongoing. Among the most difficult challenges in understanding yeast pathogenesis is identifying the yeast proteome and virulence factors that affect the course of infection. While the cell wall composition of Candida albicans, the most common opportunistic yeast found in candidiasis, has been quite extensively studied, less is known of C. glabrata’s cell wall proteins, whose key elements and interactions remain undiscovered. The cell wall proteome is the front line when contacting host cells. C. glabrata cell wall structure resembles that of C. albicans and, even more closely, of S. cerevisiae [1–5].
Glycosylphosphatidylinositol (GPI) proteins, covalently bound to the cell wall β-1,6-glucan, as well as proteins linked through a mild-alkali-sensitive linkage to β-1,3-glucan are the two main classes of C. glabrata cell wall proteins, which participate in the interaction with the host [2]. To the last group belong Candida proteins that are delivered to the cell wall from their original intracellular location, named as “moonlighting” proteins. They lack an N-terminal signal sequence guiding formed proteins into the cell wall and are transported there in extracellular vesicles; these have extracellular enzymatic activities in addition to their intracellular housekeeping roles and play multiple functions in host cell interactions. The role of these moonlighting proteins as virulence factors is complex and partially unknown.
The presented protocol describes the enzymatic separation and purification of proteases from C. glabrata cell wall attached to the glucan network (see workflow in Graphical overview). The main objective is to identify novel proteases from the C. glabrata cell wall and enable further analysis of their potential enzymatic activities in host protein degradation. The protocol retains the isolated proteases in their active forms, which is a crucial advantage compared to previous studies in proteomics, allowing the understanding of the functions and interactions with host proteins. The glycosylation of cell wall proteins poses challenges in their identification process: carbohydrates attached to proteins mask them, hampering enzymes used in assays from degrading the protein into a form that may be identified by mass analysis. By detaching the proteases from the cell wall glucan network, it is possible to identify isoenzyme forms by MS/MS. Although dithiothreitol or β-mercaptoethanol have been traditionally used in extracting yeast cell wall proteins, they may cause cell rupture or denaturation of proteins [4]. The use of the β-1,3-glucan specific enzyme lyticase (from Arthrobacter luteus) in cleaving is essential to maintain the proteases in their active form, contrary to conventional non-functional studies. Lyticase has traditionally been used in the degradation of the yeast cell wall, formation of spheroplasts, and transformation procedures, and Candida enzymatic activity may be determined by 7-amino-4-methylcoumarin (AMC)-labeled L-arginine fluorogenic substrate degradation [6].
2-Methoxy-2,4-diphenyl-3(2H)-furanone (MDPF)-gelatin zymography is a convenient method for monitoring the protein purification process. In MDPF-gelatin zymography, the C. glabrata proteases retain their enzymatic activity, because non-denaturing conditions are maintained through the process. The strength of gelatinolysis caused by C. glabrata proteases may be seen as the disappearance of the MDPF-labeled gelatin fluorescence in the SDS-PAGE gel, which may be monitored under UV light. MDPF is a more stable fluorophore compared with FITC or fluorescein [7,8]. MDPF-gelatin zymography is suitable for host protein degradation assays [9]. The lyticase extract is initially separated into coarse fractions with centrifugal devices having cut-off values of 10 and 50 kDa. Further protein charge–based ion-exchange chromatographic separation is optimized regarding pH, and a weak anion-exchange column is used to extract > 50 kDa proteins; further 10–50 kDa and > 50 kDa fractions are obtained by size-exclusion gel filtration. The refining of these fractions enables proper separation, extraction, and identification of the protein bands from silver-stained gels for further mass analyses. The methods used here are easily applied with modifications to study the cell wall proteome in other pathogenic species when searching proteins with specific linkage to the cell wall. In studying the entire proteome of the cell wall, a shotgun approach should be preferred.
Materials and reagents
Biological materials
C. glabrata T-1639, blood isolate (Helsinki University Central Hospital, Helsinki, Finland). Other Candida species may be studied by these methods if there are structural analogies with C. glabrata cell wall protein attachment molecules.
Sabouraud dextrose agar (Lab M, catalog number: LAB009)
Lyticase, 10 kU (Sigma-Aldrich, catalog number: 37340-57-1)
2-Methoxy-2,4-diphenyl-3(2H)-furanone (MDPF) (Sigma-Aldrich, catalog number: 50632-57-0)
Precision Plus ProteinTM standards (Bio-Rad, catalog number: 161-0373)
Tris base (Sigma, catalog number: T-1378)
ZnCl2 (Sigma, catalog number: 208086)
CaCl2 (Sigma, catalog number: 10035-04-8)
Tween 80, BioXtra viscous liquid (Sigma, catalog number: P8074)
BactoTM yeast extract (BD, catalog number: 212750)
Peptone, BactoTM peptone (BD, catalog number: 211677)
Glucose (Merck, catalog number: 108337)
Sodium dodecyl sulphate (SDS) (Fisher Scientific, catalog number: BP166-500)
Sodium azide (NaN3) (Merck, catalog number: 6688), for long-term preservation of ion-exchange columns
Coomassie Brilliant Blue, Serva Blue R (Serva Electrophoresis GmbH, catalog number: 35051)
Sodium tetraborate (STB) (Merck, catalog number: 6308)
Gelatin (Merck, catalog number: 1.04070)
Acetone (Merck, catalog number: 1.00658)
Na2HPO4 (Sigma, catalog number: 137036)
KCl (Sigma, catalog number: PHR1329)
NaCl (Fisher Scientific, catalog number: FLS271500)
KH2PO4 (Merck, catalog number: 104871)
β-mercaptoethanol (Merck, catalog number: M6250)
TEMED (Merck, catalog number: 411019)
Acrylamide 30% (Merck, catalog number: A3574)
EtOH, Ethanolum anhydricum, Aa (Berner, catalog number: 13110124)
HCl 1 M (Riedel-de-Haen, catalog number: 30721)
HCl 37% (Sigma, catalog number: 258148)
NaOH 1 M (Acros Organics, catalog number: 15634890)
Methanol (Fisher, catalog number: M/4057/PB17)
Acetic acid (Fisher, catalog number: A/0400/PB17)
Ammonium persulfate (APS) 10% (Sigma, catalog number: A3678), store in 100 µL aliquots at -20 °C
4x Laemmli Sample Buffer (Bio-Rad, catalog number 1610747)
Glycine (Merck, catalog number: G8898)
Solutions
Yeast extract peptone glucose (YPG) (see Recipes)
MDPF-gelatin (see Recipes)
Separating gel (lower) buffer for zymo gels (see Recipes)
Stacking gel (upper) buffer for zymo gels (see Recipes)
8% MDPF-gelatin SDS-PAGE (see Recipes)
SDS-PAGE running buffer 5× (see Recipes)
Zymo buffer 10× (see Recipes)
Zymo gel incubation buffers (see Recipes)
Coomassie Brilliant Blue (CBB) 0.1% (see Recipes)
IEC start buffer (see Recipes)
IEC elution buffer I (see Recipes)
IEC elution buffer II (see Recipes)
Phosphate-buffered saline (PBS) (see Recipes)
10% SDS (see Recipes)
Lyticase stock (see Recipes)
Recipes
Perform pH adjustments with 1 M NaOH or 1 M HCl. Prepare zymogels as needed and solutions according to Recipes beforehand. Preparing double amounts of Recipes 10–13 is advised.
Yeast extract peptone glucose (YPG)
0.5% yeast extract
1% peptone
1% glucose
Add Milli-Q water (MQ) up to 4 L
Sterilize by autoclave at 121 °C for 15 min
MDPF-gelatin
20 mM STB solution: 0.3814 g of STB per 50 mL of MQ, pH 9.2.
Add 500 mg of gelatin into STB in warm water bath (approximately 37 °C) with magnetic stirring.
Dissolve 25 mg of MDPF into -20 °C acetone on ice and add MDPF into gelatin-STB at room temperature (RT) with magnetic stirring for 1 h protected from light. Divide into 500 µL aliquots and store at -20 °C protected from light.
Separating gel (lower) buffer for zymo gels
800 mL of MQ
187 g of Tris base
Adjust pH to 8.8
4 g of SDS
Add MQ up to 1 L
Stacking gel (upper) buffer for zymo gels
800 mL of MQ
60.5 g of Tris base
Adjust pH to 6.8
4 g of SDS
Add MQ up to 1 L
8% MDPF (1 mg/mL)-gelatin SDS-PAGE (4 gels)
Prepare gels in a fume hood:
Bottom: separating gel
Separating buffer 5 mL (Recipe 3)
30% Acrylamide 5.3 mL
MQ 7.3 mL
MDPF-gelatin (1 mg/mL) 2 mL (Recipe 2)
10% SDS 200 µL
TEMED 20 µL
10% APS 200 µL
Top: stacking gel
Stacking gel buffer 2.5 mL (Recipe 4)
30% Acrylamide 1.35 mL
MQ 5.95 mL
10% SDS 100 µL
TEMED 10 µL
10% APS 100 µL
SDS-PAGE running buffer 5×
Add 4 L of MQ into a Duran bottle
723 g of glycine
150 g of Tris base
20 g of SDS
Adjust pH to 8.3
Add MQ up to 5 L
Zymo buffer 10×
Add 800 mL of MQ into a Duran bottle
60.55 g of Tris base
2 g of NaN3
33 mL of 37% HCl
Adjust pH to 7.5
Add MQ up to 1 L
Zymo gel incubation buffers for 4 gels
Zymo buffer I
360 mL of 1× zymo buffer (dilute from 10× zymo buffer with MQ)
40 mL of 25% Tween 80
Zymo buffer II
270 mL of 1× zymo buffer
30 mL of 25% Tween 80
30 µL of 10 mM ZnCl2
1530 µL of 100 mM CaCl2
Zymo buffer III
300 mL of 1× zymo buffer
30 µL of 10 mM ZnCl2
1530 µL of 100 mM CaCl2
Coomassie Brilliant Blue (CBB), 0.1% (staining and destaining in fume hood)
1 g of Coomassie Brilliant Blue, Serva Blue R
600 mL of MQ
300 mL of methanol
100 mL of acetic acid
Destaining: 40% MeOH + 10% acetic acid in MQ
IEC start buffer (20 mM Tris HCl, pH 8.2)
2.4 g of Tris base
Add 800 mL of MQ
Adjust pH to 8.2
Add MQ up to 1 L
Sterile filter and degas
IEC elution buffer I (20 mM Tris HCl-0.5 M NaCl, pH 8.2)
29.22 g of NaCl
2.4 g of Tris base
Add 800 mL of MQ
Adjust pH to 8.2
Add MQ up to 1 L
Sterile filter and degas
IEC elution buffer II (20 mM Tris HCl- 1 M NaCl, pH 8.2)
14.61 g of NaCl
0.6 g of Tris base
Add 200 mL MQ
Adjust pH to 8.2
Add MQ up to 250 mL
Sterile filter and degas
Phosphate-buffered saline (PBS), pH 7.4
800 mL of MQ
8.0 g of NaCl
1.44 g of Na2HPO4
0.2 g of KCl
0.24 g of KH2PO4
Adjust pH to 7.4 using HCl
Add MQ up to 1 L
Filter and sterilize by autoclavation
10% SDS
80 mL of MQ
10 g of SDS
Add MQ up to 100 mL
Lyticase stock
1.6 mL of MQ
Add 10 kU lyticase
Store at -20 °C in 400 µL aliquots
Laboratory supplies
Sterile plastic inoculation loops (Greiner Bio-One, catalog number: 731171)
Millipore ExpressTM Plus or Filtration unit Stericup® with MILLIPORE Express® PLUS (PES), 0.22 µm (Merck Millipore, catalog number: X342.1, optional method: centrifugation)
Millipore Millex, 0.22 µm (Merck, catalog number: 051581)
Agar plates, plastic Petri dishes, PS (Sigma, catalog number: P5731)
Glass chromatography column (9.5 mL) with stopcock and lower silicon tubing (or Econo Alpha column, Bio-Rad, catalog number: 12009430) and column stand
Glass Erlenmeyers, 2× 3 L, 2× 200 mL
Amicon® Ultra-15 Centrifugal Filter Devices, 10 K and 50 K (Merck Millipore, catalog numbers: UFC901024, UFC905024)
Glass decanters 500 mL
EppendorfTM Snap-Cap Microcentrifuge Safe-LockTM Tubes [Fisher, catalog number: 05-402-18 (1.5 mL) and 05-402-18 (0.5 mL)]
FalconTM tubes, 15 mL, 50 mL (Fisher, catalog numbers: 14-959-53A, 14-432-22)
Micropipettes and sterile pipette tips, 10 µL–5 mL
Aluminum foil
Tube racks for FalconTM and EppendorfTM tubes
HiTrap® IEX Selection Kit (Merck, GE17-6002-33)
Sephadex G-100 and G-200 (Pharmacia Fine Chemicals, catalog numbers: 01-900-1-1861-03, 01-900-1-1863-03) 1 g/40 mL of buffer
10 mL sterile plastic syringes (Fisher, catalog number: 14955459)
Ice, 2 × 5 L plastic dish
Duran bottles, 1 L, 5 L
Funnel that fits gel filtration column
Equipment
Heating cabinet, 31 °C, 37 °C, optional: Melag Incubat® 85 (or Merck My Temp Mini Digital Incubator, catalog number: Z763357) or SureTemp® Dual Convection Incubator (Merck, catalog number: Z742696)
Nanodrop 1000 (or NanoDrop Lite Plus Spectrophotometer, catalog number: NDLPLUSGL)
pH meter (VWR® phenomenal® MU 6100 H, catalog number: 665-0311)
PAGE-running device, 20 µL well volume, 4–6 combs of 10 wells (Bio-Rad, model: Mini-Protean II, catalog number: 165-2940)
Plastic containers (500 mL) for zymo buffer incubation, disposable PP, flat bottom (Marjukka, catalog number: 6410416249018)
Light box for gel visualization, LX104, Lammex (or X-ray viewer; Quirumed, catalog number: 482-6001.S)
Gel scanner, GS-700 densitometer (Bio-Rad, catalog number: 170-7601 with QUANTITY ONE PROGRAM)
Light microscope, 10–40× magnification
Vacuum suction system for degassing buffers and Sephadex gel
UV light for gel visualization and photography (Uvidoc, model: M03 0103 or GelDoc Go Gel Imaging System with Image Lab Touch Software, catalog number: 12009077)
Level mixer (Heidolph Vibramax 100, catalog number: 544-21200-00)
Centrifuge SL16R and adapters for 15 mL tubes (Thermo Scientific, catalog number: 75004030)
Autoclave (Systec, model: D45)
Fume hood (Visukaluste Ltd, model: VISU ProFocus EN 14175-2)
Procedure
Preparation of yeast cells
Transfer C. glabrata cells from -80 °C onto a Sabouraud dextrose agar (SDA) plate with a sterile inoculation loop. Incubate the plate at 37 °C for 24 h. This incubation time is optimal to obtain enough stationary-phase yeast cells (expected OD > 2), as C. glabrata does not form true hyphae. However, the 24 h incubation time might be a problem in hyphae-forming species, such as C. albicans [try overnight (o/n) culturing].
Check purity of growth visually from the culture plate and with light microscopy (10–40× magnification) regarding colony and cell morphology. Prepare a pure culture from this by picking one colony and incubate as previously described.
Transfer a loopful of cells with a sterile inoculation loop from the pure culture SDA plate (check purity as above) into 2 × 2 L of YPG (see Recipe 1) divided into two Erlenmeyers. Cap the Erlenmeyers loosely with aluminum foil, place onto a level mixer, and incubate at low speed (65 rpm) for 24 h at 37 °C.
Filter the cell suspension into 2 L aliquots with a Stericup with MILLIPORE Express PLUS 0.22 µm filter with vacuum suction. Use a second filter unit for the rest of the cell suspension, as the filter may clog. Recover the cells from the filter membranes with 150 mL of PBS/filter into eight 50 mL Falcon tubes in equal volumes.
Centrifuge at 1,753× g for 10 min at RT. Discard supernatant.
Add 40 mL of PBS onto the cells in each tube and suspend to even composition. Centrifuge at 1,753× g for 10 min at RT. Discard supernatants, add 40 mL of PBS to each tube, centrifuge at 1,753× g, and discard supernatants.
Recover all cells from the bottom of each Falcon tube with 2.5 mL of PBS by suspending cells with a pipette and transfer them into a 100 mL Erlenmeyer. Add 40 mL of PBS (total suspension volume = 80 mL).
Lyticase treatments
Add 400 µL of prepared lyticase solution onto the cell suspension (final concentration approximately 30 U/mL), cap loosely with aluminum foil, and incubate on a level shaker (at 65 rpm) for 22 h at 31 °C. (Here, the heating cabinet was placed and strapped onto a level mixer; optionally, place level mixer into a larger incubator cabinet.)
Divide cell suspension equally into two 50 mL FalconTM tubes and centrifuge at 1,753× g for 10 min at RT. Recover supernatants (total volume = 35 mL) and store at -20 °C.
Recover cells from Falcon tubes with a total of 40 mL of PBS into a 200 mL Erlenmeyer. Add 400 µL of lyticase solution, cap loosely with aluminum foil, and incubate on a level shaker (65 rpm) for 22 h at 31 °C.
Repeat step B3. Freeze supernatants or continue pooling with supernatants from the freezer. Take a 500 µL sample for zymography and protein concentration measurement and store the rest at -20 °C.
Resuspend cells in residual volume in the Falcons and verify that cells are intact with light microscopy (see Figure 1 ).
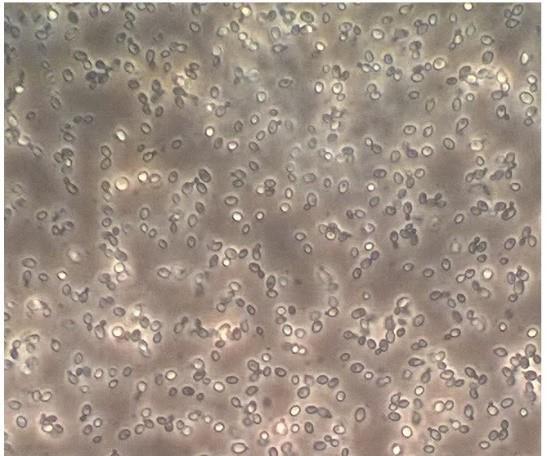
Figure 1. Intact C. glabrata spheroplasts after lyticase treatments. Light microscopy, 40× magnification.
Separation of 10–50 kDa and > 50 kDa fractions
Place PBS on ice. Thaw and filter pooled supernatants (V = 70 mL) with a Filtration unit Stericup with MILLIPORE Express PLUS 0.22 µm filter with vacuum suction or, optionally, by centrifugation. Take a 500 µL sample for protein concentration analysis and zymography. Divide the supernatant into eight Amicon® Ultra-15 Centrifugal Filter Devices 10 K. Centrifuge at 1,753× g for 20 min at 4 °C. Remove and pool flowthroughs.
Add 10 mL of cold PBS to each tube and centrifuge at 1,753× g for 20 min at 4 °C.
Pool flowthroughs with earlier flowthroughs. Take a 500 µL sample for zymography of the pooled total flowthrough and store it and the rest in 50 mL Falcon tubes at -20 °C (< 10 kDa fraction). Transfer upper supernatants equally into two Amicon® Ultra-15 Centrifugal Filter Devices 50 K. Centrifuge at 1,753× g for 20 min at 4 °C.
Recover flowthrough (10–50 kDa fraction).
Add 12 mL of PBS/filter and centrifuge at 1,753× g for 20 min at 4 °C. Pool flowthrough with the 10–50 kDa fraction (approximately 40 mL). Take a 500 µL sample and store it and the rest in 10 mL aliquots in Falcon tubes at -20 °C.
Recover > 50 kDa fraction by suspending 3 × 5 mL of PBS to obtain approximately 30 mL of > 50 kDa fraction. Take a 500 µL sample and store it and the rest in 8 mL aliquots in Falcon tubes at -20 °C.
Measure protein concentrations (A280 nm) with Nanodrop 1000 and run 8% MDPF-gelatin zymography to evaluate protein recovery. See Figure 2.
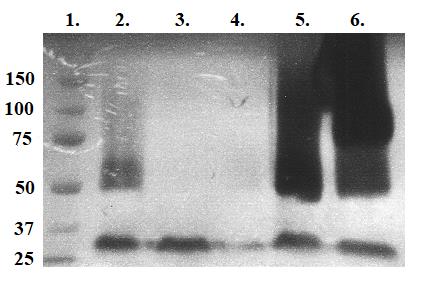
Figure 2. UV photograph of 8% MDPF-gelatin zymography after separation of 10–50 kDa and > 50 kDa proteases with centrifugation devices. Lanes: 1. Molecular weight standard in kDa. 2. Lyticase-treated fraction. 3. Lyticase-treated fraction with 1 µL of β-mercaptoethanol. 4. Lyticase-treated 10–50 kDa fraction. 5. Lyticase-treated > 50 kDa fraction. 6. Lyticase-treated > 50 kDa fraction with 1 µL of β-mercaptoethanol. Fraction in lane 5 was chosen for ion-exchange chromatography because of desired strong gelatinolytic activity of > 50 kDa sized proteases. The activating effect of β-mercaptoethanol as a reducing agent breaking disulfide bonds in the > 50 kDa fraction in lane 6 may be seen.
Preparation for gel filtration
Measure 1 g of Sephadex G-200 and swell in 40 mL of 20 mM Tris HCl, 0.5 M NaCl, pH 8.2 at RT for 3 days in a sterile decanter (covered with aluminum foil).
Degas gel and buffers with vacuum suction until no bubbling of gases. Store these at 4 °C until used.
MDPF-gelatin zymography
Prepare 8% MDPF-gelatin-labeled zymogels. Prepare bottom separating gel. Add 4.45 mL of this solution between glasses in the casting device and add MQ on top of the gel to prevent drying and to obtain a linear top level of the gel. Let it settle for 30 min. Remove MQ by pouring and gently absorbing with cellulose blotting paper before casting the stacking gel. Prepare top stacking gel. Add stacking gel and comb and cover from light and drying while setting of gels (30 min). Store at 4 °C in MQ-moistened paper wrap and plastic bag covered from light until used. Shield from light during and after setting. Gels may be stored at 4 °C wrapped in MQ-moistened paper in a plastic bag shielded from the light up to one week.
Incubate 15 µL of samples with 5 µL of 4× Laemmli sample buffer in 0.5 mL Eppendorf tubes at RT for 2 h in non-shaking conditions.
Fill the running chamber using 1× SDS-PAGE running buffer (diluted from 5× running buffer with MQ). Use pre-stained SDS–polyacrylamide gel electrophoresis standards (10 µL, Precision Plus ProteinTM standards) for evaluation of protein size, pipette the standard, and incubated samples into gel wells.
Perform electrophoresis at 110 V with the device shielded from light and on ice water.
Incubate gels in zymo buffer I for 30 min at RT and then in zymo buffer II for 30 min at RT on a platform swing. Then, incubate in zymo buffer III o/n or for 1–7 days at 37 °C in a heat cabinet, all protected from light with aluminum foil.
Follow the gelatinolytic reaction for 1–2 days with UV light (photograph), stop at the desired gelatin degradation stage, and stain the gel with 0.1 CBB, destain, scan image, and analyze. Gels may be stored in MQ until scanned.
Choose fractions with the highest gelatinolytic activity and protein concentration. Pool and/or concentrate fractions if needed for the next step (see Figure 2).
Ion-exchange chromatography (IEC)
Evaluate cation and anion exchangers with pH optimization (tested pH for the 10–50 kDa fraction was 6.4; for the > 50 kDa fraction, pH was 7.5 and 8.2) with both weak and strong anion and cation exchangers to find the best combination to capture desired proteins (HiTrap® IEX Selection Kit). This process depends on the isoelectric point of the proteins and affects the bonding to and elution from the column depending on the net charge of the proteins. Aggregation of proteins is maximal at the isoelectric point (pI). We had prior knowledge of the estimated pIs from earlier studies using isoelectric focusing and 2D-MDPF-gelatin zymography. DEAE FF (weak anion exchanger) with buffer pH 8.2 is described in the rest of the protocol as an example for obtaining best recovery of the > 50 kDa fraction.
Prepare IEC start buffer, IEC elution buffer I, and IEC elution buffer II (see Recipes). Filter buffers (0.22 µm), degas, and place on ice.
Centrifuge 8 mL of > 50 kDa fraction at 1753× g for 10 min at 4 °C (fraction C6). Discard supernatant and recover volume to 8 mL with IEC start buffer. Concentrate sample as needed by centrifugation at 1753× g for 20 min at 4 °C. Keep sample on ice.
Wash the DEAE FF column (HiTrap® IEX Selection Kit) with 5 mL of IEC start buffer at 1 mL/min, then with 5 mL of IEC elution buffer II, and then with 5 mL of IEC start buffer again. Avoid air bubbles.
Apply sample with syringe at 1 mL/min. Start gathering 0.5 mL fractions into 1.5 mL Eppendorf tubes. Wash the column with 5 mL of IEC start buffer and then add 5 mL of IEC elution buffer I.
Store the column according to manufacturer’s instructions.
Measure protein concentration with Nanodrop 1000 and run zymography gels to locate fractions with gelatinolytic activity and highest protein concentration. See Figure 3.
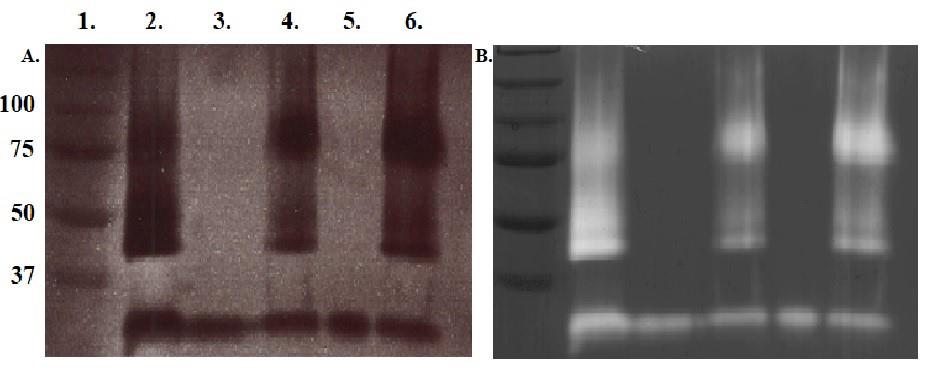
Figure 3. UV photograph of 8% MDPF-gelatin zymography (A) and after Coomassie brilliant blue stain (B) for evaluation of ion-exchange chromatography. Lanes: 1. Molecular weight standard in kDa. 2. Protease sample before ion-exchange chromatography. 3 and 5. Flowthroughs. 4 and 6. Elution fractions. Elution fraction in lane 6 was chosen for gel filtration because of strongest gelatinolytic activity and highest A 280 in the desired protein sizes.
Gel filtration
The setup of the equipment for gel filtration is shown in Figure 4.

Figure 4. Example of setting up the equipment for gel filtration. Keep samples on ice. Remember the safety loop in silicon tubing.Fill a 500 mL decanter with IEC elution buffer I. Enable buffer flow by placing the decanter so high that its silicon tubing end is at a higher level than the silicon tubing end attached to the lower part of the glass column. Ensure there is a safety loop in the tubing to avoid gel dehydration if buffer runs out (see Figure 4).
Take a 9.5 mL column (15 cm length, radius = 4.5 cm) with lower silicon tubing. Open the lower valve of the column and, with a funnel, pour gel until the column is 4/5 full in one continuous motion to avoid air bubbles. Let it settle so that the upper level of the gel does not drop. Fill the column halfway from its lower end with 10% EtOH with a syringe to exclude air bubbles.
Adjust the flow rate of the buffer with 2 × 10 mL of IEC elution buffer I to one drop per 30 s by adjusting decanter position or lower tube length. Drain the buffer to just above the top level of the gel. Shut the lower valve. The column may be stored at 4 °C for approximately one week if not immediately used. For longer storage, add 0.02% sodium azide and wash with approximately 2× column volumes before use.
Recommended sample volume is 1%–5% of bed volume. Choose best elution fraction from ion exchange chromatography (see Figure 3) and concentrate 5 mL to 500 µL with Amicon® Ultra-15 Centrifugal Filter Device 50 K.
Apply 500 µL of the > 50 kDa fraction gently along the edge, avoiding splashing onto the top of the gel bed. Open the lower valve and let the sample sink in. Fill the column all the way to the top with IEC elution buffer I, close upper cap of the column, and attach the silicon tubing from the decanter to the top of the column. Open the lower valve and observe if buffer flow is continuous.
Collect approximately 24 × 0.5 mL fractions. Number the tubes. Measure protein concentration and select the highest concentration (pool fractions if necessary). Run zymography gels to detect highest enzymatic activity for further analyses. See Figure 5. Chosen fraction may be further purified with Sephadex G-100 (as described above) and run on traditional 8% SDS-PAGE and parallel silver-stained gel for LC−MS/MS analyses (Figure 6).
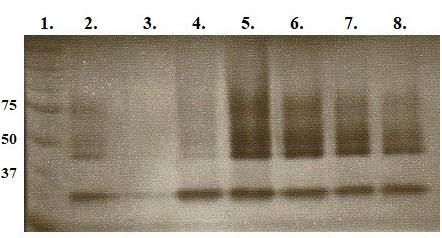
Figure 5. UV photograph of 8% MDPF-gelatin zymography after gel filtration. Lanes: 1. Molecular weight standard in kDa. 2. > 50 kDa fraction after ion-exchange chromatography. 3–8. Elution fractions 1–6 after G-200 gel filtration. Gel filtration of > 50 kDa fraction with Sephadex G-100 did not show any activity with zymography. Fraction 3 in lane 5 was further used in LC−MS/MS analyses.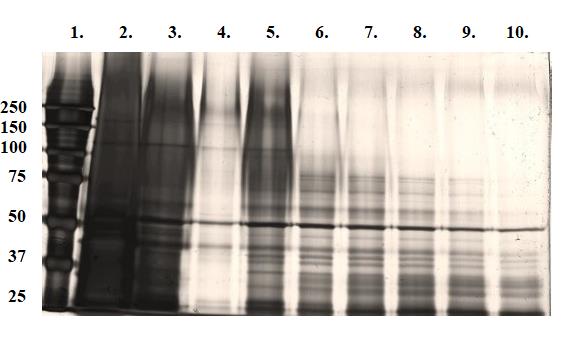
Figure 6. Overview of the purification results, 8% SDS-PAGE gel, silver stained. Lanes: 1. Molecular weight standard in kDa. 2. > 50 kDa fraction before IEC. 3. > 50 kDa fraction before gel filtration. 4−10. > 50 kDa gel filtration fractions.
Validation of protocol
This protocol or parts of it has been used and validated in the following research articles:
Pärnänen, P., Sorsa, T., Tervahartiala, T., and Nikula-Ijäs, P. (2020). Isolation, characterization, and regulation of moonlighting proteases from Candida glabrata cell wall. Microb. Pathogen. 149: 104547. https://doi.org/10.1016/j.micpath.2020.104547 (Figures 1 and 2).
Pärnänen, P., Meurman, J.H., and Nikula-Ijäs, P. (2015). A novel Candida glabrata cell wall associated serine protease. Biochem. Biophys. Res. Commun. 457: 676–680. https://doi.org/10.1016/j.bbrc.2015.01.047 (Figures 1 and 2).
Pärnänen, P., Kari, K., Virtanen, I., Sorsa, T., and Meurman, J. (2008). Human laminin-332 degradation by Candida proteinases. J. Oral Pathol. Med.37: 329-35. https://doi.org/10.1111/j.1600-0714.2008.00638.x (Figure 1).
General notes and troubleshooting
General notes
Keep fractions and samples on ice when appropriate to not lose activity.
Sterile filter or autoclave and degas buffers and store at 4 °C.
Ion exchange chromatography and gel filtration may be conducted also for the 10–50 kDa proteins in the sample with the same principles as for the > 50 kDa fraction.
Use sterile laboratory equipment to avoid contaminations.
Getting to know the principles of the methods in advance is advised.
Troubleshooting
Preparation of yeast cells
Twenty-four hours has proven to be the best incubation time to ensure enough cells for the study. C. glabrata does not form true hyphae, but in other Candida species the incubation time should be shorter to keep cells in non-hyphal form; anti-hyphae substances may have to be used in addition. The OD 600 should be well over two. Too long incubation periods often cause cells to start breaking or transforming.
Lyticase treatment
If yeast cells are not intact when visualized by light microscopy, the lyticase concentration may be too high or treatment time too long. In this case, the intracellular proteins have leaked out and new cells must be cultured.
If there is no or low gelatinolytic activity (zymography) left after lyticase treatment, check if incubation temperature is too low /too high. Other Candida species than C. glabrata may have a different bonding of the cell wall gelatinolytic proteases, which are not hydrolyzed by lyticase.
Separation of 10–50 kDa and > 50 kDa fractions
If fractions show no or low gelatinolytic activity (zymography) left after centrifugal filtering devices, the proteins might not have been recovered from the filter. Try to gently suspend the buffer with a pipette tip (avoiding foaming) on the filter to detach stuck proteins.
Too high salt concentration may precipitate proteins.
Keep fractions on ice to avoid gradual loss of enzymatic activity.
MDPF-gelatin zymography
If CBB-stained gels are not blue, check MDPF-gelatin concentration or expiration date. Keep MDPF-gelatin in the dark; otherwise, fluorescence is lost.
Do not boil zymography samples prior to electrophoresis or enzymatic activity will be lost.
Proteins < 50 kDa may be seen in Figures 3, 4, 6, and 7. This is assumed to be caused by autodegradation or dissociation of smaller subunits of proteins.
IEC
Filter and degas buffers to avoid clogging of column by impurities or air bubbles.
No or low activity in zymography: protein concentration too low (concentrate sample but remember that too much concentration aggregates proteins); wrong pH in buffer or unsuitable ion-exchange column matrix.
Gel filtration
If proper size-dependent separation does not occur, the filtration column height or diameter may be wrong. You may also have to pack the column with matrix carefully and wait until the matrix settles to a horizontal position on the top part of the column. Gentle application of the sample into the column (not disturbing the settled matrix) ensures sharper fraction separation.
If there is no buffer flowing through the column: impurities or air bubbles in gel matrix (caused by non-continuous packing of the column) or buffer vial located too low relative to the column (no hydrostatic pressure).
Collect enough fractions and run zymograms of all fractions to recover/detect desired proteins.
Competing interests
The authors declare no competing interests.
References
- Gow, N. A. R., Latge, J. P. and Munro, C. A. (2017). The Fungal Cell Wall: Structure, Biosynthesis, and Function. Microbiol. Spectr. 5. https://doi.org/10.1128/microbiolspec.FUNK-0035-2016
- de Groot, P. W. J., Kraneveld, E. A., Yin, Q. Y., Dekker, H. L., Gross, U., Crielaard, W., de Koster, C. G., Bader, O., Klis, F. M., and Weig, M. (2008). The Cell Wall of the Human Pathogen Candida glabrata: Differential Incorporation of Novel Adhesin-Like Wall Proteins. Eukaryotic Cell 7(11): 1951–1964. https://doi.org/10.1128/ec.00284-08
- Pardini, G., De Groot, P. W., Coste, A. T., Karababa, M., Klis, F. M., de Koster, C. G. and Sanglard, D. (2006). The CRH Family Coding for Cell Wall Glycosylphosphatidylinositol Proteins with a Predicted Transglycosidase Domain Affects Cell Wall Organization and Virulence of Candida albicans. J. Biol. Chem. 281(52): 40399–40411. https://doi.org/10.1074/jbc.m606361200
- Satala, D., Bras, G., Kozik, A., Rapala-Kozik, M. and Karkowska-Kuleta, J. (2023). More than Just Protein Degradation: The Regulatory Roles and Moonlighting Functions of Extracellular Proteases Produced by Fungi Pathogenic for Humans. J. Fungi 9(1): 121. https://doi.org/10.3390/jof9010121
- Klis, F. M., de Jong, M., Brul, S. and de Groot, P. W. J. (2007). Extraction of cell surface‐associated proteins from living yeast cells. Yeast 24(4): 253–258. https://doi.org/10.1002/yea.1476
- Klinke, T., Rump, A., Pönisch, R., Schellenberger, W., Müller, E. C., Otto, A., Klimm, W. and Kriegel, T. M. (2008). Identification and characterization of CaApe2-- a neutral arginine/alanine/leucine-specific metallo-aminopeptidase from Candida albicans. FEMS Yeast Res. 8(6): 858–869. https://doi.org/10.1111/j.1567-1364.2008.00411.x
- Snoek-van Beurden, P. A. and Von den Hoff, J. W. (2005). Zymographic techniques for the analysis of matrix metalloproteinases and their inhibitors. BioTechniques 38(1): 73–83. https://doi.org/10.2144/05381rv01
- O'Grady, R. L., Nethery, A. and Hunter, N. (1984). A fluorescent screening assay for collagenase using collagen labeled with 2-methoxy-2,4-diphenyl-3(2H)-furanone. Anal. Biochem. 140(2): 490–494. https://doi.org/10.1016/0003-2697(84)90199-4
- Sorsa, T., Salo, T., Koivunen, E., Tyynelä, J., Konttinen, Y. T., Bergmann, U., Tuuttila, A., Niemi, E., Teronen, O., Heikkilä, P., et al. (1997). Activation of Type IV Procollagenases by Human Tumor-associated Trypsin-2. J. Biol. Chem. 272(34): 21067–21074. https://doi.org/10.1074/jbc.272.34.21067
Article Information
Copyright
© 2024 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Pärnänen, P., Sorsa, T., Tervahartiala, T. and Nikula-Ijäs, P. (2024). Preparation and Purification of β-1,3-glucan-Linked Candida glabrata Cell Wall Proteases by Ion-Exchange Chromatography, Gel Filtration, and MDPF-Gelatin-Zymography Assay. Bio-protocol 14(6): e4958. DOI: 10.21769/BioProtoc.4958.
Category
Microbiology > Microbial biochemistry > Protein > Isolation and purification
Biochemistry > Protein > Isolation and purification
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.