- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Simultaneous Profiling of Chromosome Conformation and Gene Expression in Single Cells
(*contributed equally to this work) Published: Vol 13, Iss 22, Nov 20, 2023 DOI: 10.21769/BioProtoc.4886 Views: 3149
Reviewed by: Rajesh RanjanVartika SharmaAnonymous reviewer(s)

Original research article
The authors used this protocol in:

Jun 2023

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Rapid development in single-cell chromosome conformation capture technologies has provided valuable insights into the importance of spatial genome architecture for gene regulation. However, a long-standing technical gap remains in the simultaneous characterization of three-dimensional genomes and transcriptomes in the same cell. We have described an assay named Hi-C and RNA-seq employed simultaneously (HiRES), which integrates in situ reverse transcription and chromosome conformation capture (3C) for the parallel analysis of chromatin organization and gene expression. Here, we provide a detailed implementation of the assay, using mouse embryos and cerebral cortices as examples. The versatility of this method extends beyond these two samples, with the potential to be used in various other cell types.
Key features
• A multi-omics sequencing approach to profile 3D genome structure and gene expression simultaneously in single cells.
• Compatible with animal tissues.
• One-tube amplification of both DNA and RNA components.
• Requires three days to complete.
Graphical overview
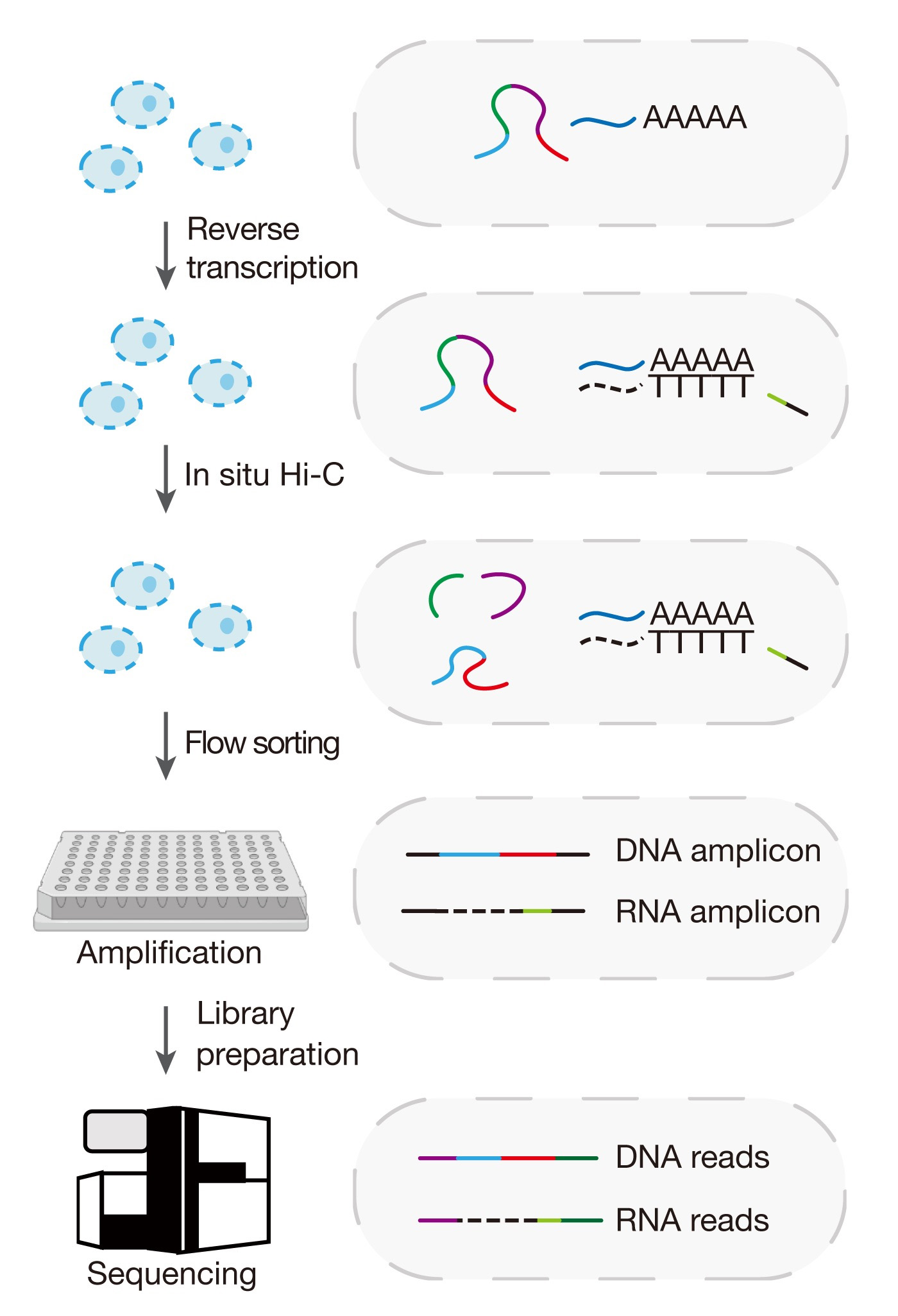
Schematic illustration for the Hi-C and RNA-seq employed simultaneously (HiRES) workflow
Background
The acquisition of distinct cellular identities in multicellular organisms during development requires precise regulation of gene expression across various levels. The progress of single-cell multi-omics sequencing technologies has provided new ways for the comprehensive profiling of the transcriptome in conjunction with various other features, including DNA methylation, cellular proteins, chromatin accessibility, and histone modifications [1–4]. Emerging evidence has suggested that transcription is also closely related with higher-order spatial organization of chromatin in the nucleus [5–7]. Nevertheless, there is a lack of techniques allowing for simultaneous profiling of chromatin conformation and gene expression in individual cells. We have recently developed a method named Hi-C and RNA-seq employed simultaneously (HiRES) [8]. Here, we provide a step-by-step guide on how to perform the assay on mouse embryos and brains. This assay can also be performed on other cell lines or tissues from which single-cell suspensions can be obtained.
Materials and reagents
Materials
Tools and supplies for animal dissection (e.g., forceps, tweezers, eye scissors)
DNA low-bind disposable tips (Axygen, catalog numbers: TF-1000-L-R-S, TF-200-L-R-S, TF-20-L-R-S, TF-300-L-R-S)
Multi-channel pipette tips (Rainin, catalog numbers: 30374581, 30374584)
DNA LoBind tube 1.5 mL (Eppendorf, catalog number: 022431021)
0.5 mL thin-wall PCR tubes (Axygen, catalog number: PCR-05-C)
Thermo-fast 96-well plates (Thermo Fisher, catalog number: AB-1400-L)
40 μm cell strainer (Falcon, catalog number: 352340)
0.22 μm filter unit, sterile (Millipore, catalog number: SLGP033RB)
Hemacytometer (INCYTO, catalog number: DHC-N015)
Dounce tissue grinder, 1 mL (Wheaton, catalog number: 357538)
DNA Clean & Concentrator-5 (Zymo Research, catalog number: D4014)
AMPure XP beads (Beckman Coulter, catalog number: A63881)
Adhesive PCR plate seal (Bio-Rad, catalog number: MSB1001)
Reagents
RNase ZapTM (Invitrogen, catalog number: AM9780)
0.4% Trypan Blue solution (Sigma, catalog number: T8154-20ML)
Nuclease-free water (not DEPC-treated) (Thermo Fisher, catalog number: AM9937)
Phosphate-buffered saline (PBS) (Thermo Fisher, catalog number: 10010049)
Fetal bovine serum (FBS) (Gibco, catalog number: 10099141C)
TrypLETM Express (Gibco, catalog number: 12604013)
Sucrose, reagent grade, 99% (Sigma, catalog number: V900116-500G)
HEPES, BioUltra (Sigma, catalog number: 54457-250G-F)
1 M potassium chloride (KCl) (Sigma, catalog number: 60142-100ML-F)
1 M magnesium chloride (MgCl2) (Thermo Fisher, catalog number: AM9530G)
0.1 M DL-Dithiothreitol solution (DTT) (Invitrogen, catalog number: 18090050)
RNase inhibitor (TaKaRa, catalog number: 2313B)
SUPERaseInTM RNase inhibitor (Invitrogen, catalog number: AM2694)
10% Igepal CA 630 (Sigma, catalog number: I8896-100ML)
Protease inhibitor cocktail (Sigma, catalog number: P8340-5ML)
16% formaldehyde solution (Thermo Fisher, catalog number: 28906)
Bovine serum albumin, powder (BSA) (Sigma, catalog number: B2064)
Bovine serum albumin (20 mg/mL BSA) (NEB, catalog number: B9000S)
Glycerol (Sigma, catalog number: 49767-250ML)
1 M Tris pH 8.0 (Invitrogen, catalog number: AM9855G)
5 M sodium chloride (NaCl) (Invitrogen, catalog number: AM9760G)
Sodium dodecyl sulfate solution, BioUltra, ~10% in H2O (10% SDS) (Sigma, catalog number: 71736-100ML)
TritonTM X-100 solution, BioUltra, ~10% in H2O (10% Triton X-100) (Sigma, catalog number: 93443)
10 mM deoxynucleotide (dNTP) solution mix (dNTPs) (NEB, catalog number: N0447L)
100 mM magnesium sulfate solution (MgSO4) (NEB, catalog number: M0259L)
Betaine solution, 5 M, PCR reagent (Sigma, catalog number: B0300-5VL)
5× SuperScriptTM IV RT buffer (Invitrogen, catalog number: 18090050)
SuperScriptTM IV reverse transcriptase (Invitrogen, catalog number: 18090050)
10× NEBuffer 2 (NEB, catalog number: B7002S)
25 U/μL MboI (NEB, catalog number: R0147M)
10× T4 DNA ligase reaction buffer (NEB, catalog number: B0202S)
T4 DNA ligase (1 U/μL) (Invitrogen, catalog number: 15224-025)
QIAGEN protease (30 AU) (QIAGEN, catalog number: 19157)
0.5 M Ethylenediaminetetraacetic acid, pH 8.0 (EDTA) (Invitrogen, catalog number: AM9260G)
10× ThermoPol® reaction buffer (NEB, catalog number: M0259L)
Deep Vent® (exo-) DNA polymerase (NEB, catalog number: M0259L)
TruePrep DNA Library Prep kit V2 for Illumina®, 5× TTBL (Vazyme, catalog number: TD501)
TruePrep DNA Library Prep kit V2 for Illumina®, TTE Mix V50 (Vazyme, catalog number: TD501)
KAPA HiFi PCR Kit, 5× KAPA HiFi GC buffer (Roche, catalog number: KK2102)
KAPA HiFi PCR Kit, KAPA HiFi DNA polymerase (1 U/μL) (Roche, catalog number: KK2102)
High Sensitivity NGS Fragment Analysis kit (Agilent, catalog number: DNF-474-0500)
i5 and i7 unique dual (UD) index (Illumina, catalog numbers: 20091654, 20091656, 20091658, 20091660)
Primers (synthesized by Sangon Biotech with PAGE purification)
GAT5-RT: GTAGGTGTGAGTGATGGTTGAGGTAGTATTGCGCAATGNNNNNNNN TTTTTTTTTTTTTTTVN
GAT5-7N: GTAGGTGTGAGTGATGGTTGAGGTAGTNNNNNNN
GAT5: GTAGGTGTGAGTGATGGTTGAGGTAGT
RNA-P7: GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGGTTGAGGTAGT ATTGCGCAATG
Solutions
2% BSA (see Recipes)
1.5 M Sucrose (see Recipes)
60 mg/mL Qiagen protease (see Recipes)
100 mM HEPES buffer pH 8.0 (see Recipes)
2× SC lysis buffer (see Recipes)
Recipes
2% BSA (5 mL)
Reagent Final concentration Quantity 1 M Tris-HCl pH 8.0
1 M KCl
500 mM EDTA
H2O
BSA (powder)
40 mM
200 mM
0.2 mM
n/a
2% (w/v)
200 μL
1 mL
2 μL
3,798 μL
0.2 g
Total n/a 5 mL * *Note: Filter through a 0.22 μm filter. Add an equal volume of glycerol and store at -20 °C (for long-term storage, keep at -80 °C).
1.5 M sucrose (10 mL)
Reagent Final concentration Quantity Sucrose
H2O
1.5 M
n/a
5.13 g
10 mL
Total n/a 10 mL * *Note: Filter through a 0.22 μm filter. Aliquot and store at -20 °C. H2O should be added in increments of 10 mL.
60 mg/mL Qiagen protease
Reagent Final concentration Quantity 7.5 AU Qiagen protease
H2O
60 mg/mL
n/a
One vial
2.78 mL
Total n/a 2.78 mL * *Note: The minimum package volume to be purchased was 7.5 AU/tube. Add water to dissolve directly. Mix well and filter through a 0.2 μm filter. Aliquot and store at 4 °C (maximum storage time is two months).
100 mM HEPES buffer pH 8.0 (10 mL)
Reagent Final concentration Quantity HEPES
H2O
100 mM
n/a
0.24 g
see Note*
Total n/a 10 mL * *Note: First, add 9 mL of H2O. Allow the solution to mix completely before adding more. At this time, the liquid is acidic; adjust the pH of the solution to 8.0 with 1 M NaOH and then bring the volume up to 10 mL with H2O. Filter through a 0.22 μm filter. Aliquot and store at -20 °C.
2× SC lysis buffer (5 mL)
Reagent Final concentration Quantity 1 M Tris-HCl pH8.0
5 M NaCl
10% Triton X-100
500 mM EDTA
1 M DTT
H2O
40 mM
100 mM
0.3% (v/v)
2 mM
50 mM
n/a
200 μL
100 μL
150 μL
20 μL
250 μL
4,280 μL
Total n/a 5 mL * *Note: Aliquot and store at -20 °C.
Equipment
Eppendorf ThermoMixer® C (Eppendorf, model: ThermoMixer C)
Vortex mixer (Scientific Industries, model: Vortex-Genie 2)
Centrifuge (Eppendorf, model: 5424)
Tube revolver rotator (Thermo, model: 88881002)
Thermocycler (Bio-Rad, model: C1000)
Magnetic separator (Invitrogen, model: DynaMagTM -2)
Fluorometer (Invitrogen, model: Qubit 4)
Flow cytometer (BD FACSAriaTM Cell Sorter, model: 65011040)
Bioanalyzer (Agilent Fragment Analyzer, model: 5200)
Software and datasets
Snakemake (v5.20.1, access date: Oct 19, 2023) [9] (Conda: https://anaconda.org/bioconda/snakemake)
hickit (v0.1.1, access date: Oct 19, 2023) (GitHub: https://github.com/lh3/hickit/)
CHARMtools (v0.1, access date: Oct 19, 2023) (GitHub: https://github.com/skelviper/CHARMtools)
Other software or packages listed in https://github.com/skelviper/HiRES/blob/main/HiRES_preprocess_pipeline/envs/main_env.yaml
Procedure
Animal maintenance
Raise Cast/EiJ male mice (purchased from Jackson Laboratory) and C57BL/6J female mice (purchased from Beijing HFK Bioscience Co., Ltd and Institutional Animal Care of Peking University) under 12:12 h brightness/darkness. Eight-week-old mice were mated naturally; the morning of plug detection was defined as embryonic day 0.5 (E0.5).
Tissue dissection and single-cell/nucleus isolation (0.5–2 h, depending on the number of mice and embryos)
Mouse embryosCollet post-implantation embryos in PBS containing 10% FBS from E7.0 to E11.5 (see General note 1).
For E7.0–E8.5 embryos, collect all embryos from one mouse as a biological replicate. Dissociate embryos in 100 μL of TrypLETM Express with 200 μL tips pipetting up and down for 5 min.
For E9.5–E11.5 embryos, collect one or two embryos from one mouse as a biological replicate. Dissociate embryos in 200 μL of TrypLETM Express with 1 mL tips for 5 min and then with 200 μL tips, pipetting for another 5 min (see General note 2).
Add a 5-fold volume of PBS (e.g., 500 μL of PBS for 100 μL of TrypLETM Express) to inactivate TrypLETM Express.
Filter the cell suspension with a 40 μm cell strainer to a new 1.5 mL tube.
Centrifuge at 300× g for 5 min at 4 °C.
Remove supernatant and wash pellet with 500 μL of PBS.
Centrifuge at 300× g for 5 min at 4 °C.
Remove supernatant and resuspend pellet with 500 μL of PBS.
Stain a 5 μL aliquot of the cell suspension with 5 μL of 0.4% Trypan Blue. Pipette to mix thoroughly.
Transfer to a disposable hemocytometer to estimate the number of intact cells.
Adjust the number of cells to ~100,000 in 200 μL of PBS.
Mouse brains
Prepare homogenization buffer (1 mL for each sample), vortex to mix, and keep on ice.
1.5 M sucrose 167 μL
100 mM HEPES buffer pH 8.0 100 μL
1 M KCl 25 μL
1 M MgCl2 5 μL
1 mM DTT 1 μL
RNase inhibitor 10 μL
SUPERaseIn 10 μL
10% Igepal CA 630 10 μL
Protease inhibitor 100 μL
H2O 572 μL
Precool the Dounce homogenizer and pestles on ice. Once cooled, fill the homogenizer with 1 mL of cold homogenization buffer and keep it on ice.
Dissected left cortices from 8–9-week-old F1 hybrid mice in ice-cold PBS.
Transfer the cortex from one mouse to a Petri dish (on ice) and cut out a ~10 mm3 section using a chilled scalpel. Immediately transfer the tissue section into the precooled Dounce homogenizer (see General note 3).
Homogenize the tissue with five strokes of the loose pestle, followed by 15 strokes of the tight pestle.
Filter the homogenate through a 40 μm cell strainer to a new 1.5 mL tube.
Centrifuge at 300× g for 5 min at 4 °C and remove supernatant.
Prepare nuclei storage buffer (1.5 mL for each sample), vortex to mix, and keep on ice.
1.5 M sucrose 166.5 μL
100 mM HEPES buffer pH 8.0 150 μL
1 M KCl 37.5 μL
1 M MgCl2 7.5 μL
H2O 1,138.5 μL
Combine 500 μL nuclei storage buffer with 5 μL of RNase inhibitor and 50 μL of protease inhibitor and resuspend pellet.
Centrifuge at 300× g for 5 min at 4 °C.
Remove supernatant and resuspend pellet with 500 μL of nuclei storage buffer.
Stain a 5 μL aliquot of the cell suspension with 5 μL of 0.4% Trypan Blue. Pipette to mix thoroughly.
Transfer to a disposable hemocytometer to estimate the number of intact cells.
Adjust the number of nuclei to ~100,000 in 200 μL with the remaining nuclei storage buffer.
Cell fixation and permeabilization (1–1.5 h)
To fix 200 μL of cell or nucleus suspension, add 24.56 μL of 16% formaldehyde (final 1.75%) directly. Rotate with the tube revolver rotator at 10 rpm for 10 min at room temperature.
Add 20 μL of 2% BSA and invert to mix to quench formaldehyde.
Centrifuge at 1,000× g for 5 min at 4 °C.
Prepare wash buffer (200 μL for each sample), vortex to mix, and keep on ice.
1 M Tris-HCl pH 8.0 2 μL
5 M NaCl 0.4 μL
20 mg/mL BSA 1 μL
RNase inhibitor 2.6 μL
Protease inhibitor 20 μL
H2O 174 μL
Remove supernatant and resuspend cells with 200 μL of ice-cold wash buffer. Mix well by pipetting.
Centrifuge at 1,000× g for 5 min at 4 °C.
Prepare Hi-C lysis buffer (200 μL for each sample), vortex to mix, and keep on ice.
1 M Tris-HCl pH 8.0 2 μL
5 M NaCl 0.4 μL
10% Igepal CA 630 4 μL
RNase inhibitor 2.6 μL
Protease inhibitor 20 μL
H2O 171 μL
Remove supernatant and resuspend cells with 200 μL of ice-cold Hi-C lysis buffer. Mix well by pipetting.
Incubate on ice for 15 min.
Centrifuge at 1,000× g for 5 min at 4 °C.
Prepare 0.5% SDS buffer (100 μL for each sample) and vortex to mix.
10% SDS 5 μL
10 mM dNTPs 5 μL
H2O 90 μL
Remove supernatant and resuspend cells with 100 μL of 0.5% SDS buffer.
Incubate at 65 °C for 10 min and then immediately place on ice for 3 min.
Prepare SDS quench buffer (100 μL for each sample), vortex to mix, and keep on ice.
10% Triton X-100 50 μL
RNase inhibitor 2.6 μL
H2O 47.4 μL
Add 100 μL of ice-cold SDS quench buffer, mix by vortex, and place on ice for 5 min.
Centrifuge at 2,500× g for 5 min at 4 °C.
Reverse transcription (~0.5 h)
Prepare reverse transcription buffer (75 μL for each sample), vortex to mix, and keep on ice.
5 M Betaine 20 μL
10 mM dNTPs 5 μL
100 mM DTT 5 μL
50 μM GAT5-RT primer 5 μL
RNase inhibitor 1.5 μL
SUPERaseIn 1.5 μL
10% Triton X-100 1 μL
100 mM MgSO4 1 μL
H2O 35 μL
Remove supernatant and resuspend nuclei with 75 μL of ice-cold reverse transcription buffer.
Caution: In this step, the cell pellet becomes transparent. Care should be taken to aspirate the supernatant from the opposite side of the pellet to avoid loss.
Critical note: If a salt-containing buffer, such as 5× SSIV buffer, is introduced immediately after SDS treatment, it will result in cell clumping, which is difficult to reverse without cell lysis. Therefore, prior to adding 5× SSIV buffer, it is recommended to resuspend the pellet as thoroughly as possible with reverse transcription buffer to achieve a single-cell suspension.
Add 20 μL of 5× SuperScriptTM IV RT buffer and 5 μL of 200 U/μL SuperScriptTM IV reverse transcriptase (final 10 U/μL). Mix thoroughly by pipetting (see General note 4).
Incubate at 55 °C for 15 min.
Centrifuge at 2,500× g for 5 min at 4 °C.
Digestion and ligation (~17 h)
Prepare digestion buffer (600 μL for each sample) and vortex to mix.
10× NEB buffer 2 60 μL
10% Triton X-100 6 μL
H2O 534 μL
Remove supernatant and resuspend nuclei with 200 μL of digestion buffer.
Centrifuge at 2,500× g for 5 min at 4 °C.
Remove supernatant and resuspend nuclei with 200 μL of digestion buffer.
Centrifuge at 2,500× g for 5 min at 4 °C.
Remove supernatant and resuspend nuclei with 100 μL of digestion buffer.
Add 5 μL of 25 U/μL MboI. Mix by pipetting.
Incubate at 37 °C overnight.
The next day, incubate at 65 °C for 20 min to inactive MboI.
Add 790 μL of water and resuspend nuclei by pipetting.
Take a 20 μL aliquot of the sample as the digested control.
Then, add 100 μL of 10× T4 DNA ligase buffer, 5 μL of 20 mg/mL BSA, and 20 μL of 1 U/μL T4 DNA ligase. Mix by pipetting.
Incubate at 16 °C for 4 h.
Take a 20 μL aliquot of the sample as the ligated control.
Single nuclei flow sorting (0.5–2 h, depending on the number of nuclei needed)
Filter approximately 1 mL of nucleus suspension through a 40 μm cell strainer.
Sort a single nucleus into each well of an empty 96-well plate with a BD FACSAriaTM cell sorter. Forward vs. side scatter (FSC vs. SSC) gating is used to identify single nucleus populations and exclude debris and clumps.
Pause point: Sorted nuclei in dry 96-well plates can be stored at -80 °C for up to six months.
Single nucleus lysis and preamplification (~8 h for each 96-well plate)
Prepare lysis buffer (420 μL for each 96-well plate, 4 μL for each well) and vortex to mix. Dilute 60 mg/mL Qiagen protease to a concentration of 6 mg/mL first.
2× SC lysis buffer 210 μL
6 mg/mL Qiagen protease 37.8 μL
50 μM GAT5 4.2 μL
H2O 168 μL
Add 4 μL of lysis buffer to each well of a 96-well plate containing sorted nuclei with a multichannel pipette (see General note 5).
Briefly spin down and lyse the nuclei by running the following PCR program in a thermocycler:
50 °C 3 h 70 °C 1 h 4 °C ∞ Prepare amplification mixture (3.6 mL for each 96-well plate, 36 μL for each well) and vortex to mix.
10× ThermoPol buffer 400 μL
10 mM dNTPs 120 μL
50 μM GAT5-7N 60 μL
50 μM GAT5 60 μL
100 mM MgSO4 30 μL
Deep Vent (exo-) 100 μL
H2O 2,830 μL
Take the 96-well plate from the thermocycler.
Add 36 μL of amplification mixture to each well of the above 96-well plate with a multichannel pipette.
Caution: Avoid touching the liquid in the plate to prevent template loss before amplification.
Briefly spin down and perform amplification by running the following PCR program in a thermocycler:
1) 95 °C 5 min 2) 4 °C 50 s 3) 10 °C 50 s 4) 20 °C 50 s 5) 30 °C 50 s 6) 40 °C 45 s 7) 50 °C 45 s 8) 65 °C 4 min 9) 95 °C 20 s 10) 58 °C 20 s 11) Go to step 2) for 9 cycles 12) 95 °C 1 min 13) 95 °C 20 s 14) 58 °C 30 s 15) 72 °C 3 min 16) Go to step 13) for 14 cycles 17) 72 °C 5 min 18) 4 °C ∞ Pause point: Amplified DNA can be stored at -20 °C for up to six months.
Measure the concentration of single-nucleus preamplification product with QubitTM 1× dsDNA HS Assay kit through random sampling to ensure the efficacy of the amplification reaction in each 96-well plate (see General note 6).
Library preparation (~4 h for each 96-well plate, ~2 h for library construction, ~2 h for post-PCR cleanup)
Prepare transposition mixture and vortex to mix (318 μL for each 96-well plate, 3 μL for each well):
5× TTBL 106 μL
TTE mix V50 26.5 μL
H2O 185.5 μL
Add 3 μL of transposition mixture to each well of an empty 96-well plate with a multichannel pipette.
Add 2 μL of diluted single-nucleus preamplification product to each well of the above 96-well plate with a multichannel pipette and mix thoroughly.
Briefly spin down and run the following PCR program in a thermocycler:
55 °C 10 min 4 °C ∞ Prepare 200 μL of 0.2 % SDS:
10% SDS 4 μL
H2O 196 μL
Take the 96-well plate from the thermocycler.
Add 1.25 μL of 0.2 % SDS to each well of the above 96-well plate with a multichannel pipette and mix thoroughly.
Briefly spin down and incubate at room temperature for 10 min.
Prepare PCR mixture (1198.5 μL for each 96-well plate, 11.75 μL for each well) and vortex to mix.
5× KAPA HiFi GC buffer 408 μL
10 mM dNTPs 61.2 μL
50 μM RNA-P7 20.4 μL
KAPA HiFi DNA polymerase 40.8 μL
H2O 668.1 μL
Add 2 μL of 5 μM i5 unique dual (UD) index to each well of the above 96-well plate with a multichannel pipette.
Add 11.75 μL of PCR mixture to each well of the above 96-well plate with a multichannel pipette.
Briefly spin down and run the following PCR program in a thermocycler for a slight enrichment of cDNA reads:
1) 72 °C 3 min 2) 98 °C 30 s 3) 98 °C 15 s 4) 60 °C 30 s 5) 72 °C 2 min 6) Go to step 3) for 5 cycles 7) 4 °C ∞ Take the 96-well plate from the thermocycler and add 2 μL of 5 μM i7 unique dual (UD) index with a multichannel pipette.
Briefly spin down and run the following PCR program in a thermocycler:
1) 4 °C 3 min 2) 98 °C 30 s 3) 98 °C 15 s 4) 60 °C 30 s 5) 72 °C 2 min 6) Go to step 3) for 5 cycles 7) 72 °C 5 min 8) 4 °C ∞ Pool the barcoded single-cell libraries and purify pooled libraries with 0.6× and 0.15× AMPure XP beads. The final libraries were sequenced with paired-end 150-bp reads on a NovaSeq 6000 (Illumina) platform.
Quality assessment (1–2 h)
Lyse the digested control and ligated control by adding 1 μL of 60 mg/mL Qiagen protease and run the following PCR program in a thermocycler:
50 °C 1 h 70 °C 15 min 4 °C ∞ Purify the digested control and ligated control with Zymo DNA Clean & Concentrator-5 columns.
To ensure that the crucial steps of the experiment were effectively carried out, we measure size distribution of the samples after digestion, ligation, single-nucleus preamplification, library construction after PCR, and size selection (as shown in Figure 1).
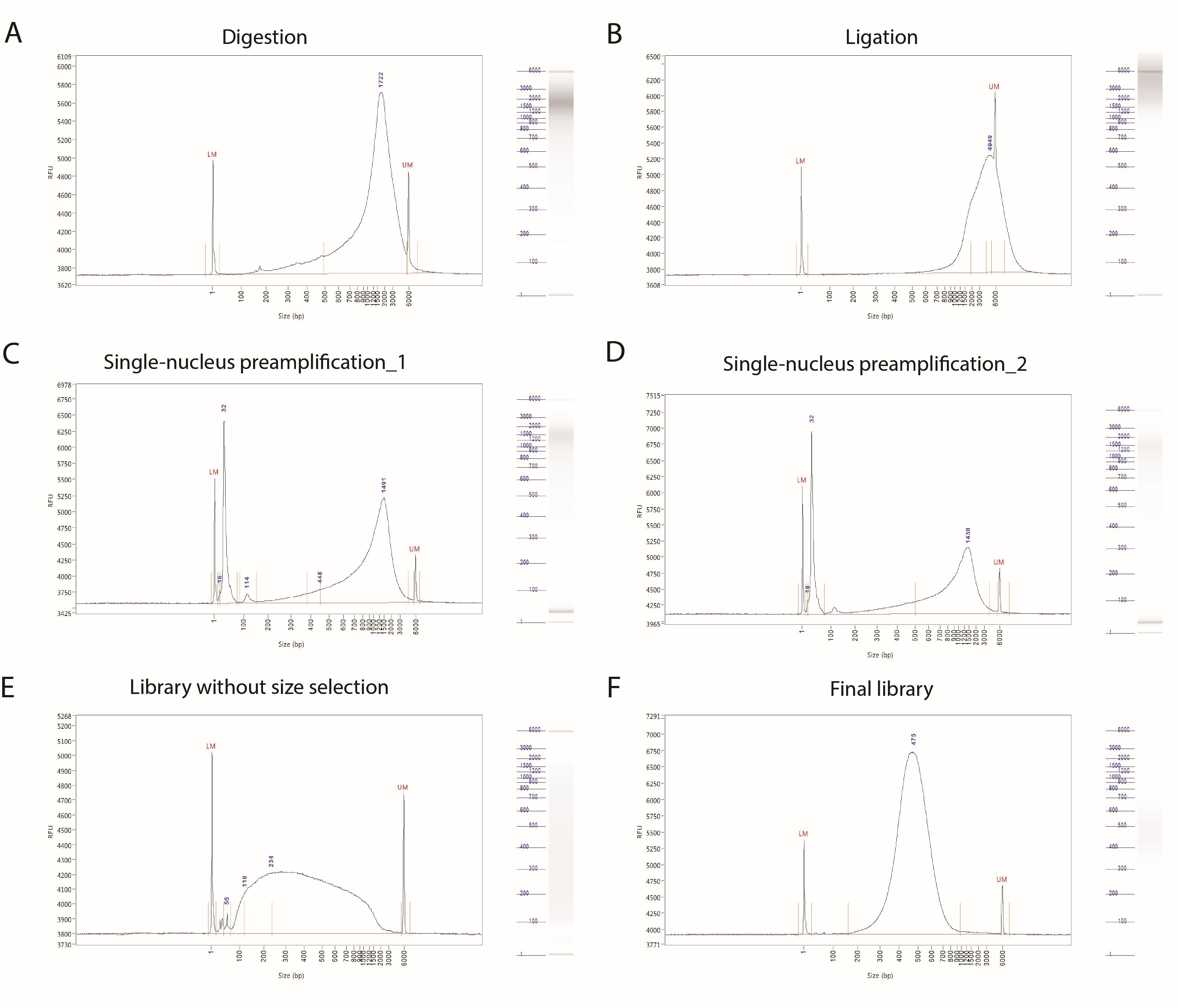
Figure 1. Representative Bioanalyzer traces for the quality control of digestion (A), ligation (B), preamplification (C–D), and library preparation steps (E–F). All samples were run with the High Sensitivity NGS Fragment Analysis Kit on Agilent 5200 Fragment Analyzer.
Data analysis
Preprocessing of HiRES data to derive single-cell RNA expression matrices, single-cell chromatin contact matrices, and three-dimensional reconstruction structures from the original FASTQ sequencing data includes the following steps:
We recommend processing HiRES raw data using our HiRES pre-processing Snakemake workflow. The code can be downloaded from https://github.com/skelviper/HiRES. Please ensure that the workflow code and the folder containing the fastq data are placed within the same working directory.
Set up the environment by executing the following command:
conda create -n hires -c conda-forge -c bioconda python=3.8 snakemake=5.20.1.
Modify the paths to the dependent files in config.yaml and select a reference genome that is appropriate for your experiment.
Execute the workflow by navigating to the HiRES_preprocess_pipeline directory and running the script with the following commands: cd HiRES_preprocess_pipeline; ./runHiRES_preprocess.sh. By default, the workflow utilizes the Slurm scheduler, but you can modify runHiRES_preprocess.sh to use a scheduler that better suits your preference.
Generate experimental statistical data by executing the Jupyter Notebook found at analysis/stat.ipynb.
Use output files for other downstream data analysis.
With HiRES data, the gene expression level can be mapped onto the reconstructed 3D genome structure for the same single cell (as shown in Figure 2).
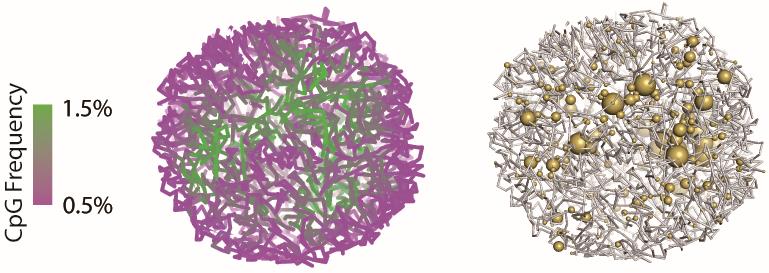
Figure 2. Representative example of a single-cell 3D genome structure and its corresponding gene expression. Compartmentalization of euchromatin (green) and heterochromatin (magenta) was visualized by CpG frequency as a proxy (left). The size of yellow balls was proportional to the gene expression level (right). Example single-cell data and figures are from reference [8].
Validation of protocol
This protocol or parts of it has been used and validated in the following research article(s):
Liu et al. (2023). Linking genome structures to functions by simultaneous single-cell Hi-C and RNA-seq. Science (Figure 1, panel B–D; Figure 2, panel A–D; Figure S2, panel C; Figure S3, panel B).
General notes and troubleshooting
General notes
Before dissection, keep the workstation and hood RNase-free by thoroughly cleaning with RNase Zap.
For steps B2 and B3, the digestion time for embryos at different stages could be slightly adjusted according to the homogeneity of the cell suspension. Pipetting should be performed softly to maintain cell integrity as much as possible.
To reduce heat caused by friction, Dounce homogenization should be performed on ice with gentle strokes, and care should be taken to avoid foaming. The mortar should be immersed in ice. The precooled homogenization buffer is an important aid in heat reduction during homogenization.
Increasing the volume of SuperScript IV Reverse Transcriptase to 10 μL led to more transcripts detected but was accompanied by a decrease in the detected number of contacts.
The lysis buffer should be carefully added along the tube wall, while ensuring that the pipette tip does not reach the bottom of the tube to prevent loss of single nuclei. If subsequent experiments involving cell lysis can be conducted immediately after flow sorting, the single nuclei can be directly sorted into a 96-well plate containing lysis buffer.
The concentration of a single-nucleus preamplification product is generally between 30 and 50 ng/μL. For convenience, we generally dilute the product by a factor of 10 using water, resulting in a diluted product concentration of approximately 3–5 ng/μL. Each diluted sample was used for library preparation.
If the assay is intended to be applied to other biological samples, it might be necessary to modify the procedures for obtaining single-cell suspensions according to the specific characteristics of different tissues.
Troubleshooting
Insufficient single nuclei available for flow cytometry sorting.
Potential solution: if a substantial loss of nuclei is observed after each centrifugation, consider extending the centrifugation duration as needed.
Cell loss or cell aggregation after SDS treatment.
Potential solution: if the transparent cell pellet cannot be found after SDS treatment, aspirate the supernatant slowly from the opposite side of the pellet to avoid loss and resuspend the pellet by pipetting at least 20 times to avoid cell aggregation.
No or low-yield single-nucleus amplification product (i.e., less than 10 ng/μL) after preamplification.
Potential solution: check reagents that are crucial for lysis and preamplification. For example, Qiagen protease must be present in lysis buffer and the final concentration must be appropriate.
Acknowledgments
This protocol is based on the original research paper of Liu et al., 2023. This work was supported by the National Natural Science Foundation of China (grant 92049110) and the funding from Beijing Advanced Innovation Center for Genomics (ICG).
Competing interests
D.X., Y.C., and Z.L. are investors on a patent that covers HiRES. H.X. declares no competing interests.
Ethical considerations
All animal maintenance and experimental procedures were carried out in accordance with the guidelines of the Institutional Animal Care and Use Committee (IACUC) of Peking University.
References
- Angermueller, C., Clark, S. J., Lee, H. J., Macaulay, I. C., Teng, M. J., Hu, T. X., Krueger, F., Smallwood, S. A., Ponting, C. P., Voet, T., et al. (2016). Parallel single-cell sequencing links transcriptional and epigenetic heterogeneity. Nat. Methods 13(3): 229–232. doi: 10.1038/nmeth.3728
- Stoeckius, M., Hafemeister, C., Stephenson, W., Houck-Loomis, B., Chattopadhyay, P. K., Swerdlow, H., Satija, R. and Smibert, P. (2017). Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods 14(9): 865–868. doi: 10.1038/nmeth.4380
- Ma, S., Zhang, B., LaFave, L. M., Earl, A. S., Chiang, Z., Hu, Y., Ding, J., Brack, A., Kartha, V. K., Tay, T., et al. (2020). Chromatin Potential Identified by Shared Single-Cell Profiling of RNA and Chromatin. Cell 183(4): 1103–1116.e20. doi: 10.1016/j.cell.2020.09.056
- Zhu, C., Zhang, Y., Li, Y. E., Lucero, J., Behrens, M. M. and Ren, B. (2021). Joint profiling of histone modifications and transcriptome in single cells from mouse brain. Nat. Methods 18(3): 283–292. doi: 10.1038/s41592-021-01060-3
- Zheng, H. and Xie, W. (2019). The role of 3D genome organization in development and cell differentiation. Nat. Rev. Mol. Cell Biol. 20(9): 535–550. doi: 10.1038/s41580-019-0132-4
- Hafner, A. and Boettiger, A. (2022). The spatial organization of transcriptional control. Nat. Rev. Genet. 24(1): 53–68. doi: 10.1038/s41576-022-00526-0
- Tan, L., Ma, W., Wu, H., Zheng, Y., Xing, D., Chen, R., Li, X., Daley, N., Deisseroth, K., Xie, X. S., et al. (2021). Changes in genome architecture and transcriptional dynamics progress independently of sensory experience during post-natal brain development. Cell 184(3): 741–758.e17. doi: 10.1016/j.cell.2020.12.032
- Liu, Z., Chen, Y., Xia, Q., Liu, M., Xu, H., Chi, Y., Deng, Y. and Xing, D. (2023). Linking genome structures to functions by simultaneous single-cell Hi-C and RNA-seq. Science 380(6649): 1070–1076. doi: 10.1126/science.adg3797
- Mölder, F., Jablonski, K. P., Letcher, B., Hall, M. B., Tomkins-Tinch, C. H., Sochat, V., Forster, J., Lee, S., Twardziok, S. O., Kanitz, A., et al. (2021). Sustainable data analysis with Snakemake. F1000Research 10: 33. doi: 10.12688/f1000research.29032.2
Article Information
Copyright
© 2023 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Chen, Y., Xu, H., Liu, Z. and Xing, D. (2023). Simultaneous Profiling of Chromosome Conformation and Gene Expression in Single Cells. Bio-protocol 13(22): e4886. DOI: 10.21769/BioProtoc.4886.
- Liu, Z., Chen, Y., Xia, Q., Liu, M., Xu, H., Chi, Y., Deng, Y. and Xing, D. (2023). Linking genome structures to functions by simultaneous single-cell Hi-C and RNA-seq. Science 380(6649): 1070–1076. doi: 10.1126/science.adg3797
Category
Systems Biology > Genomics > Sequencing
Molecular Biology > DNA > DNA sequencing
Cell Biology > Single cell analysis
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.