- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Efficient Large DNA Fragment Knock-in by Long dsDNA with 3′-Overhangs Mediated CRISPR Knock-in (LOCK) in Mammalian Cells
Published: Vol 13, Iss 20, Oct 20, 2023 DOI: 10.21769/BioProtoc.4853 Views: 3061
Reviewed by: Xin XuAnonymous reviewer(s)
Original research article
The authors used this protocol in:

May 2023
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
An efficient and precise genome-editing approach is in high demand in any molecular biology or cell biology laboratory worldwide. However, despite a recent rapid progress in the toolbox tailored for precise genome-editing, including the base editors and prime editors, there is still a need for a cost-effective knock-in (KI) approach amenable for long donor DNA cargos with high efficiency. By harnessing the high-efficient double-strand break (DSB) repair pathway of microhomology-mediated end joining, we previously showed that a specially designed 3′-overhang double-strand DNA (odsDNA) donor harboring 50-nt homology arm (HA) allows high-efficient exogenous DNA KI when combined with CRISPR-Cas9 technology. The lengths of the 3′-overhangs of odsDNA donors could be manipulated by the five consecutive phosphorothioate (PT) modifications. In this protocol, we detail the stepwise procedures to conduct the LOCK (Long dsDNA with 3′-Overhangs mediated CRISPR Knock-in) method for gene-sized (~1–3 kb) KI in mammalian cells.
Graphical overview
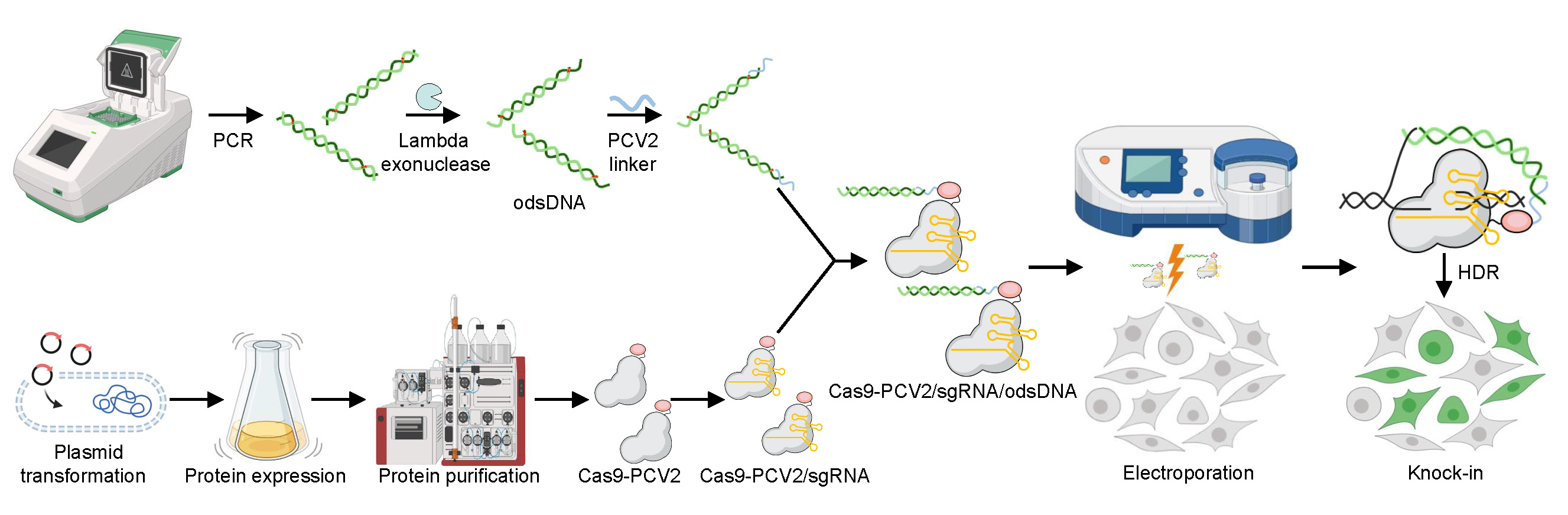
Improvement of large DNA fragment knock-in rates by attaching odsDNA donors to Cas9-PCV2 fusion protein
Background
The advent of CRISPR and its derived technologies have tremendously expanded the toolbox for genome manipulation for scientists in the fields of biomedical research and innovative biotechnological development. Compared with the ZFN or TALEN approaches, which are highly reliant on the specific recognition of the target genomic DNA (gDNA) sequence by specially engineered protein readers, the CRISPR-Cas9 method only necessitates a single crRNA (~20 nt) that uniquely recognizes and pairs with the specific gDNA locus (Hsu et al., 2014). This improvement makes genome-editing technique adoptable in any individual lab because of the low cost and simplified procedures. However, it is known that the precise integration of large donor DNA sequences into the host genome is dependent on homology-directed repair (HDR), which occurs at low frequencies, in mammalian cells. In addition, the double-strand break (DSB) sites induced by Cas9 cutting could happen at undesired genomic loci (off-target), and this might result in unexpected DSB repair outcomes, e.g., insertions/deletions (indels) and translocations, which are fundamentally detrimental to cellular health. It is, therefore, critical to increase the precise targeting efficiency while minimizing the off-target effects (Doudna, 2020).
In the past decade, the scientists have made great achievements in the improvement of the precise CRISPR-Cas9 knock-in (KI) efficiency. The phosphoric backbone of the donor DNA sequences is highly negatively charged, which naturally impedes their transportation across the cellular membrane, leading to the inaccessibility of exogenous DNA donors in the local DSB sites. This has been attributed as one of the reasons causing the low efficiencies of HDR-mediated KI. As such, versatile approaches have by far been devised to promote the nuclear delivery or the structural stability of the exogenous repair DNA donors (Yu et al., 2020). For instance, the nuclear entry of DNA donors could be promoted by adeno-associated viral (AAV) vectors, by Strep-biotin labeled tethering (Gu et al., 2018), or by chromatinized packaging (Cruz-Becerra and Kadonaga, 2020).
On the other hand, it is known that while the canonical nonhomologous end joining (c-NHEJ) repair is much more frequent at the CRISPR-Cas9-induced DSB sites, the repair pathway could be biased to the precise HDR repair outcome through artificially manipulating the cellular milieu (Maruyama et al., 2015). Indeed, accumulating examples have shown that the long ssDNA (lssDNA), as exogenous HDR donors, display superior advantages over the conventional double-strand DNA (dsDNA) donors, in terms of the HDR efficiency and off-target effect (Quadros et al., 2017; Shy et al., 2023). By leveraging the distinct advantages from both dsDNA and ssDNA donors, our previous study showed that a single hybrid “3′-overhang dsDNA” donor, termed odsDNA, has proved to significantly improve the HDR efficiencies by up to 5-fold increase with low off-target effect (Han et al., 2023). We named this Long dsDNA with 3′-Overhangs mediated CRISPR Knock-in, “LOCK” for short. In this protocol, we describe this easy technique with the stepwise procedures needed to be operational in any individual lab.
Materials and reagents
Materials
Pipette tips: 10, 200, and 1,000 μL (Yeasen, catalog numbers: 83010ES20, 83040ES20, and 83070ES08)
200 μL extended pipette tips, sterilized (Yeasen, catalog number: 83061ES50)
Pipettes: 2.5, 10, 20, 100, 200, and 1,000 μL (Eppendorf, catalog numbers: 3120000216, 3120000224, 3120000232, 3120000240, 3120000259, and 3120000267)
0.2 mL PCR strips of eight tubes (Yeasen, catalog number: 83602ES10)
1.5 mL microcentrifuge tube (Axygen, catalog number: MCT-150-C)
50 mL conical tube (Thermo Fisher, catalog number: 339652)
DNA oligonucleotide primers and PCV2 linker (Sangon Biotech)
6-well cell culture plate (BIOFIL, catalog number: TCP011006)
60 mm cell culture dish (BIOFIL, catalog number: TCD010060)
10 cm cell culture dish (BIOFIL, catalog number: TCD010100)
HEK293T cells (American Type Culture Collection, catalog number: CRL-3216)
DH5α competent cells (Sangon Biotech, catalog number: B528413)
Escherichia coli Rosetta 2 (DE3) strain (Millipore EMD, catalog number: 71397)
Isopropyl β-D-1-thiogalactopyranoside (IPTG) (Invitrogen, catalog number: 15529019)
Tryptone (Gibco, catalog number: 211705)
Yeast extract (Gibco, catalog number: 211931)
Kanamycin monosulfate (Sangon Biotech, catalog number: A600286)
Agar (Thermo Fisher Scientific, catalog number: 22700025)
Terrific broth (Thermo Fisher Scientific, catalog number: 22711022)
Glycine (Invitrogen, catalog number: 15527013)
SDS (Sigma-Aldrich, catalog number: 436143)
NaCl (Sigma-Aldrich, catalog number: S9888)
KCl (Sigma-Aldrich, catalog number: P3911)
MgCl2 hexahydrate (Sigma-Aldrich, catalog number: M2670)
Tris (Invitrogen, catalog number: A32355)
HCl (Sigma-Aldrich, catalog number: 30721)
NaOH (Sigma-Aldrich, catalog number: 221465)
Imidazole (Sigma-Aldrich, catalog number: I2399)
UltraPure glycine (Invitrogen, catalog number: 15527013)
β-mercaptoethanol (Gibco, catalog number: 21985023)
EDTA (Thermo Fisher Scientific, catalog number: 17892)
HEPES (Sigma-Aldrich, catalog number: 54457)
TCEP (Thermo Fisher Scientific, catalog number: T2556)
Glycerol (Thermo Fisher Scientific, catalog number: 17904)
HisTrap fast flow, 5 mL (GE Healthcare, catalog number: GE17-5255-01)
Amicon Ultracel-100 regenerated cellulose membrane, 50 mL (Millipore, catalog number: UFC910008)
Amicon Ultracel-30 regenerated cellulose membrane, 4 mL (Millipore, catalog number: UFC803008)
0.22 μm syringe filter, PES membrane, 33 mm diameter (Millipore, catalog number: SLGPR33RS)
0.45 μm syringe filter, PES membrane, 33 mm diameter (Millipore, catalog number: SLHPR33RS)
Nalgene Rapid-Flow with PES 500 mL (Thermo Fisher Scientific, catalog number: 566-0020)
PD-10 desalting columns (GE Healthcare, catalog number: GE17-0851-01)
BeyoGel SDS-PAGE Precast Gel, Tris-Gly, 4%–20%, 12 wells (Beyotime, catalog number: P0057A)
Agarose (Thermo Fisher Scientific, catalog number: R0491)
GeneArt Precision gRNA Synthesis kit (Invitrogen, catalog number: A29377)
Invitrogen TrueCut Cas9 Protein v2 (Thermo Fisher Scientific, catalog number: A36497)
PARAFILM sealing film (Sigma-Aldrich, catalog number: HS234526B-1EA)
Kimwipes disposable wipers (Sigma-Aldrich, catalog number: Z188956)
Reagents
ddH2O, DEPC-treated water (Sangon Biotech, catalog number: B300592)
Lambda exonuclease (New England Biolabs, catalog number: M0262L)
GeneJET PCR Purification kit (Thermo Fisher Scientific, catalog number: K0702)
SpCas9 expression plasmid (Addgene, catalog number: 47327)
Dulbecco’s modified Eagle’s medium (DMEM) (Gibco, catalog number: C11885500BT)
TrypLE express (Gibco, catalog number: 12604039)
DPBS (without Ca2+ or Mg2+) (Gibco, catalog number: 14190-144)
Fetal bovine serum (FBS) (Gibco, catalog number: 10091148)
Cell Line Nucleofector kit V (Lonza, catalog number: VCA-1003)
Q5 High-Fidelity 2× Master Mix (New England Biolabs, catalog number: M0492L)
50× TAE buffer (Sangon Biotech, catalog number: B548101)
6× Gel Loading Dye Purple (New England Biolabs, catalog number: B7024S)
SDS-PAGE Protein Loading Buffer, 5× (Beyotime, catalog number: P0286)
BeyoColor Prestained Color Protein Marker, 10–170 kDa (Beyotime, catalog number: P0077)
Imperial protein stain solution (Thermo Fisher Scientific, catalog number: 24615)
Annealing buffer for DNA Oligos, 5× (Beyotime, catalog number: D0251)
LB medium (see Recipes)
50 mg/mL kanamycin (see Recipes)
LB agar plate with kanamycin (see Recipes)
Terrific broth (TB) medium (see Recipes)
1 M IPTG (see Recipes)
Buffer A: start buffer (see Recipes)
Buffer B: elution buffer (see Recipes)
Buffer C: storage buffer (see Recipes)
Buffer D: cleavage buffer (see Recipes)
0.1 M NiSO4 (see Recipes)
0.1 M EDTA, 1 M NaCl (see Recipes)
1 M NaOH (see Recipes)
1 M HCl (see Recipes)
SDS-PAGE running buffer (see Recipes)
Low-EDTA TE buffer (see Recipes)
Recipes
LB medium (1,000 mL)
Reagent Final concentration Quantity Tryptone
Yeast extract
NaCl
10 g/L
5 g/L
10 g/L
10 g
5 g
10 g
ddH2O n/a ~980 mL Total n/a 1,000 mL Adjust the pH to 7.4 with NaOH. Autoclave at 121 °C for 15 min. Store at RT.
50 mg/mL kanamycin (10 mL)
Reagent Final concentration Quantity Kanamycin 50 mg/mL 0.5 g ddH2O n/a 10 mL Total n/a 10 mL Sterilize the solution with a 0.22 μm syringe filter. Store at 4 °C.
LB agar plate with kanamycin (1,000 mL)
Reagent Final concentration Quantity Tryptone
Yeast extract
NaCl
10 g/L
5 g/L
10 g/L
10 g
5 g
10 g
Agar 15 g/L 15 g 50 mg/mL Kanamycin 50 μg/mL 1 mL ddH2O n/a ~960 mL Total n/a 1,000 mL Adjust the pH to 7.4 with NaOH.
Autoclave at 121 °C for 15 min. Agar is added after sterilization.
Stir the medium on a stir plate and allow to cool to 30–40 °C.
Add 1 mL of 50 mg/mL kanamycin solution. Stir well.
Transfer 25 mL of the solution to 10 cm Petri dishes with a sterile pipette.
Allow the agar plates to cool and dry in a laminar flow hood for 30 min.
Store the plates at 4 °C in a plastic bag.
Terrific broth medium (1,000 mL)
Reagent Final concentration Quantity Terrific broth
Glycerol
47.6 g/L
0.4% (v/v)
47.6 g
4 mL
ddH2O n/a ~960 mL Total n/a 1,000 mL Adjust the pH to approximately 7.4 with NaOH. Autoclave at 121 °C for 15 min. Store at RT.
1 M IPTG (10 mL)
Reagent Final concentration Quantity IPTG 1 M 2.38 g ddH2O n/a ~9.8 mL Total n/a 10 mL Sterilize the solution with a 0.22 μm syringe filter. Store at -20 °C.
Buffer A: start buffer (1,000 mL)
Reagent Final concentration Quantity Tris
NaCl
20 mM
500 mM
2.4228 g
29.22 g
ddH2O n/a ~960 mL Total n/a 1,000 mL Adjust the pH to approximately 8.0 with HCl. Sterilize the solution with Nalgene Rapid-Flow. Store at 4 °C.
Buffer B: elution buffer (1,000 mL)
Reagent Final concentration Quantity Tris
NaCl
20 mM
500 mM
2.4228 g
29.22 g
imidazole 500 mM 34.04 g ddH2O n/a ~960 mL Total n/a 1,000 mL Adjust the pH to approximately 8.0 with HCl. Sterilize the solution with Nalgene Rapid-Flow. Store at 4 °C.
Buffer C: storage buffer (1,000 mL)
Reagent Final concentration Quantity Tris
KCl
20 mM
200 mM
2.4228 g
14.91 g
MgCl2·6H2O 10 mM 2.03 g Glycerol 10% (v/v) 100 mL ddH2O n/a ~860 mL Total n/a 1,000 mL Adjust the pH to approximately 8.0 with HCl. Sterilize the solution with Nalgene Rapid-Flow. Store at 4 °C.
Buffer D: cleavage buffer (1,000 mL)
Reagent Final concentration Quantity HEPES
KCl
20 mM
150 mM
4.77 g
11.18 g
MgCl2·6H2O 1 mM 0.2 g TCEP 1 mM 0.25 g Glycerol 10% (v/v) 100 mL ddH2O n/a ~860 mL Total n/a 1,000 mL Adjust the pH to approximately 8.0 with HCl. Sterilize the solution with Nalgene Rapid-Flow. Store at -20 °C.
0.1 M NiSO4 (100 mL)
Reagent Final concentration Quantity NiSO4·6H2O 0.1 M 2.63 g ddH2O n/a ~96 mL Total n/a 100 mL Sterilize the solution with a 0.22 μm syringe filter. Store at RT.
0.1 M EDTA, 1M NaCl (100 mL)
Reagent Final concentration Quantity EDTA 0.5 M 2.92 g NaCl 1 M 5.84 g ddH2O n/a ~96 mL Total n/a 100 mL Adjust the pH to 8.0 with NaOH. Sterilize the solution with a 0.22 μm syringe filter. Store at RT.
1 M NaOH (100 mL)
Reagent Final concentration Quantity NaOH 1 M 4 g ddH2O n/a ~96 mL Total n/a 100 mL Store at RT.
1 M HCl (120 mL)
Reagent Final concentration Quantity 12 M HCl 1 M 10 mL ddH2O n/a 110 mL Total n/a 120 mL Store at RT.
SDS-PAGE running buffer (1,000 mL)
Reagent Final concentration Quantity Tris
Glycine
125 mM
1.25 M
15.1 g
94 g
SDS 0.5% (w/v) 5 g ddH2O n/a ~800 mL Total n/a 1,000 mL Store at RT.
Low-EDTA TE buffer, 1× (for DNA) (100 mL)
Reagent Final concentration Quantity Tris
EDTA
10 mM
0.2 mM
0.1211 g
0.0012 g
ddH2O n/a ~100 mL Total n/a 100 mL Adjust the pH to 8.0 with HCl. Sterilize the solution with a 0.22 μm syringe filter. Store at RT.
Equipment
Amaxa Nucleofector IIb device (Lonza, model: AAB-1001)
BD Accuri C6 flow cytometer (BD Biosciences, catalog number: 23432)
Heracell VIOS 160i CO2 copper chamber incubator (Thermo Fisher Scientific, catalog number: 51030476)
Biological safety cabinet (Esco Airstream, model: AB2-4S8-CN)
Sonicator Q125 (Qsonica, model: Q125-220)
1/4 diameter sonica probe (Qsonica, catalog number: 4435)
Protein purification system (AKTA pure, catalog number: 29046665)
Fluorescence spectrophotometer F-7000 (HITACHI, model: F-7000)
IX71 inverted microscope (Olympus, model: IX71)
Snowflake ice machine (XUEKE, model: IMS-20)
Water bath (EYELA, model: NTT-2200)
Mini gel DNA electrophoresis system (Thermo Fisher, model: B1)
Gel imaging system (Azure Biosystems, model: C300)
Veriti 96- Well PCR thermal cycler (Applied Biosystems, catalog number: 4375786)
TAdvanced Twin PCR thermocycler (Biometra, catalog number: 2070212)
NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, model: ND-2000)
pH meter (Mettle Toledo, catalog number: 51302807)
Milli-Q water purification system (Millipore, model: MP0024)
Eppendorf centrifuge 5424 R (Eppendorf, catalog number: 5404000090)
Eppendorf high-speed refrigerated centrifuge 5910 R (Eppendorf, catalog number: 5942000598)
Thermo shaker (Yeasen, catalog number: 80440ES03)
Software
Prism v8, for statistics and data visualization (GraphPad, version 8.0.1.244)
Snapgene, view DNA sequence and primer design (version 4.2.4)
NUPACK: Nucleic Acid Package (https://nupack.org/)
Tm Calculator (version 1.16.5, https://tmcalculator.neb.com/#!/main)
CHOPCHOP, a web-based tool for sgRNA design. The website is for non-profit and academic use only (https://chopchop.cbu.uib.no/)
Expasy, conversion of DNA sequence to amino acid sequence (https://web.expasy.org/translate/)
AAT Bioquest: Protein Concentration Calculator (https://www.aatbio.com/tools/calculate-protein-concentration)
NEBioCalculator (https://nebiocalculator.neb.com/#!/dsdnaamt)
Procedure
Preparation of dsDNA and odsDNA as DNA donors to achieve green fluorescent protein KI at varying genomic loci in HEK293T cells
Design target sites for Lamin A/C, GAPDH, and AAVS1 loci, as examples. The donor DNA sequences contain the to-be-inserted sequence flanked by 50 nt homology arm.
The target sequences were selected by the online pipeline CHOPCHOP; an example is shown in Figure 1.
Note: Target sequences with fewer mismatches and higher scores.
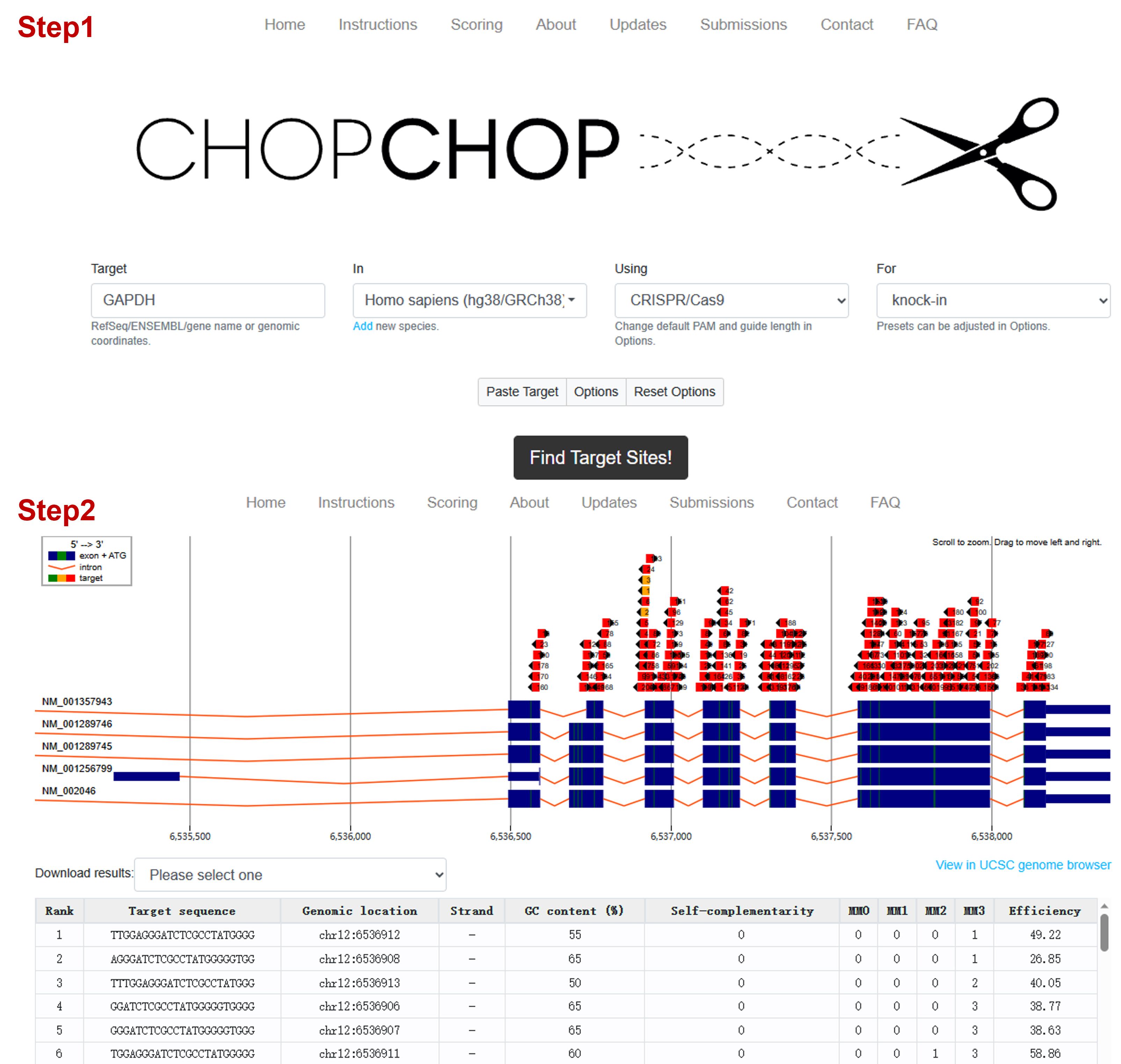
Figure 1. Screenshot example of the CHOPCHOP website when selecting the target sequenceAll the 1,010 and 2,500 bp gene fragments as donors were cloned into plasmids following general cloning procedures, including promoter region and EGFP sequence. HA sequences are underlined. The template sequences are shown in Table 1.
Table 1. Sequences of the target insert in donor plasmids used as PCR templates
Lamin A/C locus donor sequence (EF-1α core promoter-EGFP 1110bp) ctttggtttttttcttctgtatttgtttttctaagagaagttattttctataggtcttgaaaggagtgggtcaattggctccggtgcccgtcagtgggcagagcgcacatcgcccacagtccccgagaagttggggggaggggtcggcaattgatccggtgcctagagaaggtggcgcggggtaaactgggaaagtgatgtcgtgtactggctccgcctttttcccgagggtgggggagaaccgtatataagtgcagtagtcgccgtgaacgttctttttcgcaacgggtttgccgccagaacacaggaagcttgccaccatggtgagcaagggcgaggagctgttcaccggggtggtgcccatcctggtcgagctggacggcgacgtaaacggccacaagttcagcgtgtccggcgagggcgagggcgatgccacctacggcaagctgaccctgaagttcatctgcaccaccggcaagctgcccgtgccctggcccaccctcgtgaccaccctgacctacggcgtgcagtgcttcagccgctaccccgaccacatgaagcagcacgacttcttcaagtccgccatgcccgaaggctacgtccaggagcgcaccatcttcttcaaggacgacggcaactacaagacccgcgccgaggtgaagttcgagggcgacaccctggtgaaccgcatcgagctgaagggcatcgacttcaaggaggacggcaacatcctggggcacaagctggagtacaactacaacagccacaacgtctatatcatggccgacaagcagaagaacggcatcaaggtgaacttcaagatccgccacaacatcgaggacggcagcgtgcagctcgccgaccactaccagcagaacacccccatcggcgacggccccgtgctgctgcccgacaaccactacctgagcacccagtccgccctgagcaaagaccccaacgagaagcgcgatcacatggtcctgctggagttcgtgaccgccgccgggatcactctcggcatggacgagctgtacaagtaagactctggtcagagatacctcagtggttttatactgaaggaaaaacacaagcaaaaaaaaaaaaaaagca GAPDH locus donor sequence (EF-1α core promoter-EGFP 1110bp) atggcctccaaggagtaagacccctggaccaccagccccagcaagagcactaggtcttgaaaggagtgggtcaattggctccggtgcccgtcagtgggcagagcgcacatcgcccacagtccccgagaagttggggggaggggtcggcaattgatccggtgcctagagaaggtggcgcggggtaaactgggaaagtgatgtcgtgtactggctccgcctttttcccgagggtgggggagaaccgtatataagtgcagtagtcgccgtgaacgttctttttcgcaacgggtttgccgccagaacacaggaagcttgccaccatggtgagcaagggcgaggagctgttcaccggggtggtgcccatcctggtcgagctggacggcgacgtaaacggccacaagttcagcgtgtccggcgagggcgagggcgatgccacctacggcaagctgaccctgaagttcatctgcaccaccggcaagctgcccgtgccctggcccaccctcgtgaccaccctgacctacggcgtgcagtgcttcagccgctaccccgaccacatgaagcagcacgacttcttcaagtccgccatgcccgaaggctacgtccaggagcgcaccatcttcttcaaggacgacggcaactacaagacccgcgccgaggtgaagttcgagggcgacaccctggtgaaccgcatcgagctgaagggcatcgacttcaaggaggacggcaacatcctggggcacaagctggagtacaactacaacagccacaacgtctatatcatggccgacaagcagaagaacggcatcaaggtgaacttcaagatccgccacaacatcgaggacggcagcgtgcagctcgccgaccactaccagcagaacacccccatcggcgacggccccgtgctgctgcccgacaaccactacctgagcacccagtccgccctgagcaaagaccccaacgagaagcgcgatcacatggtcctgctggagttcgtgaccgccgccgggatcactctcggcatggacgagctgtacaagtaagactctggtcagagatacctaagaggaagagagagaccctcactgctggggagtccctgccacactcagt AAVS1 locus donor sequence (EF-1α core promoter-EGFP 1110bp) gttctgggtacttttatctgtcccctccaccccacagtggggccactaggtaggtcttgaaaggagtgggtcaattggctccggtgcccgtcagtgggcagagcgcacatcgcccacagtccccgagaagttggggggaggggtcggcaattgatccggtgcctagagaaggtggcgcggggtaaactgggaaagtgatgtcgtgtactggctccgcctttttcccgagggtgggggagaaccgtatataagtgcagtagtcgccgtgaacgttctttttcgcaacgggtttgccgccagaacacaggaagcttgccaccatggtgagcaagggcgaggagctgttcaccggggtggtgcccatcctggtcgagctggacggcgacgtaaacggccacaagttcagcgtgtccggcgagggcgagggcgatgccacctacggcaagctgaccctgaagttcatctgcaccaccggcaagctgcccgtgccctggcccaccctcgtgaccaccctgacctacggcgtgcagtgcttcagccgctaccccgaccacatgaagcagcacgacttcttcaagtccgccatgcccgaaggctacgtccaggagcgcaccatcttcttcaaggacgacggcaactacaagacccgcgccgaggtgaagttcgagggcgacaccctggtgaaccgcatcgagctgaagggcatcgacttcaaggaggacggcaacatcctggggcacaagctggagtacaactacaacagccacaacgtctatatcatggccgacaagcagaagaacggcatcaaggtgaacttcaagatccgccacaacatcgaggacggcagcgtgcagctcgccgaccactaccagcagaacacccccatcggcgacggccccgtgctgctgcccgacaaccactacctgagcacccagtccgccctgagcaaagaccccaacgagaagcgcgatcacatggtcctgctggagttcgtgaccgccgccgggatcactctcggcatggacgagctgtacaagtaagactctggtcagagatacctgacaggattggtgacagaaaagcccccatccttaggcctcctccttccta Lamin A/C locus donor sequence (EF-1α promoter-EGFP 2600bp) ctttggtttttttcttctgtatttgtttttctaagagaagttattttctatgcccggcgagagatcacgtggggcgcggaggcggtgctgctggggcacggccgtccagcctcggcggccatatttttgaggggctgttcatctcgttcacacgctctgtccgccatgtttgtgagtggaagcgccattaccccttcaagcgactgaaggctgcagggcctctggtggcccgcatggggagaccagacccgccaggcccgcctttccgcactcagtccgggcttactttattttgtgagacagggtctcgcctagaggctccggtgcccgtcagtgggcagagcgcacatcgcccacagtccccgagaagttggggggaggggtcggcaattgaaccggtgcctagagaaggtggcgcggggtaaactgggaaagtgatgtcgtgtactggctccgcctttttcccgagggtgggggagaaccgtatataagtgcagtagtcgccgtgaacgttctttttcgcaacgggtttgccgccagaacacaggtaagtgccgtgtgtggttcccgcgggcctggcctctttacgggttatggcccttgcgtgccttgaattacttccacgcccctggctgcagtacgtgattcttgatcccgagcttcgggttggaagtgggtgggagagttcgaggccttgcgcttaaggagccccttcgcctcgtgcttgagttgaggcttggcctgggcgctggggccgccgcgtgcgaatctggtggcaccttcgcgcctgtctcgctgctttcgataagtctctagccatttaaaatttttgatgacctgctgcgacgctttttttctggcaagatagtcttgtaaatgcgggccaagatctgcacactggtatttcggtttttggggccgcgggcggcgacggggcccgtgcgtcccagcgcacatgttcggcgaggcggggcctgcgagcgcggccaccgagaatcggacgggggtagtctcaagctggccggcctgctctggtgcctggcctcgcgccgccgtgtatcgccccgccctgggcggcaaggctggcccggtcggcaccagttgcgtgagcggaaagatggccgcttcccggccctgctgcagggagctcaaaatggaggacgcggcgctcgggagagcgggcgggtgagtcacccacacaaaggaaaagggcctttccgtcctcagccgtcgcttcatgtgactccacggagtaccgggcgccgtccaggcacctcgattagttctcgagcttttggagtacgtcgtctttaggttggggggaggggttttatgcgatggagtttccccatactgagtgggtggagactgaagttaggccagcttggcacttgatgtaattctccttggaatttgccctttttgagtttggatcttggttcattctcaagcctcagacagtggttcaaagtttttttcttccatttaaggtgtcgtgaaaactaccccaagctggcctctgaggccaccatggctgtgagcaagggcgaggagctgttcaccggggtggtgcccatcctggtcgagctggacggcgacgtaaacggccacaagttcagcgtgtccggcgagggcgagggcgatgccacctacggcaagctgaccctgaagttcatctgcaccaccggcaagctgcccgtgccctggcccaccctcgtgaccaccctgacctacggcgtgcagtgcttcagccgctaccccgaccacatgaagcagcacgacttcttcaagtccgccatgcccgaaggctacgtccaggagcgcaccatcttcttcaaggacgacggcaactacaagacccgcgccgaggtgaagttcgagggcgacaccctggtgaaccgcatcgagctgaagggcatcgacttcaaggaggacggcaacatcctggggcacaagctggagtacaactacaacagccacaacgtctatatcatggccgacaagcagaagaacggcatcaaggtgaacttcaagatccgccacaacatcgaggacggcagcgtgcagctcgccgaccactaccagcagaacacccccatcggcgacggccccgtgctgctgcccgacaaccactacctgagcacccagtccgccctgagcaaagaccccaacgagaagcgcgatcacatggtcctgctggagttcgtgaccgccgccgggatcactctcggcatggacgagctgtacaagtaaaagcttggggatcaattctctagagctcgctgatcagcctcgactgtgccttctagttgccagccatctgttgtttgcccctcccccgtgccttccttgaccctggaaggtgccactcccactgtcctttcctaataaaatgaggaaattgcatcgcattgtctgagtaggtgtcattctattctggggggtggggtggggcaggacagcaagggggaggattgggaagacaatagcaggcatgctggggatgcggtgggctctatggcttctgaggcggaaagaaccagctgggcccagtggttttatactgaaggaaaaacacaagcaaaaaaaaaaaaaaagca
DNA donor PCR primer design and synthesis
Principle for design of dsDNA donor primers:
Only two homology arms (HAs) with 50 nt sequence are designed at the 5′ end of the synthesized forward and reverse primers.
Note: Longer HA sequences generally lead to higher HDR efficiency, but also likely increase the PCR difficulties.
Principle of design for odsDNA donor primers:
i. Likewise, two 50 nt HA sequences are designed at the 5′ ends of the synthesized forward and reverse primers, respectively.
ii. Specify the five consecutive phosphorothioate modifications in the synthesized 50 nt primers at designated positions, as needed. “*” represents phosphorothioate (PT) modification. Lower-case letters are HA sequences and upper-case letters depict exogenous KI sequences. We used the first PCR product as a template in order to save the cost of primer synthesis; long primers are not needed. The primer sequences are shown in Table 2.
Table 2. Synthesis of PCR primer sequences for dsDNA and odsDNA donors
Lamin A/C locus EF-1α core promoter-EGFP (1,110 bp) primers or
EF-1α promoter-EGFP-ploy A signal (2,600 bp) primers (5′→3′)
L-50-F ctttggtttttttcttctgtatttgtttttctaagagaagttattttctaTAGGTCTTGAAAGGAGTGGG L-50-R tgctttttttttttttttgcttgtgtttttccttcagtataaaaccactgAGGTATCTCTGACCAGAGTC L-30S-F ctttggtttttttcttctgtatttgtttttc*t*a*a*g*agaagttattttcta L-30S-R tgctttttttttttttttgcttgtgtttttc*c*t*t*c*agtataaaaccactg L-20S-F ctttggtttttttcttctgta*t*t*t*g*tttttctaagagaag L-20S-R tgctttttttttttttttgct*t*g*t*g*tttttccttcagtat L-15S-F ctttggtttttttctt*c*t*g*t*atttgtttttctaag L-15S-R tgcttttttttttttt*t*t*g*c*ttgtgtttttccttc L-10S-F ctttggttttt*t*t*c*t*tctgtatttgttttt L-10S-R tgctttttttt*t*t*t*t*tttgcttgtgttttt L-5S-F ctttgg*t*t*t*t*tttcttctgtatttg L-5S-R tgcttt*t*t*t*t*ttttttttgcttgtg GAPDH locus EF-1α core promoter-EGFP primers G-50-F atggcctccaaggagtaagacccctggaccaccagccccagcaagagcacTAGGTCTTGAAAGGAGTGGG G-50-R actgagtgtggcagggactccccagcagtgagggtctctctcttcctcttAGGTATCTCTGACCAGAGTC G-30S-F atggcctccaaggagtaagacccctggacca*c*c*a*g*ccccagcaagagcac G-30S-R actgagtgtggcagggactccccagcagtga*g*g*g*t*ctctctcttcctctt G-20S-F atggcctccaaggagtaagac*c*c*c*t*ggaccaccagcccca G-20S-R actgagtgtggcagggactcc*c*c*a*g*cagtgagggtctctc G-15S-F atggcctccaaggagt*a*a*g*a*cccctggaccaccag G-15S-R actgagtgtggcaggg*a*c*t*c*cccagcagtgagggt G-10S-F atggcctccaa*g*g*a*g*taagacccctggacc G-10S-R actgagtgtgg*c*a*g*g*gactccccagcagtg G-5S-F atggcc*t*c*c*a*aggagtaagacccct G-5S-R actgag*t*g*t*g*gcagggactccccag AAVS1 locus EF-1α core promoter-EGFP primers A-50-F gttctgggtacttttatctgtcccctccaccccacagtggggccactaggTAGGTCTTGAAAGGAGTGGG A-50-R taggaaggaggaggcctaaggatgggggcttttctgtcaccaatcctgtcAGGTATCTCTGACCAGAGTC A-30S-F gttctgggtacttttatctgtcccctccacc*c*c*a*c*a A-30S-R taggaaggaggaggcctaaggatgggggctt*t*t*c*t*g A-20S-F gttctgggtacttttatctgt*c*c*c*c*t A-20S-R taggaaggaggaggcctaagg*a*t*g*g*g A-15S-F gttctgggtactttta*t*c*t*g*t A-15S-R taggaaggaggaggcc*t*a*a*g*g A-10S-F gttctgggtac*t*t*t*t*atctg A-10S-R taggaaggagg*a*g*g*c*ctaag A-5S-F gttctg*g*g*t*a*cttttatctg A-5S-R taggaa*g*g*a*g*gaggcctaag Lamin A/C loucs EF-1α promoter-EGFP-ploy A signal (2,600 bp) primers for esgRNA L-12S-F ctttggttttttt*c*t*t*c*tgtatttgtttttctaagag L-12S-R attgagatagatgagatagatgctttttttttt*t*t*t*t*t Primers’ synthesis by Sangon Biotech. The dry powder for each primer was first diluted to 100 μM (stock solution) using low-EDTA TE buffer (10 mM Tris and 0.2 mM EDTA, pH 8.0), and then to 10 μM (working solution) using nuclease-free ddH2O.
PCR amplification of the donor DNA:
i. Set up the following PCR assembly reaction in a 400 μL mastermix volume. Reagents and volumes are shown in Table 3. Vortex for 5 s, spin down quickly, and make 50 μL aliquots using 0.2 mL strips of eight tubes.
Table 3. Components of PCR reaction for DNA donor
Reagent Volume Q5 High-Fidelity 2× Master Mix 200 μL 10 μM forward primer 20 μL 10 μM reverse primer 20 μL Template DNA (plasmids or PCR product) 80 ng Nuclease-free water to 400 μL ii. Add the reaction components in the order given. Aliquot 400 μL of the reaction solution in 50 μL portions into 0.2 mL strips of eight tubes.
iii. Perform assembly PCR using the cycling parameters below, as shown in Table 4.
Table 4. PCR reaction for DNA donor preparation
Cycle step Temperature Time Cycles Initial denaturation 98 °C 30 s 1× Denaturation 98 °C 5 s 35×
Annealing 65 °C 15 s Final extension 72 °C 1 min/2 min 20 s 1× Hold 4 °C Hold 1× Note: Set up the extension time for 1 min for 1,110 bp or 2 min 20 s for 2,600 bp.
Purification of PCR DNA donors using GeneJET PCR Purification kit.
Add a 1:1 volume of binding buffer to completed PCR mixture (for every 400 μL of reaction mixture, add 400 μL of binding buffer). Mix thoroughly by pipetting up and down gently.
Transfer up to 800 μL of the solution from step A3a to the GeneJET purification column. Centrifuge at 12,000× g for 60 s. Discard the flowthrough.
Add 700 μL of wash buffer to the GeneJET purification column. Centrifuge at 12,000× g for 60 s. Discard the flowthrough and place the purification column back into the collection tube.
Centrifuge the empty GeneJET purification column for an additional 1 min to completely remove any residual wash buffer.
Transfer the GeneJET purification column to a clean 1.5 mL microcentrifuge tube. Add 25 μL of nuclease-free water to the center of the GeneJET purification column membrane and centrifuge at 12,000× g for 1 min. To increase the yield, the effluent can be added to the GeneJET purification column membrane and the operation repeated.
Measure the DNA concentration using NanoDrop 2000. Wipe NanoDrop 2000 with Kimwipes disposable wipers before and after use. Store the purified dsDNA donor in -20 ℃ refrigerator.
Note: The PT-modified DNA product (odsDNA precursor) requires a subsequent Lambda exonuclease digestion treatment to prepare odsDNA donor template.
Preparation of odsDNA donor:
Set up the following digest reaction in a 300 μL volume a clean 1.5 mL microcentrifuge tube. Reagents and volumes are shown in Table 5.
Table 5. Components to odsDNA donor digestion reaction
Reagent Volume Lambda exonuclease reaction buffer (10×) 30 μL Lambda exonuclease 6 μL Purified odsDNA precursor (3.f) 18 μg Nuclease-free H2O to 300 μL Digest at 37 °C for 60 min in a dry-bath incubator.
Purification of odsDNA donors using GeneJET PCR Purification kit:
Add a 1:1 volume of binding buffer to the PCR mixture (for every 300 μL of reaction mixture, add 300 μL of binding buffer). Mix thoroughly by pipetting up and down gently.
Transfer up to 600 μL of solution from step A5a to the GeneJET purification column. Centrifuge for 60 s. Discard the flowthrough.
Add 700 μL of wash buffer to the GeneJET purification column. Centrifuge for 60 s. Discard the flowthrough and place the purification column back into the collection tube.
Centrifuge the empty GeneJET purification column for an additional 1 min to completely remove any residual wash buffer.
Transfer the GeneJET purification column to a clean 1.5 mL microcentrifuge tube. Add 25 μL of nuclease-free water to the center of the GeneJET purification column membrane and centrifuge for 1 min. To increase the yield, the eluent can be added back to the GeneJET purification column membrane and centrifuge again.
Measure the DNA concentration using NanoDrop 2000. Store the odsDNA donor in -20 ℃ refrigerator.
Expression and purification of Cas9 and Cas9-PCV2 fusion proteins in E. coli cells
Transform the competent E. coli (Rosetta) cells with the expression plasmid: pET28b-3NLS-Cas9-3NLS-His (pET 28b-3NLS-Cas9-PCV2-3NLS-His) and grow on LB plates (kanamycin+) overnight.
The next day, inoculate one colony into 25 mL of fresh LB medium (kanamycin+) from the LB plates (kanamycin+) and incubate in a shaker (200 rpm) at 37 ℃ overnight.
Inoculate 10 mL of overnight culture to a flask containing 1L of fresh Terrific Broth (TB) medium (kanamycin+) and shake at 200 rpm at 37 ℃. Grow the bacteria until the OD600 reaches ~0.6–0.8.
Note: The optimal growth time for TB is different, which requires OD600 to be more than 1.0–1.5 prior to IPTG induction.
Cool on ice or in a cold room for 20 min, then add IPTG to a final 0.2 mM concentration. Continue the shaking at 18 ℃ overnight (18–20 h).
The next day, pellet the culture medium and weigh the pellet. Resuspend the cells with buffer A at ~6 mL/g.
Add 50 μL of β-mercaptoethanol to per 50 mL of bacterial suspension. Sonication: set up at 35% amplitude, 1.5 s ON, 8.5 s OFF, to get the whole ON time for ~10 min, for a total time of ~66.7 min.
Note: The parameters vary depending on the sonicator; the goal is to attain a homogeneous lysate (more whitish in appearance).
Spin the lysate at 16,000× g for 30 min at 4 ℃ and transfer supernatant to a new 50 mL tube.
Filter the supernatant through a 0.45 μm filter.
Equilibrate a 5 mL His-trap column (GE) with buffer A (at least 25 mL).
Note: If using a previously used column, it can be recovered through the washing cycle by NaOH, ddH2O, and HCl, and then keep the column submerged in 5 mL of ddH2O.
Cleaning of His-trap column:
Note: This step is only required for recovery of old columns.
Before use, first wash with 10 mL of 0.1 M NiSO4, 10 mL of ddH2O, and finally with 20 mL of buffer A.
In use: Slowly compress the pipette to ensure that the filter filters out drop by drop (not forming a liquid flow).
After use: ddH2O wash 10 mL
0.1 M EDTA, 1 M NaCl wash 10 mL
ddH2O wash 10 mL
1 M NaOH wash 10 mL
ddH2O wash 10 mL
1 M HCl wash 10 mL
ddH2O wash 10 mL
Note: The column must be kept in water.
Filter to ensure that there are no air bubbles in the column. Store at 4 ℃ .
Next purification step using FPLC protocol.
Note: The buffer needs to be prepared in advance! The filtered column should not be sucked by water and cannot contact other solutions (so ensure that all connections are correct). Instrument settings: place the start buffer in Pump A1, place the elution buffer in Pump B1, select the column connection line of position 2, place the connection point of A1 upstream, and place the connection point of B1 downstream.
Wash step:
i. Open the AKTA Explorer UNICORN software, select Execute... in Manual, select Pumps B in wash, and then click Inlet.
ii. Select position 2 for the middle circle and waste for the filtered interface.
iii. Select Pumps A wash after the automatic stop. The steps are as above.
iv. Finally, clean the whole system flow, select the flow rate of 5 mL/min, pressure OFF, wash for 10 min.
Note: Use all buffer A and gradient options as 0% buffer B.
All tubes after elution need to be replaced with new ones. Starting from No. 1, approximately forty 1.5 mL tubes are placed in order. Lift the outflow tip up, move directly onto the first tube, and position the column in place.
Set up the flow rate and channel in the Manual option.
i. Flow rate: 1.5 mL/min
ii. Pressure: System pressure
iii. Gradient: Target 4% buffer B
iv. Fraction collection-fraction volume: 5 mL
v. Alarms: Alarm system pressure: High clarm-1.00 Mpa
Note: After completing the setting, double-check whether the elution system functions automatically in order.
Once the mAU value stabilizes, change the target of gradient to 10% buffer B and set the time to 15 min, in order to wash possible non-specifically bound protein. This gradient takes ~15 min to go from 4% buffer B to 10% buffer B.
Wait again until it stabilizes and the peaks disappear; then, change the gradient to 20% buffer B and set the time to 15 min.
Note: The safety imidazole concentration for Cas9 protein ranges between 50 and 250 mM. After reaching 250 mM, it indicates substantial absence of impurities if no peak present.
Collect the eluent at peaks approximately within the range of 50–250 mM imidazole.
Run SDS-PAGE for gel verification of the protein molecular weight.
In the peak effluent, take 8 μL of crude protein product from each tube, add 2 μL of SDS-PAGE protein loading buffer (5×), and denature at 95 ℃ for 10 min.
Note: The first flowthrough serves as control. Marker: add 5 μL BeyoColor Prestained Color Protein marker 10–170 kDa. Running conditions: 150 V, 30 min.
Stain in protein staining solution after completion.
Note: Microwave for 15 s or shake for 5 min.
Then, shake for 30 min.
Check the protein sizes: Cas9 is ~163 kDa and Cas9-PCV2 is ~179 kDa. Gels are shown in Figure 2.
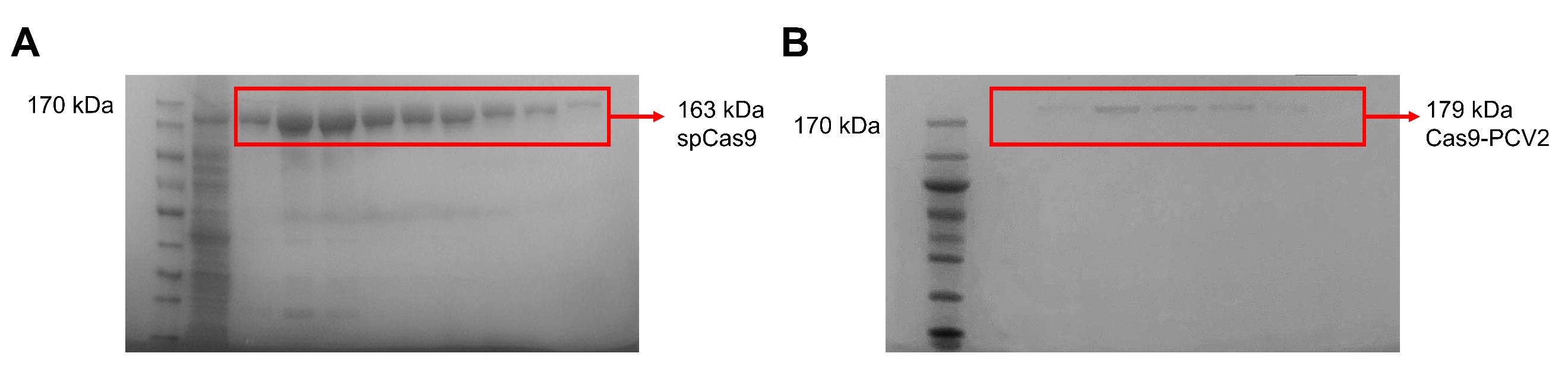
Figure 2. Purification and activity detection in vitro of Cas9 and Cas9-PCV2 fusion proteins. (A) Verification of purified spCas9 protein by SDS-PAGE. (B) Verification of purified Cas9-PCV2 fusion protein by SDS-PAGE.Compare the results to find 5–6 brightest fractions and conduct the protein concentration subsequently as below.
Note: During the running process, the desktop centrifuge is precooled at 4 ℃.
Protein concentration:
Centrifuge filters (pre-cool the 10 K 50 mL and 3 K 15 mL filters on ice).
Add the selected 5–6 fractions of the sample to the pre-cooled 10 K 50 mL filter.
Spin down in the pre-cooled centrifuge for 30 min, observe the upper liquid, and stop the centrifuge any time when the leftover is ~2.5 mL volume.
Using PD-10 column to desalt the concentrated samples.
Approximately 10 tubes of 100 μL of Coomassie Brilliant Blue are required to detect in real time when elute has protein presence and to compare protein concentrations.
Wash the PD-10 column with Cas9 storage buffer three times for 5 min every time.
Then, add 2.5 mL of concentrated solution to PD10, followed by centrifugation. Add 500 μL of storage buffer, collect the elute with a 1.5 mL centrifuge tube, add 500 μL each time for a total of seven times, and collect seven tubes for each 500 μL product.
Take out 3 μL of the product and mix with Coomassie Brilliant Blue, distinguish the blue brightness, select the brightest five tubes of product, transfer to the second PD-10, which has been exchanged with Cas9 buffer, and perform the second desalting.
Measure the brightness, pick up four tubes with the highest blue staining, and proceed to the next protein concentration.
Note: Perform these steps in a cold room; load PD-10 column with less than or equal to 2.5 mL volume.
Concentrate the desalted protein.
Add 2 mL of the desalted product to a 3 K 15 mL pre-cooled centrifuge filter and then centrifuge in a pre-cooled centrifuge at 4,000× g for approximately 30 min. Pay attention to the remaining volume and stop centrifugation when the leftover volume reaches 500 μL.
Prepare 20 μL aliquots in the 1.5 mL tubes in a cold room.
Store at -80 ℃ freezer.
Keep one tube of product for concentration determination.
The absorption peak of protein at 280 nm was measured by NanoDrop 2000.
Calculate final concentration in software Expasy and AAT Bioquest: Protein Concentration Calculator.
Testing enzymatic activity in vitro.
Examine the activity of Cas9 protein, with an in vitro cleavage assay as below.
i. Set up the following cleavage reaction in a 20 μL volume. Reagents and volumes are shown in Table 6; add the reaction components in the order given.
Table 6. Reaction mixture for in vitro cleavage assay
Reagent Volume Cleavage buffer (10×) 2 μL Substrate DNA 100 ng Cas9 (or Cas9-PCV2) 0.3 μg sgRNA 130 ng Nuclease-free water up to 20 μL ii. Incubate at 37 °C for 30 min.
iii. Visualize the cleaved bands by 3% agarose gel electrophoresis with 1× TAE buffer. Gel image is shown in Figure 3.
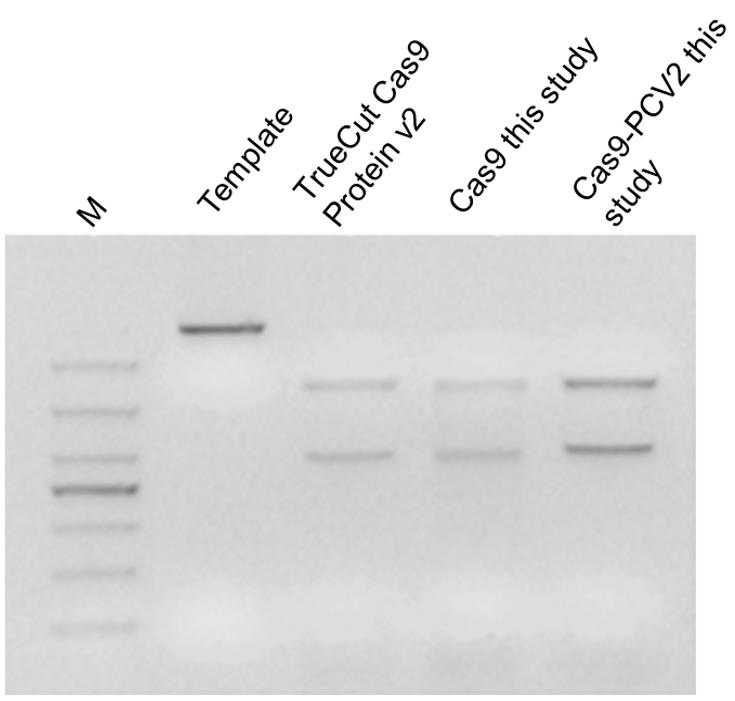
Figure 3. In vitro cleavage assay for examination of the Cas9 and Cas9-PCV2 activityMeasurement of the enzymatic activity for the Cas9-PCV2 fusion protein.
i. The ssDNA probe was synthesized by Sangon Biotech:
PCV2-linker Sequence (5′→3′): BHQ1-TAAGTATTACCAGAAA/i6FAMdT/cctcttgtcccacagat atccagaaccctgaccctgccgtgtaccagct
ii. Dilute the PCV2 linker with ddH2O to a final 0.02, 0.06, and 0.10 μM, respectively.
iii. The ssDNA probe was co-incubated with Cas9-PCV2 at 37 °C, and the fluorophore intensity was detected using 0.02 μM Cas9-PCV2 protein along with varying concentrations of ssDNA probes by fluorescence spectrophotometer F-7000.
Note: When the ssDNA probe is cleaved by the Cas9-PCV2 fusion protein, the FAM fluorophore is released from the quenching group BHQ1 and begins to emit fluorescence, which is monitored in real time by a fluorescence spectrometer.
Synthesis and purification of sgRNA to target gDNA in HEK293T cells (GeneArt Precision gRNA Synthesis kit)
Design sequences as below for the Target F1 forward and Target R1 reverse primers required for synthetic sgRNA template assembly. Sequences are shown in Table 7:
Target F1: TAATACGACTCACTATAG + target sequence (20 nt)
Target R1: TTCTAGCTCTAAAAC + target sequence reverse complement (20 nt)
Note: If the target sequence already contains a 5′ G, you can keep it, which will result in an extra G being added from the T7 promoter primer. Alternatively, you can remove the first G from the target sequence, which will be added back by the T7 promoter primer.
Table 7. Primers for in vitro translation of sgRNA
Locus Primer sequence (5′→3′) Lamin A/C TAATACGACTCACTATAGAGAGAAGTTATTTTCTACAG TTCTAGCTCTAAAACCTGTAGAAAATAACTTCTCT GAPDH TAATACGACTCACTATAGAGCCCCAGCAAGAGCACAAG TTCTAGCTCTAAAACCTTGTGCTCTTGCTGGGGCT AAVS1 TAATACGACTCACTATAGACAGTGGGGCCACTAGGGAC TTCTAGCTCTAAAACGTCCCTAGTGGCCCCACTGT Primers were synthesized by Sangon Biotech and were diluted using nuclease-free water to final 0.3 μM concentration (working solution).
sgRNA transcription in vitro using GeneArt Precision gRNA Synthesis kit:
Set up the following PCR assembly reaction in a 25 μL volume and prepare the reaction components as below; reagents and volumes are shown in Table 8.
Table 8. Components to make gRNA DNA template
Reagent Volume Phusion High-Fidelity PCR Master Mix (2×) 12.5 μL Tracr Fragment + T7 Primer Mix 1 μL 0.3 μM Target F1/R1 primer mix 1 μL Nuclease-free water 10.5 μL Perform assembly PCR using the cycling parameters below, as shown in Table 9.
Table 9. PCR parameters to generate gRNA DNA template
Cycle step Temperature Time Cycles Initial denaturation 98 °C 10 s 1× Denaturation 98 °C 5 s 32×
Annealing 55 °C 15 s Final extension 72 °C 1 min 1× Hold 4 °C Hold 1× In vitro transcription (IVT) of gRNAs; reagents and volumes are shown in Table 10.
Table 10. Components of IVT reaction
Reagent Volume NTP mix (100 mM each of ATP, GTP, CTP, UTP) 8 μL gRNA DNA template (from PCR assembly) 6 μL 5× TranscriptAidTM Reaction Buffer 4 μL TranscriptAidTM Enzyme Mix 2 μL Mix the tubes thoroughly and spin down quickly. Set up the IVT reaction for 3 h at 37 °C.
Note: All operations are preferably conducted in RNase-free workstation.
Removal of the DNA template by DNase I digestion:
Incubate the IVT reaction mix with 1 μL of DNase I (1 U/μL) immediately following the IVT reaction and incubate at 37 °C for 15 min.
Purification of sgRNA using GeneArt gRNA Clean-up kit.
Dilute the IVT reaction to 200 μL with nuclease-free water and add 100 μL of binding buffer. Mix by pipetting.
Add 300 μL of ethanol (> 96%) and mix by pipetting.
Transfer the mixture to the GeneJET RNA Purification Micro Column (preassembled with a collection tube) and centrifuge for 30–60 s at 14,000× g. Discard the flowthrough.
Wash the bound RNA with 700 μL of wash buffer 1 and wash buffer 2.
Centrifuge the empty purification column for an additional 60 s at 14,000× g to completely remove any residual wash buffer.
Transfer the purification column to a new 1.5 mL RNase-free EP tube.
Add 10 μL of nuclease-free water to the center of the column filter and centrifuge at 14,000× g for 60 s to elute the sgRNA.
Measure the concentration of sgRNA using NanoDrop 2000.
Store the sgRNA at -80 °C.
Note: All operations are preferably conducted in RNase-free workstation.
Preparation of the RNP complex for odsDNA attaching to Cas9-PCV2 fusion protein
Design and synthesis of the PCV2 linker.
Because PCV2 protein recognizes DNA sequence (AAGTATT^ACCAGAAA) in 5′ to 3′ direction, odsDNA has 3′ overhang, and a PCV2 linker is needed as a bridge to attach odsDNA to Cas9-PCV2 fusion protein.
Note: “^” represents the cleavage position of the PCV2 protein.
The PCV2 linker is divided into two parts: the 5′ end is a fixed sequence recognized by the PCV2 protein for cleavage, followed by subsequent covalent linkage to PCV2 protein, and the 3′ end is a sequence that anneals to the 3′ overhang of odsDNA.
The PCV2 linker sequence for the Lamin A/C locus is shown in Table 11.
Table 11. PCV2 linker sequence for the Lamin A/C locus
Lamin A/C locus Cas9-PCV2 linker sequence(5′→3′) PCV2 linker AAGTATTACCAGAAAtgcttttttt Note: The upper-case letters are the set sequences recognized by the PCV2 protein and the lower-case letters are the sequences that anneal to the odsDNA of the Lamin A/C locus.
Synthesis of PCV2 linker was by Sangon Biotech and diluted to 10 μM with nuclease-free water.
PCV2 linker annealing to odsDNA.
We use odsDNA with 10 bases-overhang to anneal to PCV2 linker with 10 nt paired bases.
Set up the following annealing reaction in a 10 μL volume; reagents and volumes are shown in Table 12.
Table 12. Components of annealing reaction
Reagent Molar amount Volume PCV2 linker 7.5 pmol 0.75 μL odsDNA (1 μg/μL) 7.5 pmol 5 μL 5× annealing buffer - 2 μL Nuclease-free water - to 10 μL Perform annealing reaction using the cycling parameters as shown in Table 13.
Table 13. Annealing reaction to generate odsDNA/PCV2 linker complex
Cycle step Temperature Time Cycles Denaturation 65 °C 5 min 1× Annealing 65 °C (-0.1 ℃/8 s) to 25 °C ~60 min 1× Hold 4 °C Hold 1×
Prepare RNP complex composed of odsDNA/PCV2 linker/Cas9-PCV2 fusion protein.
In order to verify that odsDNA/PCV2 linker can be effectively attached to the Cas9-PCV2 protein, different molar ratios of 0:1, 1:1, 2:1, 3:1, and 4:1 were incubated with Cas9-PCV2 protein and then verified by 2% agarose gel. As shown in Figure 4, when the molar ratio of Cas9-PCV2 fusion protein to odsDNA/PCV2 linker was 3:1, the free odsDNA/PCV2 linker below was almost completely reacted, and all odsDNA/PCV2 linker was attached to the Cas9-PCV2 fusion protein.
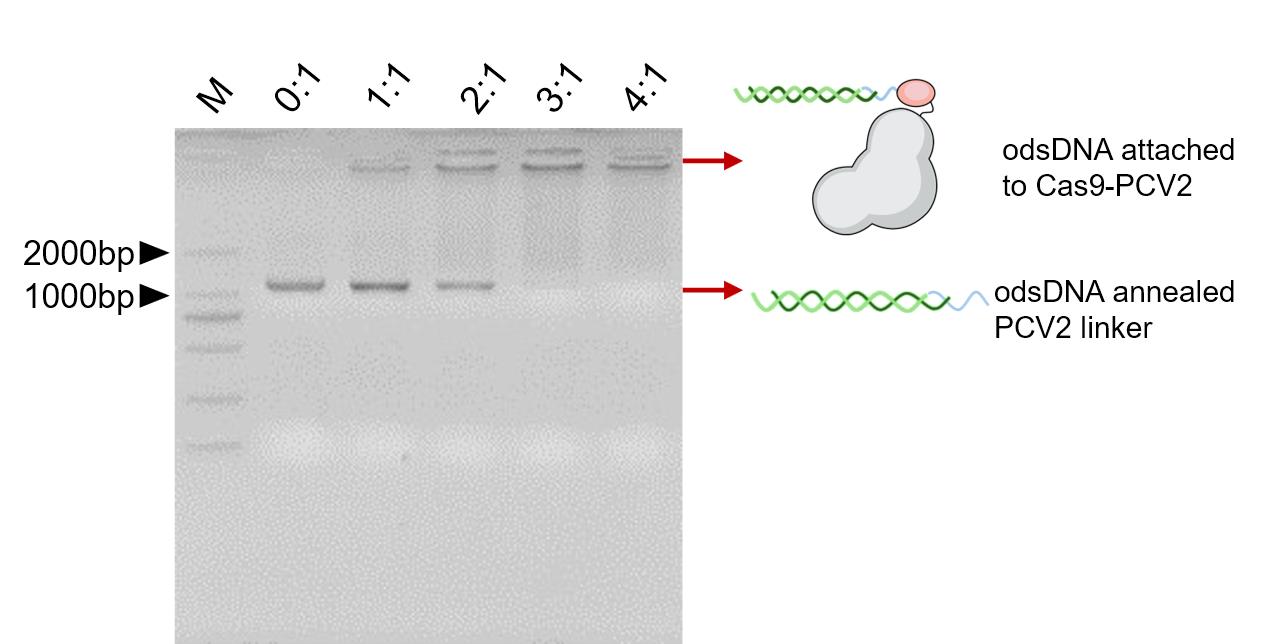
Figure 4. Verification for the attachment of odsDNA to Cas9-PCV2 protein.Efficient tethering of odsDNA donors to Cas9-PCV2 when the ratio of Cas9-PCV2 to pre-annealed odsDNA/PCV2 linker reaches > 3:1.
Add 30 pmol Cas9-PCV2 fusion protein to the odsDNA/PCV2 linker complex from step D2.
Because Cas9-PCV2 fusion protein is much more abundant than odsDNA, almost all odsDNA is reacted completely attached to Cas9-PCV2 fusion protein.
The working condition of PCV2 protein requires the presence of at least 1 mM Mg2+. Since the buffer C: storage buffer of Cas9-PCV2 protein has 10 mM MgCl2, no additional Mg2+ is needed here.
Nucleofection of the odsDNA/Cas9-PCV2/sgRNA RNP complex into HEK293T cells
Before nucleofection, sub-culture the HEK293T cells three days prior to nucleofection experiments using Dulbecco’s modified Eagle’s medium and 10% fetal bovine serum.
Nucleofection of HEK293T cells with odsDNA/Cas9-PCV2/sgRNA:
Cell preparation
i. Optimal cell confluency for nucleofection: 80%–90%. Higher cell densities may cause lower Nucleofection® efficiencies.
ii. Remove the medium from the cultured cells and wash cells once with 1× PBS. Wash with at least the same volume of 1×PBS as culture media.
iii. For harvesting, incubate the cells for ~5 min at 37 °C with TrypLE Express reagent.
iv. Neutralize the trypsinization reaction with culture medium containing 10% FBS. The majority of the cells (>90%) will be detached. The culture medium, PBS, and TrypLE are opened and sealed using PARAFILM sealing film.
Preparation for odsDNA/Cas9-PCV2/sgRNA RNP complex:
i. To prepare the Cas9-PCV2 RNPs complex, mix the odsDNA/PCV2 linker/Cas9-PCV2 fusion protein complex with 90 pmol sgRNA and incubate for 10 min at 37 °C.
ii. Perform nucleofection immediately as below.
Nucleofection
i. Ensure that the entire supplement is added to the Nucleofector® solution. The ratio of Nucleofector® solution to supplement is 4.5:1. For a single reaction, use 82 μL of Nucleofector® solution plus 18 μL of supplement to make 100 μL of total reaction volume.
ii. Prepare 6-well plates by filling the appropriate number of wells with 1 mL of supplemented culture media and pre-incubate/equilibrate plates in a humidified 5% CO2 incubator at 37 °C.
iii. Harvest the cells by trypsinization.
iv. Count the cell numbers with a hemacytometer or an automated cell counter as preferred.
v. Centrifuge the required number of cells (1 × 106 cells per sample) at 200× g for 10 min at room temperature. Remove supernatant completely.
vi. Resuspend the cell pellet carefully in 100 μL of room-temperature Nucleofector® solution per sample.
Note: Do not leave the cells in Nucleofector® solution for an extended time (i.e., longer than 15 min), as this may reduce both the cell viability and the gene transfer efficiency.
i. Combine 100 μL of cell suspension with odsDNA/Cas9-PCV2/sgRNA complex. Table 14 shows the different samples.
Table 14. One Nucleofection® sample contains
odsDNA sample odsDNA sample dsDNA sample 1 × 106 HEK293T cells 1 × 106 HEK293T cells 1 × 106 HEK293T cells odsDNA/Cas9-PCV2/sgRNA complex odsDNA, Cas9/sgRNA complex dsDNA, Cas9/sgRNA complex 100 μL Cell Line Nucleofector® Solution V 100 μL Cell Line Nucleofector® Solution V 100 μL Cell Line Nucleofector® Solution V ii. Transfer cell/DNA suspension into a certified cuvette (sample must cover the bottom of the cuvette without air bubbles). Close the cuvette with the cap.
iii. Select the appropriate Nucleofector® Program D-032 for Nucleofector® 2b Device.
iv. Insert the cuvette with cell/DNA suspension into the Nucleofector® Cuvette Holder and apply the selected program by pressing the X-button.
v. Take the cuvette out of the holder once the program is finished.
vi. Immediately replenish with ~500 μL of the pre-equilibrated culture medium to the cuvette and gently transfer the mixture into the prepared 6-well plate (final volume 1.5 mL media per well). Use the supplied pipettes and avoid repeated aspiration of the sample.
Post nucleofection:
Incubate the cells in a humidified 37 °C/5% CO2 incubator until further analysis at defined time points. Nucleofection of odsDNA with Cas9/sgRNA into HEK293T cells showed that universal odsDNA donors had higher KI rates than dsDNA donors in Figure 5. When Cas9-PCV2 was used to attach to odsDNA, the KI rate was 5-fold higher than dsDNA in Figure 6.
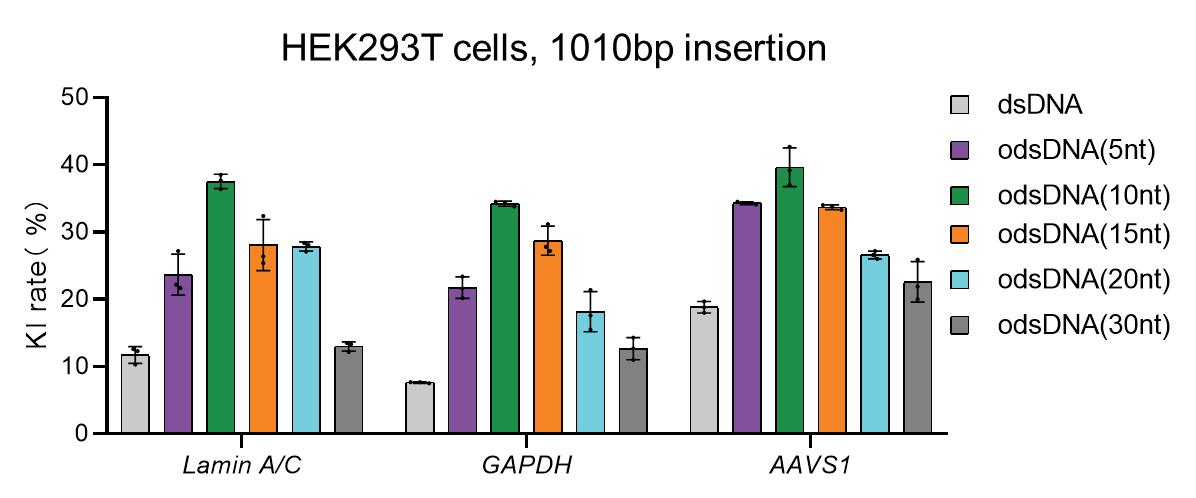
Figure 5. Knock-in (KI) rates in HEK293T cells with different donors. A panel of three different genomic loci were selected for the 1,010 bp odsDNA donor KI in HEK293T cells. The dsDNA donors were used as controls. Data were collected 15 days after nucleofection. The KI rates were calculated as the averaged percentages for the total number of EGFP-positive cells (donor KI) divided by the total number of RNP-electroporated cells.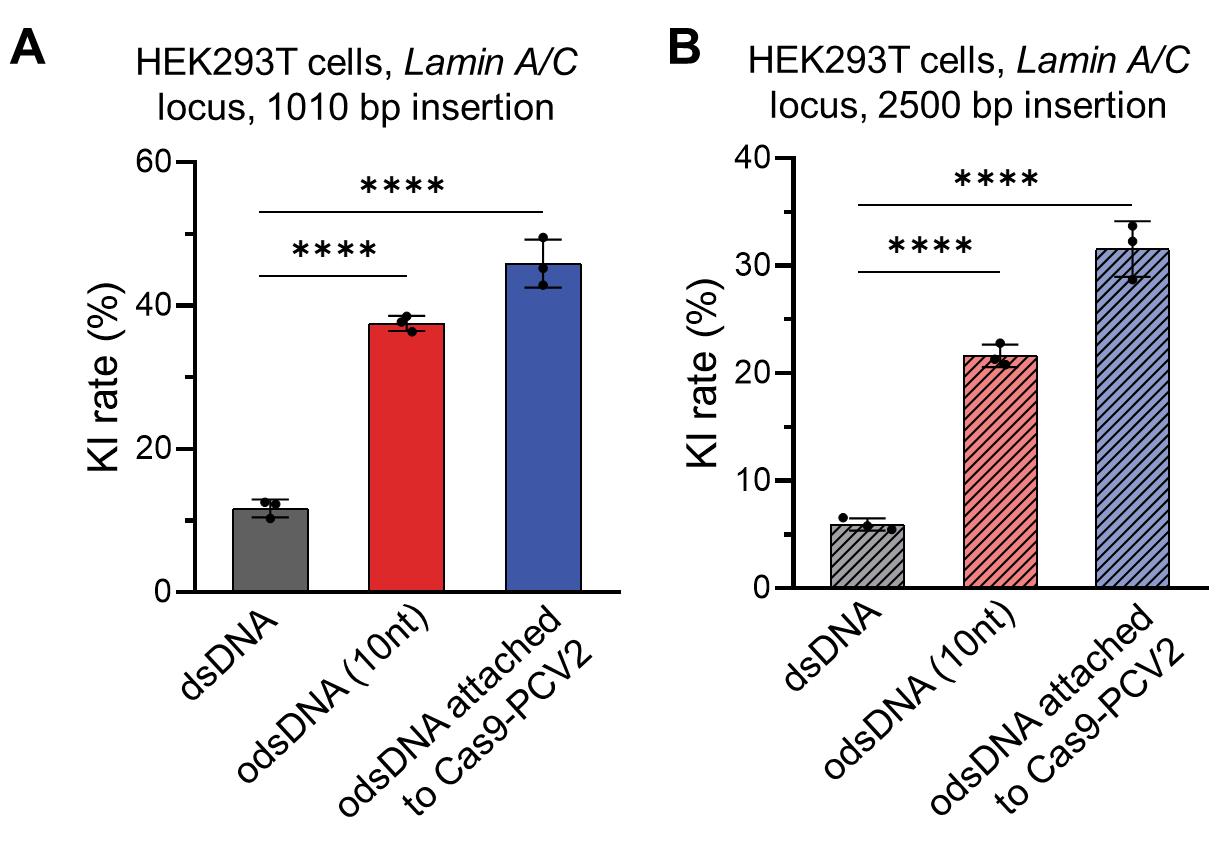
Figure 6. Different lengths of DNA fragment knock-in (KI) using the odsDNA attach to Cas9-PCV2 protein strategy. (A) The 1,010 bp donor templates, including Cas9-bound dsDNA and 10 nt odsDNA as well as Cas9-PCV2-bound 10 nt odsDNA, were electroporated to target the Lamin A/C locus in HEK293T cells. The KI rates were plotted for side-by-side comparison. Data were collected 15 days after nucleofection. (B) The 2,500 bp donor templates, including Cas9-bound dsDNA and 10-nt odsDNA as well as Cas9-PCV2-bound 10-nt odsDNA, were electroporated to target the Lamin A/C locus in HEK293T cells. The KI rates were plotted for side-by-side comparison. Data were collected 21 days after nucleofection.
Data analysis
The KI rates of different donors across varying loci in HEK293T cells were measured by BD Accuri C6 flow cytometer and plotted in Prism 8 software.
The KI rates of odsDNA attached to Cas9-PCV2 fusion protein at Lamin A/C locus in HEK293T cells was measured by BD Accuri C6 flow cytometer and plotted in Prism 8 software.
Validation of protocol
This protocol or parts of it has been used and validated in the following research article(s):
Han et al. (2023). Efficient precise integration of large DNA sequences with 3′-overhang dsDNA donors using CRISPR/Cas9. Proc. Natl. Acad. Sci. U.S.A. (Figure 4, panel B, C and D).
Tei et al. (2023). Comparable analysis of multiple DNA double-strand break repair pathways in CRISPR-mediated endogenous tagging. bioRxiv. (Figure 3, panel C and D).
Acknowledgments
This work was supported by the Ministry of Science and Technology of China (2019YFA0802600 and 2020YFA0710700), the National Natural Science Foundation of China (Nos. 21991132, 52033010, 52021002, 31970793, and 32170856). The current protocol is mainly derived from Han et al. (2023).
Competing interests
The authors declare no competing financial interests.
References
- Cruz-Becerra, G. and Kadonaga, J. T. (2020). Enhancement of homology-directed repair with chromatin donor templates in cells. eLife 9: e55780.
- Doudna, J. A. (2020). The promise and challenge of therapeutic genome editing. Nature 578(7794): 229–236.
- Gu, B., Posfai, E. and Rossant, J. (2018). Efficient generation of targeted large insertions by microinjection into two-cell-stage mouse embryos. Nat. Biotechnol. 36(7): 632–637.
- Han, W., Li, Z., Guo, Y., He, K., Li, W., Xu, C., Ge, L., He, M., Yin, X., Zhou, J., et al. (2023). Efficient precise integration of large DNA sequences with 3′-overhang dsDNA donors using CRISPR/Cas9. Proc. Natl. Acad. Sci. U. S. A. 120(22): e2221127120.
- Hsu, P. D., Lander, E. S. and Zhang, F. (2014). Development and Applications of CRISPR-Cas9 for Genome Engineering. Cell 157(6): 1262–1278.
- Maruyama, T., Dougan, S. K., Truttmann, M. C., Bilate, A. M., Ingram, J. R. and Ploegh, H. L. (2015). Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat. Biotechnol. 33(5): 538–542.
- Quadros, R. M., Miura, H., Harms, D. W., Akatsuka, H., Sato, T., Aida, T., Redder, R., Richardson, G. P., Inagaki, Y., Sakai, D., et al. (2017). Easi-CRISPR: a robust method for one-step generation of mice carrying conditional and insertion alleles using long ssDNA donors and CRISPR ribonucleoproteins. Genome Biol. 18(1): e1186/s13059-017-1220-4.
- Shy, B. R., Vykunta, V. S., Ha, A., Talbot, A., Roth, T. L., Nguyen, D. N., Pfeifer, W. G., Chen, Y. Y., Blaeschke, F., Shifrut, E., et al. (2023). High-yield genome engineering in primary cells using a hybrid ssDNA repair template and small-molecule cocktails. Nat. Biotechnol. 41(4): 521–531.
- Yu, Y., Guo, Y., Tian, Q., Lan, Y., Yeh, H., Zhang, M., Tasan, I., Jain, S. and Zhao, H. (2020). An efficient gene knock-in strategy using 5′-modified double-stranded DNA donors with short homology arms. Nat. Chem. Biol. 16(4): 387–390.
Article Information
Copyright
© 2023 The Author(s); This is an open access article under the CC BY license (https://creativecommons.org/licenses/by/4.0/).
How to cite
Han, W., Liang, H. and Bao, J. (2023). Efficient Large DNA Fragment Knock-in by Long dsDNA with 3′-Overhangs Mediated CRISPR Knock-in (LOCK) in Mammalian Cells. Bio-protocol 13(20): e4853. DOI: 10.21769/BioProtoc.4853.
Category
Molecular Biology > DNA > DNA modification
Cell Biology > Cell engineering > CRISPR-cas9
Cancer Biology > General technique > Molecular biology technique
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.