- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Isolation and Culture of Neural Stem/Progenitor Cells from the Hippocampal Dentate Gyrus of Young Adult and Aged Rats
Published: Vol 13, Iss 19, Oct 5, 2023 DOI: 10.21769/BioProtoc.4843 Views: 2510
Reviewed by: Vivien J. Coulson-ThomasMunenori IshibashiAnonymous reviewer(s)
Advertisement




Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Adult neural stem/progenitor cells (NSPCs) in two neurogenic areas of the brain, the dentate gyrus and the subventricular zone, are major players in adult neurogenesis. Addressing specific questions regarding NSPCs outside of their niche entails in vitro studies through isolation and culture of these cells. As there is heterogeneity in their morphology, proliferation, and differentiation capacity between these two neurogenic areas, NSPCs should be isolated from each area through specific procedures and media. Identifying region-specific NPSCs provides an accurate pathway for assessing the effects of extrinsic factors and drugs on these cells and investigating the mechanisms of neurogenesis in both healthy and pathologic conditions. A great number of isolation and expansion techniques for NSPCs have been reported. The growth and expansion of NSPCs obtained from the dentate gyrus of aged rats are generally difficult. There are relatively limited data and protocols about NSPCs isolation and their culture from aged rats. Our approach is an efficient and reliable strategy to isolate and expand NSPCs obtained from young adult and aged rats. NSPCs isolated by this method maintain their self-renewal and multipotency.
Key features
• NSPCs isolated from the hippocampal dentate gyrus of young adult and aged rats, based on Kempermann et al. (2014) and Aligholi et al. (2014).
• Maintenance of NSPCs isolated from the dentate gyrus of aged rats (20–24 months) in our culture condition is feasible.
• According to our protocol, maximum growth of primary neurospheres obtained from isolated NSPCs of young and aged rats took 15 and 35 days, respectively.
Graphical overview

Isolation and expansion of neural stem/progenitor cells
Background
Neural stem/progenitor cells (NSPCs) in neurogenic areas of the adult mammalian brain, i.e., the subventricular zone (SVZ) of the lateral ventricle and the subgranular zone of the hippocampal dentate gyrus (DG), divide symmetrically and asymmetrically to preserve their lineage (Walker and Kempermann, 2014; Gage, 2019). In a symmetric division, the number of progenitor cells increases, as two identical daughter cells originate from a progenitor cell or stem cell. The asymmetric division of progenitor cells produces two distinct types of cells. For example, one radial-glial cell gives rise to one radial-glial cell and one neuron (Zhao and Moore, 2018). Neurogenesis in the dentate gyrus contributes to learning, memory, and mood regulation, which decline during aging. Isolation of NSPCs from rat dentate gyrus provides valuable adult-cell-based models for identifying cellular and molecular mechanisms of neurogenesis in the hippocampus and to assess the effects of extrinsic factors and drugs on their functions outside their niches. There are different intrinsic regulatory mechanisms for NSPCs in the DG and SVZ (Takei, 2019). With increasing age, a repertoire of intrinsic and extrinsic factors drives most of the NSPCs to enter the senescent stage, limiting their proliferative capacity, and leading to subsequent dysfunctions (Obernier and Alvarez-Buylla, 2019). Slow proliferation and declining differentiation capacity are features of NSPCs in aging, which make finding strategies for improvement of neurogenesis during aging important (Kase et al., 2020).
The study of NSPCs function and their responses to aging, regeneration purposes, and molecular and cellular mechanisms of neurogenesis in various cell-culture-based models of different neurological disorders, including neurodegenerative and neurovascular diseases, requires isolation and culture of NSPCs from adult neurogenic niches (Akers et al., 2014; Christian et al., 2014; Bond et al., 2015). Among various approaches, neurosphere culture provides a 3D environment-like niche of neural stem cells, increases cell division, and offers conditions permissible for more passage numbers (Azari et al., 2010; Guo et al., 2012; Aligholi et al., 2014). However, recognition of cell morphology is difficult in the neurosphere culture system. Cells in the center of neurospheres may differentiate slowly with low survival and yield (Sun et al., 2011). In monolayer cell culture, cells have easy access to nutrients and their morphology could be easily detected. However, cells in this method might differentiate spontaneously, and self-renewal ability declined after more passages (Reynolds and Rietze, 2005; Ray and Gage, 2006).
As the proliferation and differentiation potential of the hippocampal DG differs from the SVZ, developing a complete protocol to obtain high-yield NSPCs from rat DG is needed. There is controversy regarding hippocampus-derived stem cells from adult rats and whether they have unlimited self-renewal or limited proliferative and differentiating potential (Gobbel et al., 2003; Chen et al., 2007). In this method, the expansion of isolated cells from aged rats has been introduced and discussed.
Using this isolation and culture method, we produced a robust and reproducible number of DG neurospheres from young adult and aged rats after 14–18 days and 25–30 days, respectively. In our method, 1.5 × 105 neural progenitor cells yield 120 spheres of 200–250 µm for young adult rats, and 7 × 104 neural progenitor cells yield 90 spheres of 100 µm for aged rats on average. An appropriate approach providing optimal regional-specific NSPCs could improve utilizing cells in a clinical setting for the treatment of neurological disorders as well as boost experimental aging research.
Materials and reagents
Animals
Animal housing was performed in accordance with the Guidelines of the Shefa Neuroscience Research Center, Tehran, Iran. Young adult male Wistar rats between 2 and 3 months of age (180–230 g) and aged rats between 20 and 24 months (400–450 g) were used.
General materials
Ketamine (alfasan, catalog number: 36408/3000)
Xylazine (alfasan, catalog number: 36408/3007)
Glass Petri dish (Nest Biotechnology, catalog number: 704001)
Phosphate buffer saline (PBS) (tablet) (Thermo Fisher Scientific, GibcoTM, catalog number: 18912014)
0.2, 0.5, and 1.5 mL Eppendorf tubes (Nest Biotechnology, catalog number: 613111)
6, 12, 24, 48, and 96 adherent well plates (treated) (JetBiofil, catalog number: TCP011096, TCP011012, TCP011048)
Non-adherent well plates: 6 wells (Falcon, catalog number: 353046) and 12 wells (Falcon, catalog number: 351143)
Adherent T-25 flask (JetBiofil, catalog number: TCF012050)
Non-adherent T-25 flask (JetBiofil, catalog number: TCF012600)
0.2 μm filter (PES membrane) (BioFil, catalog number: FMC 2011018)
Parafilm (Sigma, catalog number: P7793)
96% ethanol (Merck, catalog number: 100971)
70 μm cell strainer (BD Falcon, catalog number: 352340)
Petri dish culture (JetBiofil, Germany, MCD000035)
15 and 50 mL Falcon tubes (JetBiofil, catalog number: CFT011500)
Pipette tips blue 1,000 μL (Universal, catalog number: 2451-1K0), yellow 100 μL (Universal, catalog number: 200-B-YL), and 10 μL (Labcon Eclipse, catalog number: 490007-952)
Reagents
0.05% Trypsin/EDTA (Biowest, catalog number: L0931-100)
Digestive enzymes and materials:
Papain (Roche, catalog number: 108014)
Dispase (Gibco, catalog number: 17105-041)
DNase (Roche, catalog number: 11284932001)
Accutase (Sigma, catalog number: A6964)
L-Cysteine (Sigma, catalog number: 52904)
EDTA (Sigma, catalog number: 17892)
Hanks’ balanced salt solution (HBSS) (Thermo Fisher Scientific, GibcoTM, catalog number: 24020117)
Fetal bovine serum (FBS) (Thermo Fisher Scientific, GibcoTM, catalog number: 10270106)
Trypan blue solution 0.4% (Sigma-Aldrich, catalog number: T8154)
4% Paraformaldehyde (powder) (Sigma-Aldrich, catalog number: P6148)
Triton X-100 (Sigma-Aldrich, catalog number: T8787)
Bovine serum albumin (BSA) (powder) (Sigma-Aldrich, catalog number: A2058)
Normal goat serum (Thermo Fisher Scientific, catalog number: 50197Z)
Mouse anti-Nestin (Merck Millipore, catalog number: MAB 353)
Rabbit anti-Sox2 (Santa Cruz, catalog number: SC20088)
Mouse anti-GFAP (Sigma, catalog number: G3893)
Goat anti-rabbit IgG (Abcam, catalog number: ab9717)
Goat anti-mouse (Abcam, catalog number: ab6785)
4’,6-Diamidino-2-phenylindole (DAPI) (Thermo Fisher Scientific, catalog number: 62248)
Laminin (Sigma, catalog number: L2020)
Poly-L-ornithine solution (Sigma, catalog number: P4957)
Culture medium (see Recipes)
Dulbecco’s modified Eagle’s medium (DMEM)/F12 (Gibco, catalog number: 11320033)
N2 supplement (Gibco, catalog number: 17501), used for serum-free culture condition of neuroblastoma and primary cells of the central nervous systems. It is composed of vitamins, hormones, insulin, and transferrin. These components maintain neural stem cells in an undifferentiated state and support the proliferation of cells in a serum-free culture
B27 supplement (Gibco, catalog number: 12513-010), a supplement as a cocktail of antioxidants, vitamins, and proteins to reduce reactive oxygen damage and for the long-term survival of neural stem cells. It is a widely used supplement for serum-free culture conditions. Therefore, this supplement contains defined components necessary for maintaining the health of the culture and reducing variability
Heparin (Sigma, catalog number: H3149)
Penicillin/Streptomycin (Pen/Strep) (Sigma, catalog number: P4333)
Amphotericin B (Biowest, catalog number: L0009-050)
Glutamax (Gibco, catalog number: 35050-061)
Basic fibroblast growth factor (bFGF) (Sigma, catalog number: F0291)
Epidermal growth factor (EGF) (Sigma, catalog number: E4127)
Solutions
70% ethanol (see Recipes)
Digestive solution (sterile, 0.2 μm filtered) (see Recipes)
Blocking solution (see Recipes)
Antibody solution (see Recipes)
Growth medium (culture medium) (see Recipes)
Coating solution (sterile, 0.2 μm filtered) (see Recipes)
Dissection solution (see Recipes)
Ketamine (80 mg/mL) and xylazine (10 mg/mL) working solutions (see Recipes)
PBS (1×) (see Recipes)
Recipes
70% ethanol
Dilute 365 mL of 96% ethanol with 135 mL of distilled water to make up a volume of 500 mL of 70% ethanol.
Digestive solution (sterile, 0.2 μm filtered)
Dissolve 40 μL of Dispase (2 mg/mL), 80 μL of DNase (0.2 mg/mL), 100 μL of Papain (0.5 mg/mL), 1 mg of L-cysteine, and 100 μL of EDTA in 2 mL of DMEM/F12. This volume of solution is appropriate for one rat.
Preparation of papain (0.5 mg/mL): add 100 μL of papain stock solution (10 mg/mL) to 1.95 mL of DMEM/F12 basic medium to prepare 2 mL of 0.5 mg/mL papain working solution.
Preparation of Dispase (2 mg/mL): add 100 μL of Dispase stock solution (10 mg/mL) to 1.9 mL of DMEM/F12 basic medium to prepare 2 mL of 2 mg/mL Dispase working solution.
Preparation of DNase (0.2 mg/mL): add 80 μL of DNase stock solution (5 mg/mL) to 1.96 mL of PBS (1×) to prepare 2 mL of 0.2 mg/mL DNase working solution.
Add 1 mg of L-cysteine and 100 μL of EDTA to the solution.
Blocking solution
Dissolve 10% normal goat serum, 1% BSA, and 0.01% Tween in PBS (1×). To prepare 500 µL of blocking solution, add 50 µL of normal goat serum, 5 µL of BSA, and 0.5 µL of Tween to 444.5 µL of PBS (1×).
Antibody solution
Dilute primary antibodies in blocking solution:
To prepare 350 μL of 1:200 Anti-Nestin solution, add 1.75 μL of Nestin antibody to 348 μL of blocking solution. Prepare before use.
To prepare 350 μL of 1:100 Sox2 solution, add 3.5 μL of Sox2 antibody to 346.5 μL of blocking solution. Prepare before use.
To prepare 250 μL of 1:250 GFAP solution, add 1 μL of GFAP antibody to 249 μL of blocking solution. Prepare the solution before use.
Antibody Stock concentration Dilution range Final concentration (dilution used for staining) Sox2 200 μg/0.1 mL 1:50–1:500 1:100 Nestin 1.25 μg/mL 1:20–1:200 1:200 GFAP 2.5–5 μg/mL Not available for immunocytochemistry 1:250
Growth medium (culture medium)
DMEM/F12
2% B27 supplement
1% N2 supplement
20 ng/mL bFGF
20 ng/mL EGF
2 μg/mL heparin
1% Penicillin/streptomycin
1% Glutamax
Coating solution (sterile, 0.2 μm filtered)
PLO working solution (20 μg/mL):
Dissolve 2 mL of PLO stock solution (0.1 mg/mL) in 8 mL of PBS (1×) to prepare 10 mL of PLO with the final concentration of 20 μg/mL (store at 4 °C).
Laminin working solution (5 μg/mL)
Dissolve 250 μL of stock solution (1 mg/mL) in 49.75 mL of PBS (1×) to get 50 mL of laminin solution with a final concentration of 5 μg/mL.
Dissection solution
To prepare PBS or HBSS or DMEM/F12 containing 3% Pen/Strep and 1% Amphotericin B: add 0.3 mL of Pen/Strep and 0.1 mL of Amphotericin B to 9.7 mL of PBS (1×).
Ketamine (80 mg/mL) and xylazine (10 mg/mL) working solutions
To prepare 3 mL (concentration of 80 mg/mL) of ketamine working solution:
Add 2.4 mL of ketamine stock solution (100 mg/mL) to 0.6 mL of PBS (1×).
To prepare 3 mL (concentration of 10 mg/mL) of xylazine working solution:
Add 1.5 mL of xylazine stock solution (20 mg/mL) to 1.5 mL of PBS (1×).
PBS (1×)
One tablet of PBS in 500 mL of distilled water used to produce PBS (1×) solution. Then the solution is autoclaved.
Equipment
Laminar hood (Jal Tajhiz, catalog number: 1229)
37 °C incubator shaker (IKA, model: KS 4000i Control)
Centrifuge machine (Hettich Lab Technology, model: Universal 320R)
Hemocytometer (Sigma-Aldrich, catalog number: Z359629)
CO2 cell culture incubator (Memmert, model: INC108 T2T3)
Inverted fluorescence microscope (Optika, model: XDS-2FL)
Small forceps surgical tools (Fine Science Tools, catalog number: 11050-10)
Stereomicroscope (Olympus, catalog number: 7L19549)
Dissection tools
Large scissors (Fishersci, catalog number: 10633652)
Small scissors (Fishersci, catalog number: 15207266)
Forceps (Fishersci, catalog number: 10098140)
Curved forceps (Fishersci, catalog number:10213941)
Angled forceps (Fishersci, catalog number: 22-079-762)
Spatula (Fishersci, catalog number: 11750229)
Autoclave (Kavoosh, catalog number: 2589224)
Oven (Memert, catalog number: 1-100-800)
Rodent guillotine (DCAP, Germany)
Rongerus (Fine Science Tools, catalog number: 16004-16)
Software
Infinity software (version 4.6)
Procedure
A schematic diagram depicting the whole experimental procedure is shown in Figure 1.
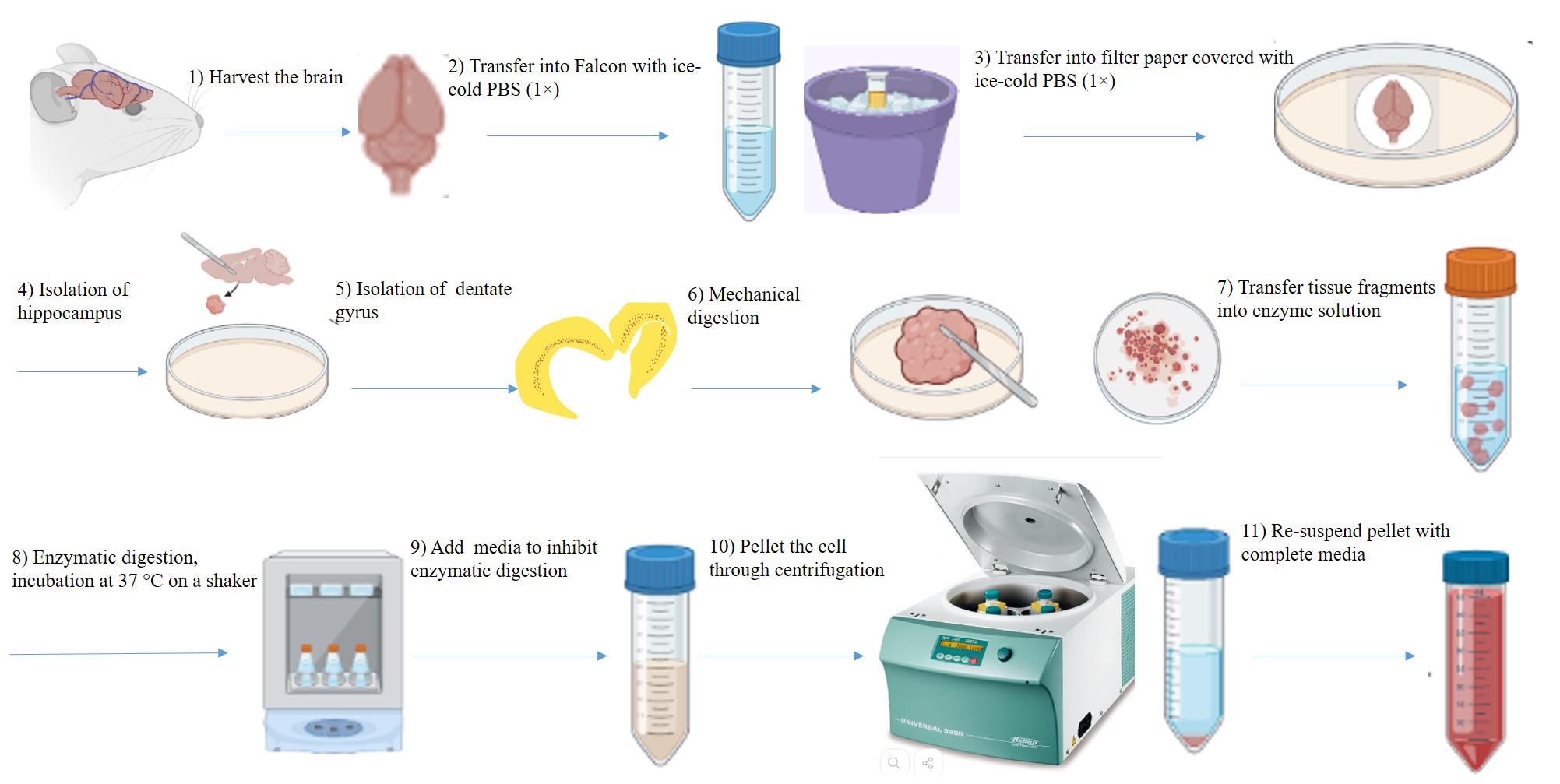
Figure 1. Picture demonstrating the brain removal, dentate gyrus (DG) dissection, and DG digestion
Poly-L-ornithine/Laminin coating of cell culture plates layer
Add the appropriate volume of Poly-L-ornithine (PLO) solution (20 μg/mL) into cell culture well plates to completely cover the surface (1.5 mL for 6-well plates, 450 mL for 24-well plates, and 60 μL for 96-well plates) (see Recipes).
Incubate at 37 °C for 2 h.
Tilt the plates and remove the PLO by pipetting at the corner of each well, then add the appropriate volume of PBS (1×) to each well (2 mL for 6-well plates, 1 mL for 24-well plates, and 100 μL for 96-well plates), and incubate for 2 min. Wash and incubate with PBS (1×) three times.
Add laminin so that its concentration in each well is 5 μg/mL, and incubate overnight (16 h) at 37 °C.
Use the plates immediately before letting them dry. It is optional to wash each well with PBS (1×) before seeding cells. Seed the cells 1–2 min after removal of laminin or after washing with PBS (1×).
Harvest rat brain tissue (Figure 2)
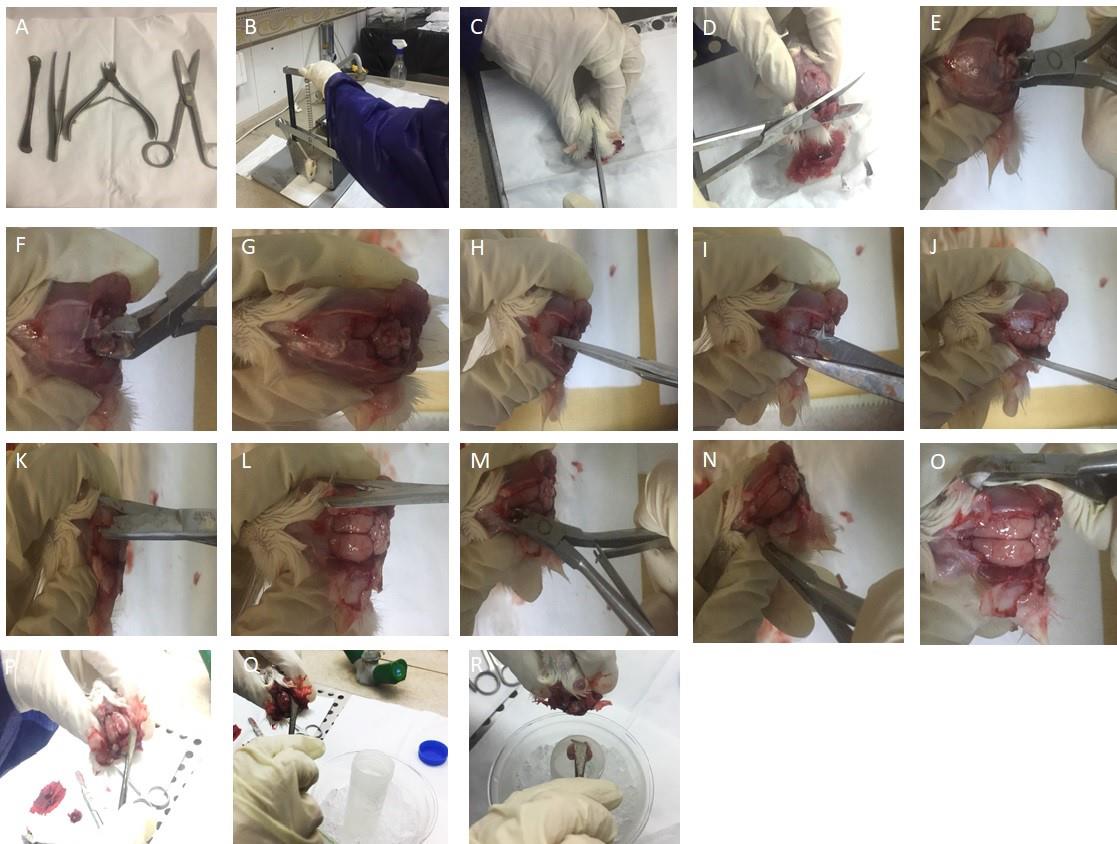
Figure 2. Harvest of the adult rat brain. A. Surgical tools. B. A rodent guillotine was used to cut the head. C. 70% alcohol was sprayed onto the head and a skin incision was made along the midline using fine scissors. D. Cutting of the remaining muscle tissues was performed by large scissors. E and F. Removal of occipital bones using rongeur is presented. G. Exposed cerebellum is presented. H. Access to the hemispheres was achieved by cutting the bone throughout the midline. I. Placing the flat blade of the scissors beneath the right parietal plate and pressing against the skull’s inner surface. J. Exposed right hemisphere is shown. K. The same procedure for the left one was performed. L. Two hemispheres are depicted. M and N. Removal of frontal bones using rongeur is shown, starting at the site of the interfrontal suture and continuing to cut small fragments of bones. O. Frontal parts of the brain have been accessed. P and Q. Separation of the brain from the skull was carried out by inserting a round spatula at the edges of the exposed brain and moving laterally in backward and forward motions and detaching the nerves through sliding the spatula beneath the brain. R. Scooping the brain out, it was transferred into a 50 mL Falcon tube containing 3–5 mL of cold PBS (1×) containing 3% Pen/Strep and 1% Amphotericin B kept on icy water.Use two separate sets of sterile surgical tools to reduce the risk of contamination with hair and blood. One set for harvesting brains from adult rats and another for microdissection of the dentate gyrus from the brain. If the following steps are performed correctly, the brain is isolated from the skull without any damage.
Anesthetize rats using intraperitoneal (i.p.) injection of a mixture of ketamine (80 mg/mL, i.p.) and xylazine (10 mg/mL, i.p.) (see Recipes) according to the following stages:
Prepare the anesthesia solution.
Lift the rat from the home cage and cover its head with a towel. Place it quietly on the cage lid to grasp its bars and become immobilized.
Grasp with one hand near the neck of the rat under the forelimb while holding the tail with the other hand. In this situation, the rat’s head is kept immobilized. Lift upward the shoulder and expose the abdomen, then inject anesthesia solution (i.p.) on the lower right quadrant of the abdomen.
Cut the neck close to the head (decapitation) with a rodent guillotine.
Sterilize the head, dissection area, and surgical tools by spraying 70% alcohol.
Hold the head in place with the lateral parts (temporal bones) between two fingers.
By using fine scissors, cut the skin out at the middle of head by a single transverse cut and retract the skin. Separate remaining muscles and tissues at the base of the skull with large scissors. Start removing bones from occipital to frontal parts.
Remove the supraoccipital bones over the cerebellum with the rongeur until it is exposed.
Now open the bones over the hemispheres in sequential steps: firstly, place the flat blade of a pair of scissors beneath the right parietal plate and force from the inside to drive away from the brain. Separate the left parietal plate in the same way.
Proceed rostrally, and remove the frontal skull plates by rongeur, starting from the site of the interfrontal suture. Remove bones to expose frontal parts of the brain (see Note 1).
To remove meninges, pierce fine scissors into the interhemispheric fissure and with a fine forceps take away the meninges over the hemispheres.
Loosen the exposed brain laterally in forward and backward motions with a round spatula.
Insert spatula inside of the skull floor and slide it to push brain away from the skull, cutting attached nerves with the same spatula, then invert brain immediately into a 50 mL Falcon tube containing 3–5 mL of cold PBS (1×) with 3% Pen/Strep and 1% Amphotericin B on water ice. Observation of the whole brain without any damage indicates its intact structure.
DG microdissection (Figure 3)
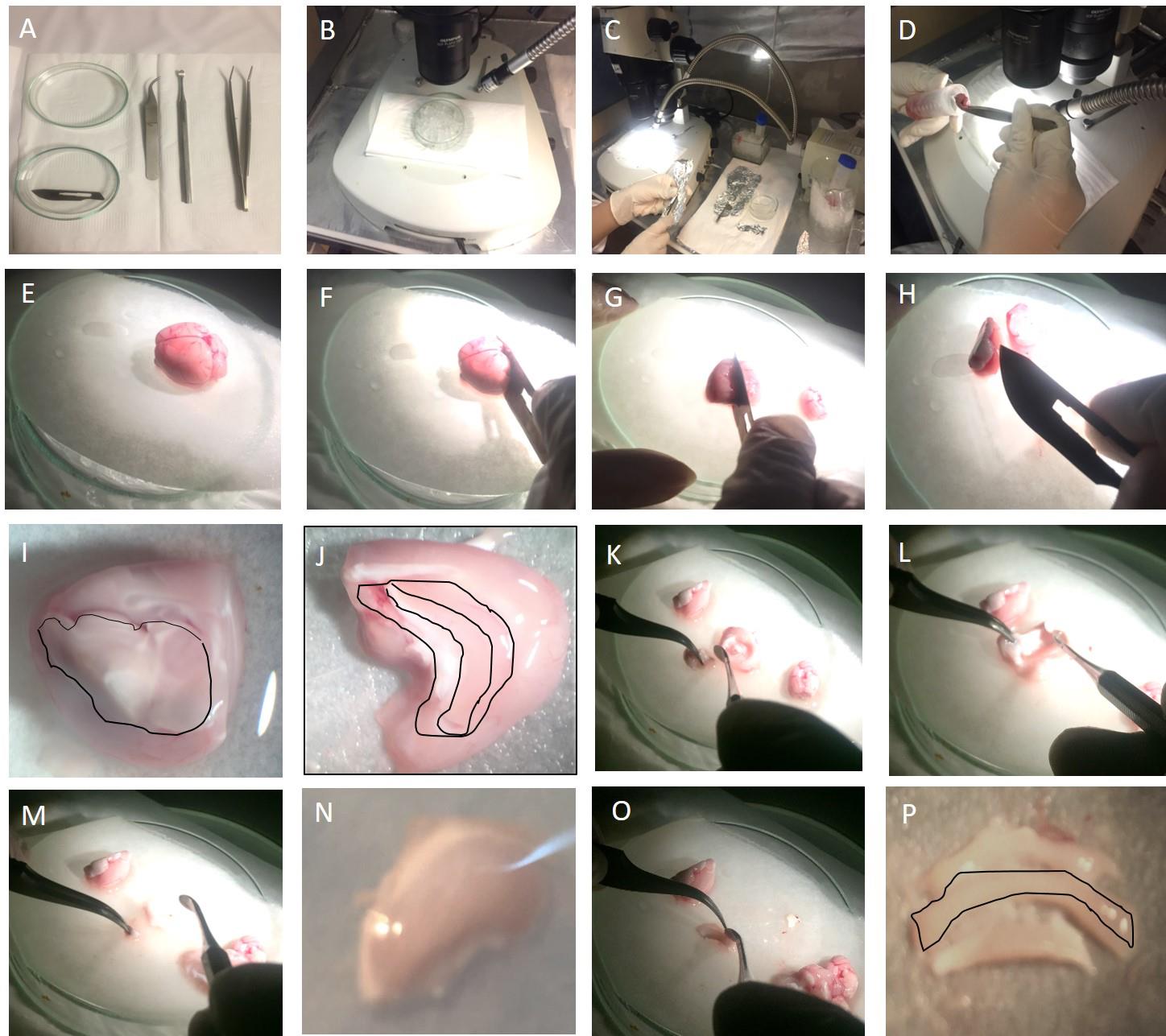
Figure 3. Dentate gyrus microdissection. A. Surgical tools. B and C. Dissection area. D. Transfer of isolated brain onto filter paper with curved forceps. E. Intact brain under a stereomicroscope. F. Cutting the cerebellum off with a scalpel. G. Splitting the brain into two hemispheres along the midline. H. Turning over each hemisphere onto the medial surface with forceps. I. Removal of the diencephalon inside of each hemisphere (indicated by a black line). J. The hippocampus inside of the cortex is visible. Middle part indicated by a black line is the dentate gyrus. K, L, and M. Hippocampus was separated from brain tissues by inserting a spatula at the isolating border and releasing into the dissection area. N. Hippocampus under stereomicroscope. O. Insertion of the spatula into the hippocampal fissure to isolate the dentate gyrus. P. Dentate gyrus (indicated by a black line) is observed in the center.Place a Petri dish lid on the top of ice under a stereomicroscope.
Cover the Petri dish with filter paper and rinse it completely with 2 mL of cold dissection solution. Place an illuminator on the right side and focus it on the dissection area.
Transfer the isolated brain from a 50 mL Falcon tube onto the dissection area using curved forceps and rinse it with dissection solution (2 mL for each brain).
Roll the brain onto its ventral surface. Firstly, by using a scalpel, cut off the cerebellum while holding the brain with forceps, and then cut the brain along the sagittal plane into two hemispheres.
Turn over each hemisphere upward to view the medial side and hold them in place using forceps.
Initially, remove the diencephalon inside of each hemisphere at the center by inserting a spatula at the inner border of the fornix and diencephalon, and then sliding the spatula from posterior to anterior, finally removing the diencephalon.
Now the hippocampus is visible on the medial side. Separate cortex overlays from the hippocampus throughout the corpus callosum with spatula. Isolate the hippocampus from the septo-temporal direction and detach it from other brain tissues with spatula, then turn over the entire hippocampus onto the dissection area. Cut the remaining fragments of cortex and white matter tissue.
To dissect DG from the hippocampus, refocus the microscope, and observe the hippocampal fissure (in some rats it is not as visible).
In most brains, a blood vessel in the hippocampal fissure, the hippocampal arteriole, is apparent and serves as a landmark to separate the DG from the CA1 area. The DG is in the middle area when the hippocampus is observed from the medial plane. While holding the hippocampus in place using forceps, slide along the hippocampal fissure with a fine spatula to cut the DG off, then transfer it to the microtube containing 1 mL of cold PBS (1×) or DMEM/F12 (1:1).
DG tissue dissociation (Figure 4)

Figure 4. Dentate gyrus (DG) dissociation. A. Transfer the microtube containing DG tissues under a laminar hood. B. Add 1 mL of PBS to wash the tissue. C. Use a sterile scalpel to cut the tissue up. D. Mince completely. E. Add enzyme solution. F. Collect tissue fragments into a Falcon tube. G. Add enzyme solution according to the amount of tissue fragments. H. Keep on the shaker. I. Cell suspension is visible, looking milky. J. Pellet of single cells after centrifugation is shown.Transfer the microtube containing DG tissues under a laminar hood.
Empty the pieces of DG into a Petri dish under the laminar hood and add 1 mL of PBS (1×) containing 1% Pen/Strep antibiotic for two dentate gyruses to wash the tissue twice, aspirate the solution out of the Petri dish, and add 500 μL of PBS (1×) to cover the tissue. Keep tissue fragments wet.
Use a sterile scalpel to cut the tissue up and mince it completely for 2–3 min according to the tissue amount. Rotation of the Petri dish during mechanical digestion makes dissociation more uniform. Cut 1 mm of the initial segment of 1 mL pipette tip with a scalpel to broaden the hole of the tip and fill it with tissue fragments.
Transfer minced tissue into a 50 mL Falcon tube containing pre-warm Papain (0.5 mg/mL), Dispase (2 mg/mL), DNase (0.02 mg/mL) (PDD) enzyme solution and incubate at 37 °C under shaking (150 rpm). Use 1 mL of enzyme solution for each animal.
Incubate in an enzyme solution as follows: for young adult rats, 10–15 min incubation time is enough. Longer times lead to greater debris and lower cell survival. For aged rats, an incubation time of 15–20 min works efficiently. After 10 min, transfer the tube under the laminar hood and slowly pipette up and down 5–7 times with a P1000 pipette. Every 5 min, observe the Falcon tube; if the tissues get soft and the solution milky, stop digestion.
After enzyme incubation, monitor the single-cell suspension. If cells are suspended, the solution will appear milky. If further large tissues remain, it is better to remove the upper liquid and continue onto the next steps. To dissociate large particles, add more enzymes to the Falcon tube and incubate for 5 min.
To stop enzyme activity, add basic DMEM/F12 medium at three times the volume of the enzymatic solution. Pipette suspension up and down 6–7 times, then centrifuge at 95× g for 5 min.
Discard the supernatant, re-suspend the cell pellet in growth medium (1 mL), and gently pipette up and down 3–4 times. Avoid bubble formation and rapid pipetting; it reduces cell yield and produces more debris. Optional: It is possible to add more medium up to 10 mL and filter through a 70 µm cell strainer. Place cell strainer on a 50 mL Falcon tube and strain the solution through it; this removes un-dissociated tissue clumps, white matter, and connective tissue. However, it might be at the expense of losing some NSPCs.
Cell counting and plating
Perform cell counting on obtained single cells.
In some culture conditions, the isolation procedure fails, and no single cell is obtained. The bright round cells are not visible, and the culture is full of undigested tissue and dead cells. Therefore, this culture is not appropriate for plating. However, the presence of debris and dead cells is inevitable in primary culture. The experimenter must recognize single cells that are small, bright cells. Cell debris have uneven shapes with one layer observable under the microscope, but live cells have three layers and round shapes. Debris appears as a dot-like spot in the culture. Also, it is possible to use DAPI staining and trypan blue staining to distinguish debris from isolated neural stem cells.
Dilute 10 μL of the cell suspension with 10 μL of 0.4% trypan blue in a microtube.
Transfer 10 μL from the mixture onto hemocytometer chambers.
Count four individual squares (WBC cell count squares).
Divide the total number by 4, multiply by 2 (dilution factor) and 104.
The area of each square is 1 mm × 1 mm. The space between the coverslip and slide is 0.1 mm. Therefore, the volume of a large square is 1 mm3 or 0.0001 mL. According to the properties of the hemocytometer chamber, to measure cell count per milliliter, the average number of cells is multiplied by the dilution factor and 104.
By this calculation, the total number of cells in 1 mL of cell suspension is measured.
Measure and optimize cell densities of your interest in specific cell culture vessels (see Note 2). 2 × 105 cells for a T-25 flask, 1.5 × 105 cells per well for 6-well plates, 1 × 104 per well for 12-well plates. It depends on the number of animals used for each condition. (Maintenance and growth of NSPCs in DMEM/F12 require plating in high density. Complete media prepared by basic DMEM/F12 medium, not neurobasal A.)
For aged rats, plate cells as following densities: 2.5 × 105 cells for the T-25 flask, 1.5 × 105 cells per well for 6-well plates, and 1 × 105 cells per well for 12-well plates. Use adherent flasks and well plates. For neurosphere culture use 1.5 × 105 cells per well in non-adherent 6-well plates.
Generation of adherent monolayer culture (Figures 5, 6)
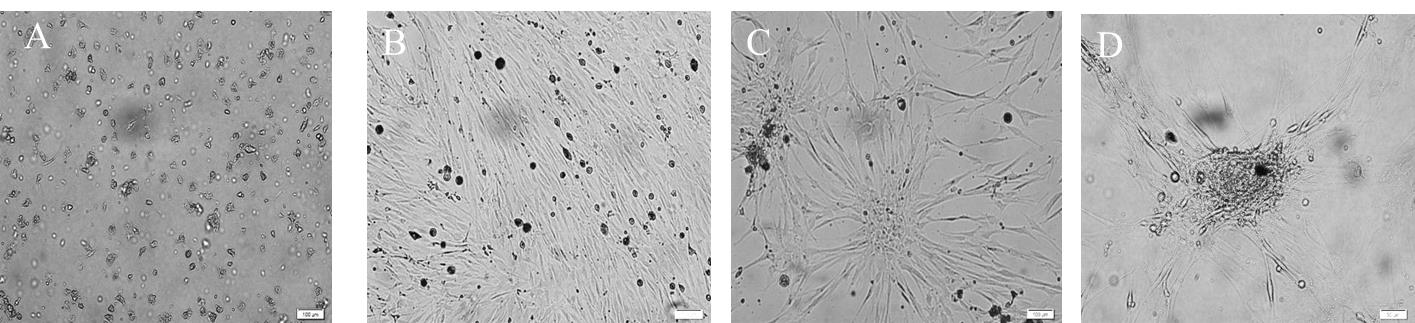
Figure 5. Culture of neural stem/progenitor cells (NSPCs) isolated from the dentate gyrus of young adult rats as a monolayer at different days in vitro. A. Day 2: neural stem/progenitor cells are visible as bright small round cells; these cells start attaching to the surface by day 3, and by day 5 most of the proliferating cells developed processes with small cell bodies. B. Day 18: represents proliferating NSPCs, which were growing continuously to occupy the culture vessel. C. By day 28, NSPCs display a substantial increase in numbers and were inclined to make connections. D. On day 38, rosette-like structures appeared. All scale bars are 100 μm with a 10× objective lens.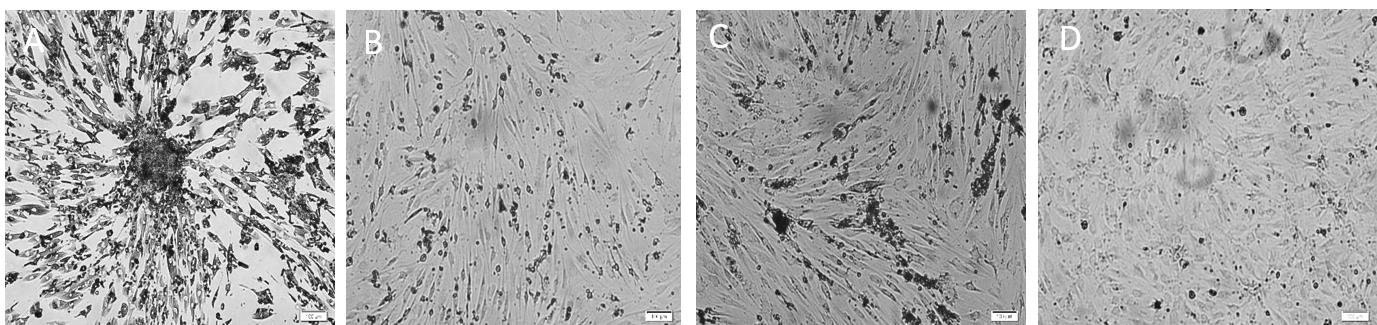
Figure 6. Culture of neural progenitor cells from dentate gyrus of adult aged rats as monolayer on different days in vitro. A. Day 12: single cells broaden their processes and grow rapidly. B. Day 19: proliferating neural progenitor cells as spindle-like shape are shown and proliferated to confluent vessel. C. Day 21: proliferated neural progenitor cells at the 80% confluency are visible. D. Day 51: most of the cells present smaller processes. All scale bars are 100 µm with objective lens 10×.Plate isolated cells onto a PLO/laminin-coated surface of a T-25 flask, which provides an adequate substrate for the maintenance and growth of NSPCs.
Allow cells to expand without disturbance. Leave cells in an incubator for up to 4 days for young adult rats and 7 days for aged rats. Monitor the culture 3–4 days after plating for young adult rats and 7 days for aged rats. Once cells adhere to the surface, remove all the medium above cells using a P1000 pipette and empty it into a 15 mL Falcon tube.
After the initial days since attached cells start expanding, add growth factors (20 ng/mL) and heparin (2 µg/mL) into the culture every 2 days.
After 7 days for young adult rats and 10 days for aged rats, remove 2.5 mL of medium from the T25 flask and add 2.5 mL of fresh medium.
Check the confluency.
At 90% confluency, tap the flask slowly with the palm of your hand to dissociate these cells for biochemical assays.
It takes 10–15 days to reach 80% confluency in the T25 flask for NSPCs obtained from young adult rats. For aged NSPCs, it takes 20–25 days to reach 80% confluency (see Figures 5 and 6).
If there are non-adherent cells or floating spheres in the medium, carry out the following steps:
Remove medium above cells using a P1000 pipette and empty it into a 15 mL Falcon tube.
Slowly pipette up and down 5–6 times with a P1000 pipette to separate the neurospheres and non-adhered cells from debris attached to them. For some cases, you can centrifuge at 95× g for 5 min and get a pellet and then add 300 μL of accutase for 3 min at room temperature.
Centrifuge at 95× g for 5 min at room temperature.
Remove the supernatant and add the same volume of complete serum-free medium to the pellet. Pipette up and down 3–4 times, then return them to their wells (see Note 3).
Subculture of adherent monolayer cultures
When cells reach approximately 80% confluency, they must be subcultured to avoid cell detachment and overgrowth, as well as to maintain cells in a healthy condition.
Perform the following steps for subculturing adherent monolayer cultures.
Remove old medium completely, then add 2 mL of PBS (1×) into the T25 flask. Slowly rinse the flask by slightly leaning it from side to side. Remove the PBS (1×) after 30 s.
Add 1 mL of accutase to the T25 flask; incubate at 37 °C for 2–3 min while observing the T25 flask under the microscope, and check for detachment of cells. If they are not detached, gently tap each side and bottom of the flask to assist in the detachment of cells. Critical: It is efficient to detach cells mechanically with a minimal volume of an enzyme.
Longer incubation time in enzyme solution causes the low attachment of single cells to the surface after subculture. Severe triturating contributes to low cell yield. Accutase is an enzyme solution of proteolytic and collagenolytic enzymes. It can be used as a direct replacement of trypsin.
To inactivate enzymatic activity, add three times the volume of the enzyme solution as DMEM/F12 medium into the T25 flask.
Pipette up and down several times, then collect the whole medium, which now contains single cells, into a 15 mL Falcon Tube.
Centrifuge at 95× g for 5 min at room temperature
Re-suspend the pellet in 1 mL of complete serum-free medium very gently. Try not to form bubbles. Immerse pipette tips in the solution thoroughly to the bottom of the Falcon tube and touching the wall of the tube, then start pipetting slowly. Seed the 100,000–150,000 cells in a 6-well plate (Table 1).
Seed the cells with high density on the coated well plate. At first passage, you can use either 100,000 cells per well of 6-well plates or 50,000 cells per well of 24-well plates. In the next passages, it is better to increase cell density.
Table 1. Recommended cell densities
Cell density Cell culture plate 100,000–150,000 Per well of 6-well plates 50,000–70,000 Per well of 24-well plates 10,000–20,000 Per well of 96-well plates
Neurosphere culture (Figures 7, 8)
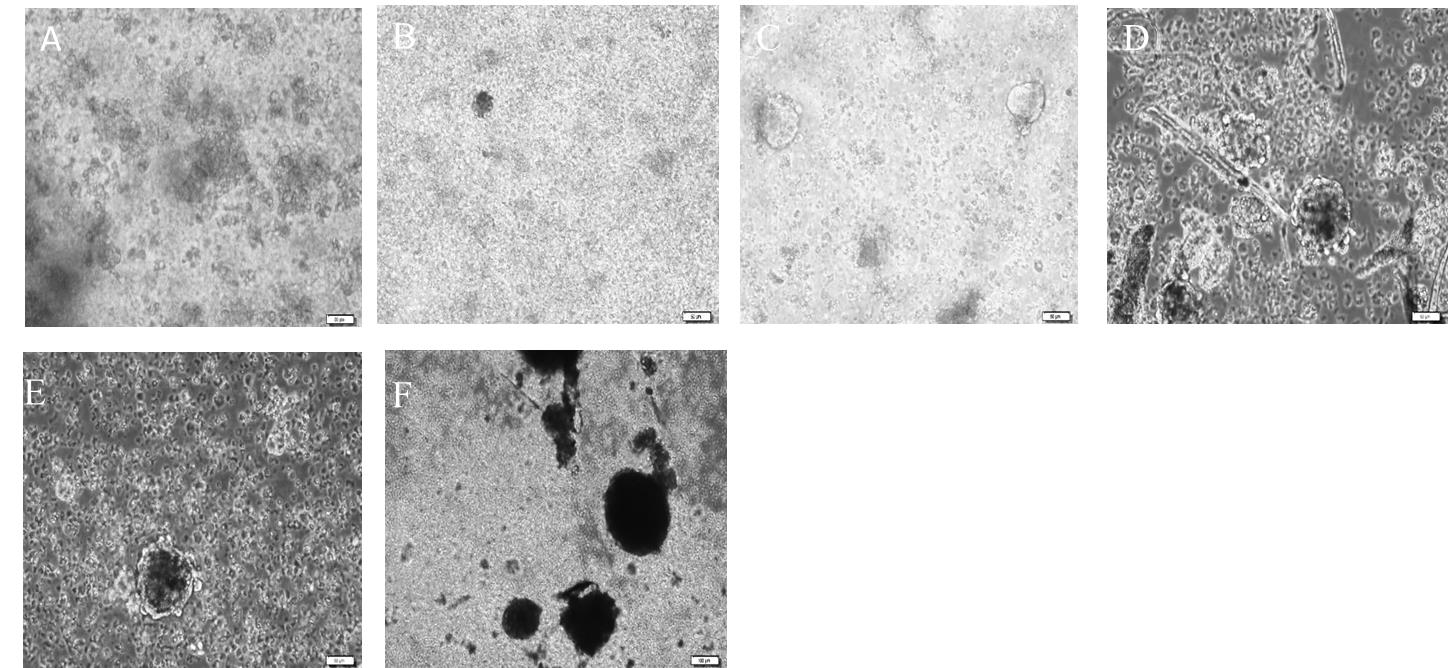
Figure 7. Culture of neural progenitor cells from the dentate gyrus of young adult rats as neurospheres on different days in vitro. A. Day 4. B. Day 8: clusters of cells were formed. C. Day 11: neurospheres show round morphology. D. Day 22: size of spheres increased. E. Day 25: spheres grew larger, and their centers appeared dark. F. Day 27: most of the neurospheres with dark centers were apparent. All scale bars are 50 μm with a 20× objective lens.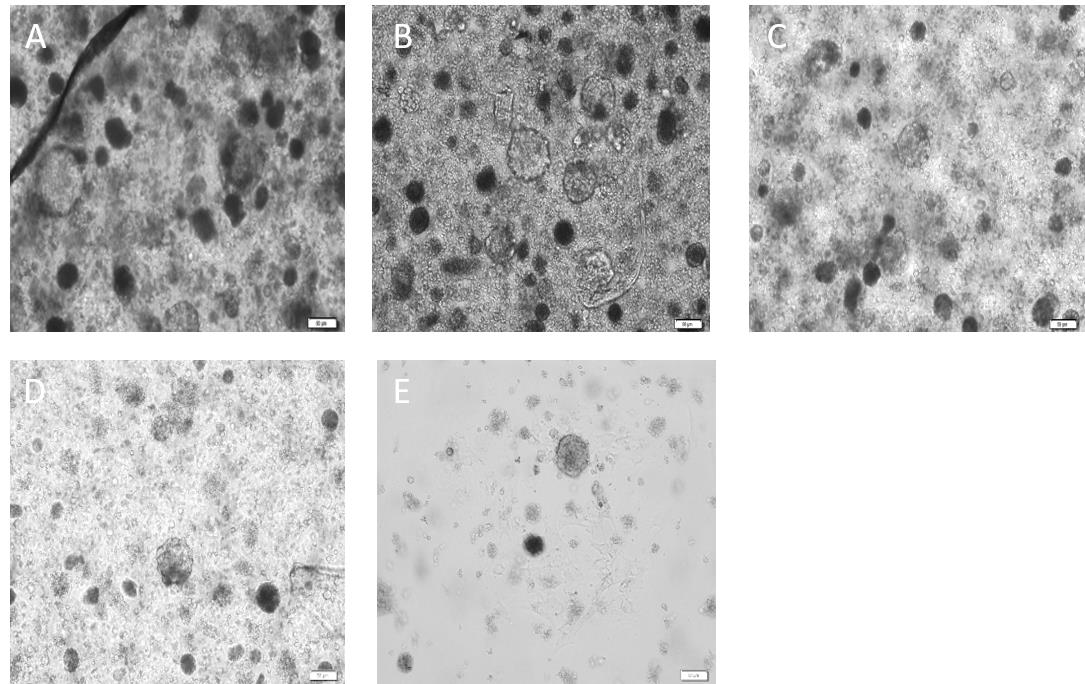
Figure 8. Culture of neural progenitor cells from dentate gyrus of adult aged rats as neurospheres on different days in vitro. A. Day 25: neurospheres are formed as their spherical shapes take form and their size increases. B. Day 32: neural stem/progenitor cells (NSPCs) proliferated, and diameter of spheres increased sharply. C. Day 36: neurospheres proliferated rapidly as their centers get a dark appearance. D. Day 41: large and dark neurospheres appeared. E. Day 48: neurospheres begin to attach to the surface. All scale bars are 50 μm with a 20× objective lens.Seed the obtained single cell suspension (2.8–3.5 × 105 cells per milliliter) from the digestion of DG tissue for neurosphere culture as follows:
Re-suspend the cell pellet in 1 mL of serum-free growth medium, then count the cells and seed them per recommended numbers (5 × 105 cells for aged rats and 3–4 × 105 cells for young adult rats) in a non-adherent T25 flask. Then, incubate at 37 °C with 5% CO2 (see Note 4). Critical: Seeding the cells at a higher density than recommended leads to cell aggregation.
It is necessary not to disturb the cells after plating until 4–5 days for the young adult group and 7 days for the aged group. Let cells adapt to culture medium for 4–5 days and start formation of spheres. Avoid any disturbances or movement of the flask (see Note 5).
After this initial incubation period, re-plate cell culture as follows: collect all the medium inside the T25 flask (which contains cells and small cell clusters starting the formation of neurospheres) into a 15 mL Falcon tube.
Add 1–1.5 mL of PBS (1×) to the T25 flask, then pipette the entire surface of the flask, and discard into a 15 mL Falcon tube.
Centrifuge at 95× g for 5 min at room temperature.
Re-suspend the cell pellet in 1 mL of neurosphere medium and pipette up and down 2–3 times slowly.
Transfer to one well of a 6-well plate.
Add growth factors and heparin every 3–4 days; growth factors (20 ng/mL) and heparin (2 μg/mL).
After 14–16 days, spheres are formed for young adult rats. Neurosphere of aged rats begin to grow from 10 days post-plating onward. After 28–30 days aged spheres are fully formed (see Note 3 as well as Figures 7 and 8). If the medium becomes yellow, add 1 mL of serum-free medium after 3 days from beginning of culture.
Quantification of the number of neurospheres
The number of neurospheres serves as an index of NSPCs proliferation. At the stage (optimal time) that the neurospheres reached maximum growth without a dark appearance in the center, estimate the number of spheres as follows:
Place the cell culture well plate under an inverted microscope, objective lens 20×.
Count spheres field-by-field, starting from a corner and going along the well diameter up to the next corner.
Adjust the focus to prevent the detection of cell aggregates instead of healthy spheres.
Quantify the size of the neurospheres
The diameter of neurospheres is used as an index of neurosphere growth.
With a 20× objective lens, take non-overlapped random images from an entire well (8–9 fields), as most of the spheres are included in the field. Use Infinity software to measure diameters of neurospheres. Average of two measured diameters is representative of neurosphere diameter. Then, express different diameters for each image. For example, if there are seven neurospheres in an image, calculate the diameter of each one. Now you have seven neurospheres with different diameters as there are three neurospheres with a diameter of 58 ± 9 micrometers, two neurospheres with a diameter of 89 ± 13 micrometers, and two neurospheres with a diameter of 120 ± 9 micrometers. This provides valuable quantification data to indirectly evaluate the proliferation rate of NSPCs on different days of in-vitro culture.
Use Infinity software as following steps:
Open Infinity software > select file > open image > adjust > micrometer > select a ruler from tool bar > place in the length of scale bar and right click > open a page > write 1 in the magnification box and write the length of scale bar in the length box > draw a horizontal line and a vertical line, and right click > length is shown (Figures 9, 10, and 11).
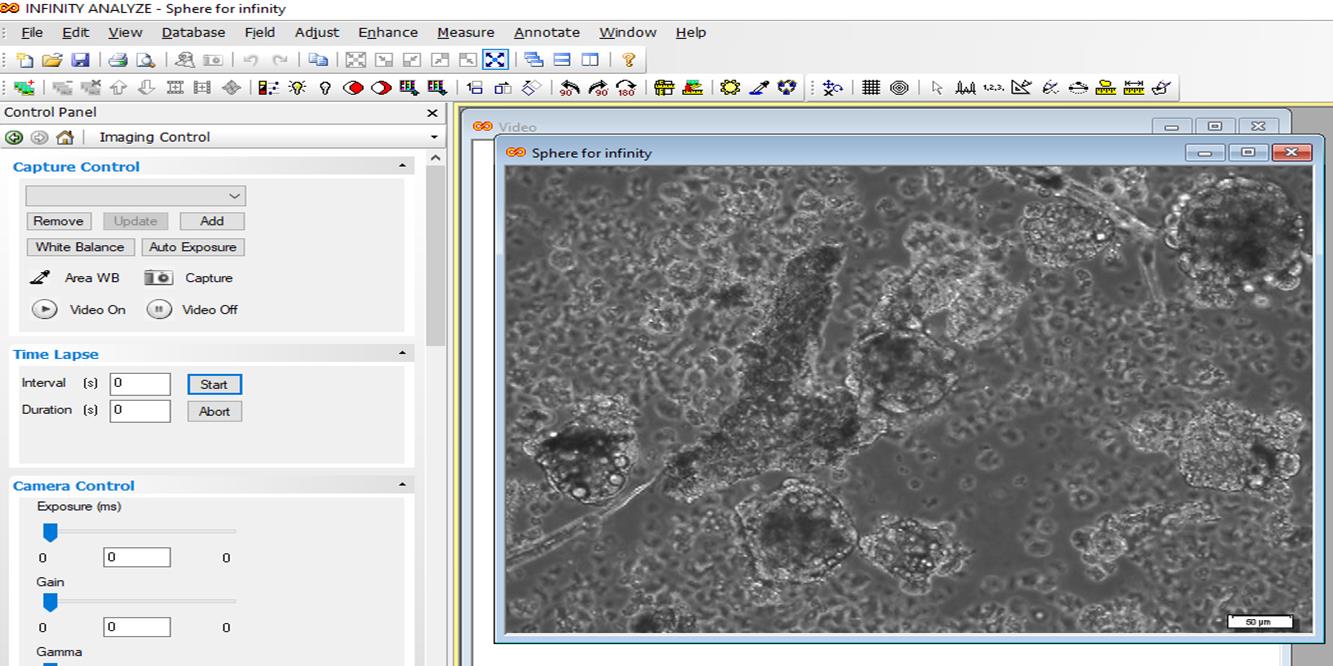
Figure 9. Quantification of neurosphere size by Infinity software. Open the image.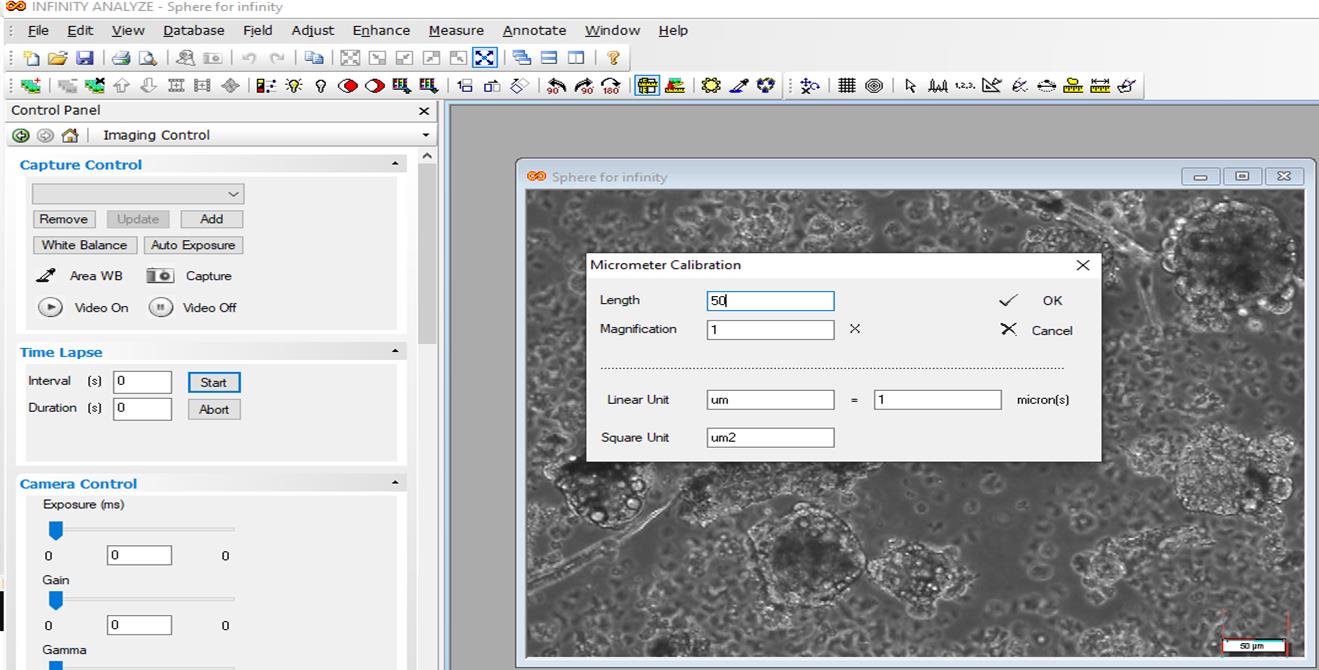
Figure 10. Open the file, adjust the micrometer, write scale bar length in the length box, and write 1 in the magnification box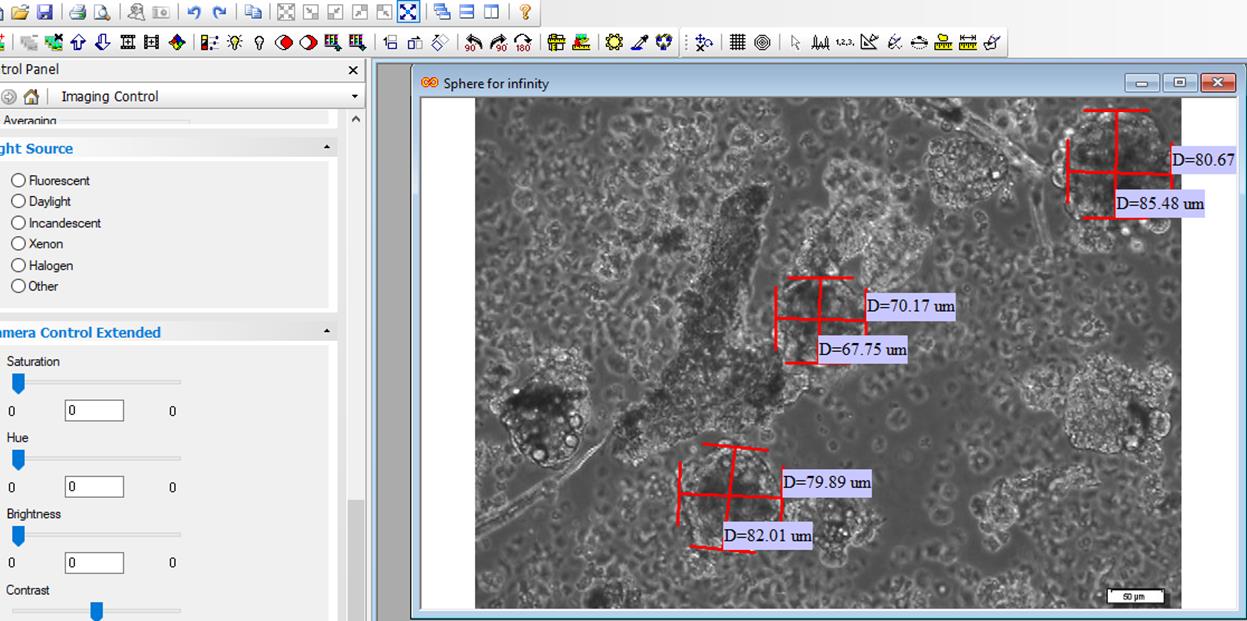
Figure 11. Draw two lines and calculate the average two numbers to acquire diameter of each spherePassage of neurospheres
Remove the content of the plate and transfer it to a 15 mL Falcon tube.
Tapping the plate and pipetting with DMEM/F12 medium to dislodge the attached spheres.
Centrifuge at 95× g for 5 min.
Remove supernatant and add 300–500 μL of accutase accurately onto the pellet (neurospheres).
Incubate at 25 °C for 4–8 min, based on the size and number of neurospheres. Critical: During enzyme incubation, it is possible to pipette up and down slowly 4–5 times without generating bubbles. Observe under light to see whether neurospheres are dissociated.
Add complete serum-free medium two times the volume of the enzyme and mix gently.
Centrifuge at 95× g for 5 min.
Re-suspend the pellet in 1 mL of neurosphere medium.
Count the cells and plate at the same density of 5 × 105 cells/mL. Critical: The number of neurosphere passages is important to determine cell density. As in our culture system the proliferation rate decreases with increasing passage numbers, it is essential to plate cells at higher density in stages following the first passage.
Immunostaining (Figure 12)
Seed 1–2 × 104 cells per well into a 96-well plate (see Note 6).
Aspirate medium from wells and wash with 100 μL PBS (1×).
Aspirate PBS completely and add 100 μL of 4% paraformaldehyde (PFA) in PBS (PH 7.4), then incubate for 20 min at room temperature (it is advisable not to use cold PFA).
Aspirate PFA and wash each well with 100 μL of PBS (1×) (three times).
Incubate for 10 min with 70–100 μL of 0.2% Triton X-100 to make cell membranes permeable.
To block non-specific antigens, add 100 μL of blocking solution (blocking solution is composed of 10% normal goat serum, 1% BSA, 0.1% Tween in PBS) (see Recipes) and incubate for 60 min.
Aspirate blocking solution.
Wash with 100 μL of PBS (1×) (three times).
Dilute primary antibodies in blocking solution as follows:
To prepare 350 μL of 1:200 Anti-Nestin solution, add 1.75 μL of Nestin antibody to 348 μL of blocking solution. Prepare before use.
To prepare 350 μL of 1:100 Sox2 solution, add 3.5 μL of Sox2 antibody to 346.5 μL of blocking solution. Prepare before use.
To prepare 250 μL of 1:250 GFAP solution, add 1 μL of GFAP antibody to 249 μL of blocking solution. Prepare before use.
Add 70 μL of primary antibodies to each well, seal the edges of the plate with parafilm to prevent evaporation of antibody solution, and incubate for 3 h at room temperature or overnight at 4 °C, preferentially inside a humid chamber.
Aspirate primary antibodies and wash with 100 μL of PBS (1×) (three times) every 5 min.
Dilute secondary antibodies with PBS (1×) (1:1,000 for both secondary antibodies). To prepare 2 mL secondary antibody solution, add 2 µL of secondary antibody into 1,998 µL of PBS (1×).
Incubate cells with 70 μL of secondary antibodies for 2 h at room temperature in the dark.
Add 100 μL of PBS (1×) into each well and incubate for 5 min, remove PBS, repeat the procedure three times, then cover the entire 96-well plate with aluminum foil to prevent light penetration.
Add 70 μL of DAPI solution per well and incubate for 1 min in the dark at room temperature.
Remove the DAPI solution and wash each well with 100 μL of PBS (1×) twice.
Capture images immediately using an inverted fluorescence microscope. For negative control, primary antibodies were not added (see Note 7).
Count and report the percentage of immunopositive cells (Figure 12).
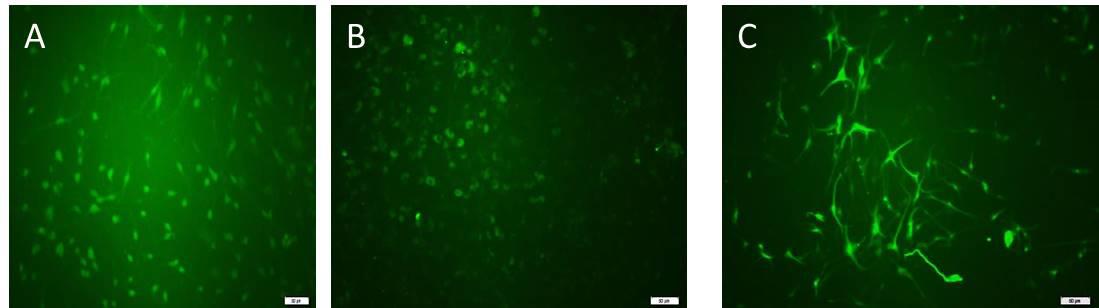
Figure 12. Immunocytochemistry to detect antigenic markers of neural progenitor cells cultured as a monolayer. Neural progenitor cells expressed (A) Sox2 antigen, (B) Nestin antigen, and (C) GFAP (marker of astroglial cells). All scale bars are 100 μm with a 10× objective lens.
Data analysis
For monolayer culture, multiple wells (4-well plates or 6-well plates) containing cells derived from a primary culture or a passage serve as technical replicates to balance variations.
Independent repeats of different experimental settings or passages serve as biological replicates. Each experimental condition and cells derived from it are generally regarded as a biological replication. Four to six biological replicates were used for each condition to reduce false positive data and make data credible. The number of N conditions is defined as biological replicates and the number of wells for each N to repeat the same condition is defined as a technical replicate. Numbers of N for sphere numbers and their sizes in two young adult and aged rats in days in vitro are represented in the following tables (Tables 2–5).
Table 2. Size of sphere (µm) in young group in days in vitro for each N
| Young group/sphere size | D5 | D10 | D12 | D14 | D15 | D17 | D18 | D20 |
| N1 | 37 | 54 | 49 | 87 | 153 | 127 | 114 | 183 |
| N2 | 49 | 47 | 50 | 49.54 | 104 | 108.50 | 129 | 137 |
| N3 | 39 | 64 | 101 | 105 | 190 | |||
| N4 | 30 | 109 |
Table 3. Size of sphere (µm) in aged group in days in vitro for each N
| Aged group/sphere size | D5 | D10 | D15 | D17 | D18 | D31 | D39 |
| N1 | 0 | 39 | 45 | 42 | 37 | 68 | 77 |
| N2 | 45 | 42 | 39 | 73 | 60 | 79 | |
| N3 | 33 | 32 | 29 | 63 | 99 | ||
| N4 | 55 | 78 | 152 |
Table 4. Number of spheres in young group per passage for each N
| Young group/sphere number | P1 | P2 |
| N1 | 70 | 79 |
| N2 | 178 | 86 |
| N3 | 145 | 129 |
| N4 | 89 | 28 |
| N5 | 59 | 137 |
Table 5. Number of spheres in aged group per passage for each N
| Aged group/sphere number | P1 | P2 |
| N1 | 56 | 79 |
| N2 | 98 | 65 |
| N3 | 78 | 87 |
| N4 | 25 | 12 |
Number of spheres and size of spheres are presented in the following figures (Figures 13, 14, and 15).
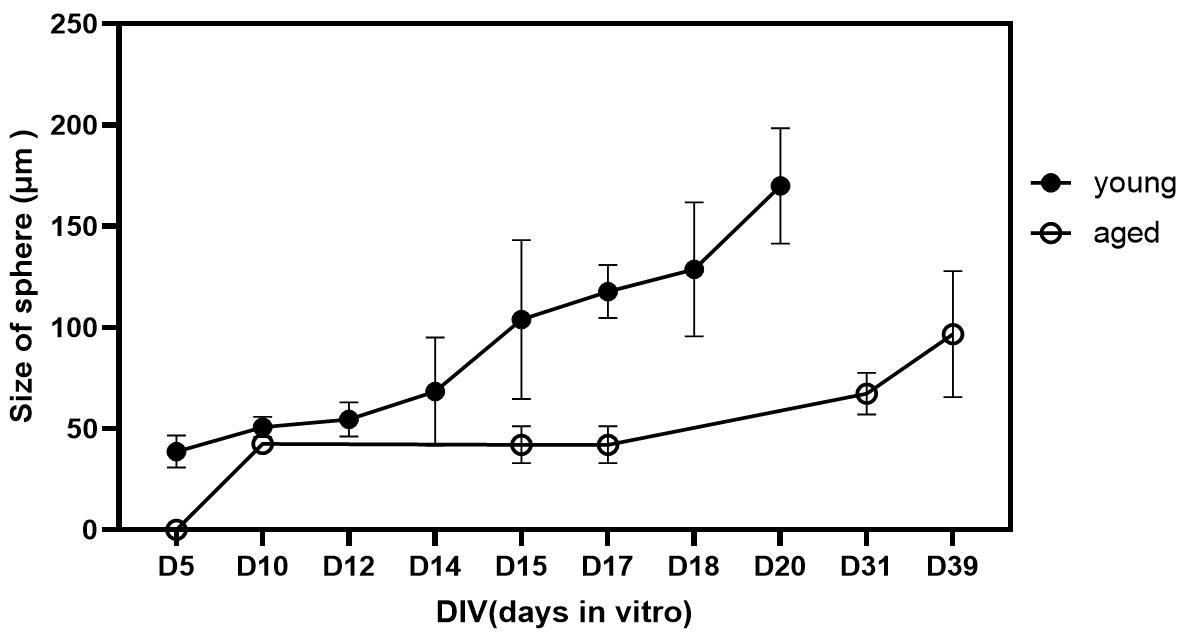
Figure 13. Comparison of size of spheres in days in vitro between young adult and aged rats. Size of spheres were greater in young adult group (not significant). Data was presented as mean ± SEM.
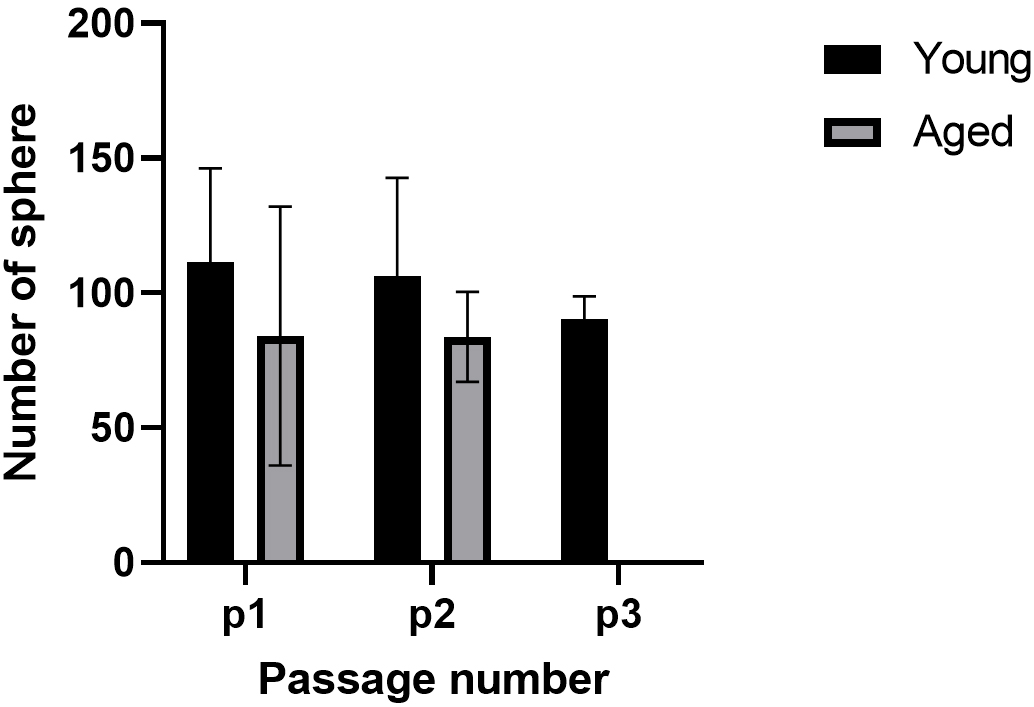
Figure 14. Comparison of number of spheres per passage between young adult and aged rats
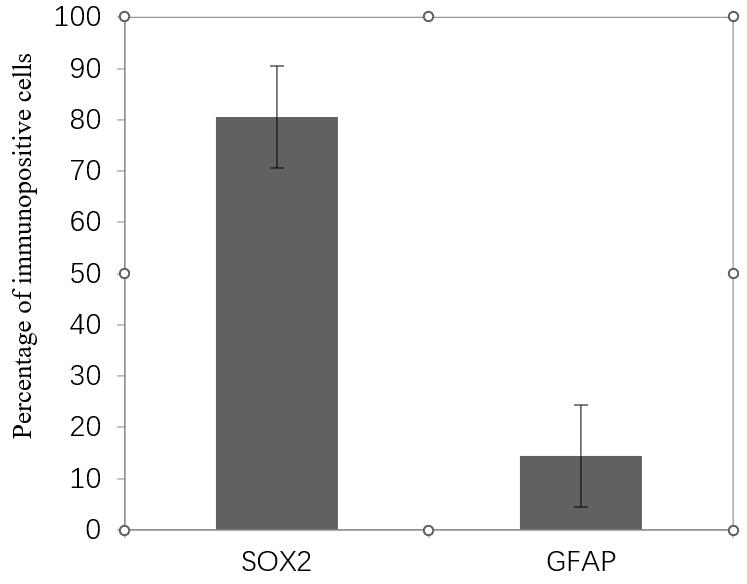
Figure 15. Percentage of positive cells for neural stem/progenitor cells (NPCs) markers per dentate gyrus (DG). Approximately 80% of total cells are SOX2 positive. Data presented as mean ± SEM.
Through this method, DGs from four to six adult rats yield 90% confluency in a monolayer system after 15 days. For aged rats, DGs from six rats produced appropriate confluency in monolayer culture after 25 days.
Cells isolated through this approach represent the identifiable morphology of adult NSPCs. These cells also represent typical markers of NSPCs. Sox2 was expressed in 80% of cells. Nestin was expressed in most of the cells, and GFAP was positive in a few cells. For each well, 6–8 non-overlapping microscopic fields were captured with a camera. The percentage of immunopositive cells was quantified as the following equation:
Number of immunopositive cells/number of DAPI-labeled nuclei × 100 = percentage of immunopositive cells.
Percentage of SOX2 and GFAP positive cells for each N was measured (Table 6). Calculated Sox2 and GFAP positive cells are shown in following bar graph (Figure 15).
In this protocol, we expanded cells as a neurosphere culture, which is a well-known culture method for NSPCs with high purity. Actually, according to this protocol, neurospheres that are cultured on the coated plates proliferate as monolayers and are mostly Sox2 positive.
Mature neurons were present in the culture, recognized by their long processes. Therefore, the presence of antigenic markers on the obtained cells, which are specific for NSPCs, was confirmed. This is an efficient and reliable protocol for the culture and expansion of aged rat NSPCs (20–24 months), which was previously reported to be challenging and difficult.
Table 6. Percentage of SOX2 and GFAP positive cells for each N
| Sample | Percentage of Sox2 positive cells | Percentage of GFAP positive cells |
| N1 | 82.30452675(200 of 243) | 12.23684 (93 of 760) |
| N2 | 68.02721088 (300 of 441) | 16.94444 (61 of 360) |
| N3 | 91.18329466 (393 of 431) | 8.333333 (42 of 504) |
| N4 | 20.13889(58 of 288) | |
| Mean | 80.50501077 | 14.41338 |
| Standard deviation | 11.68245468 | 5.192688 |
Validation of protocol
According to the data obtained from monolayer and neurosphere cultures and their immunostaining, we introduced a reliable and reproducible protocol for isolating and expanding adult rat neural progenitor cells derived from the DG of young adult and aged rats. These cells carry specific markers of neural stem cells and reliably reproduce across all experiments and assays. Through the neurosphere cell culture conditions, the stem cells from young adult rats produced free-floating neurospheres up to four passages, and proliferation slowed down at passages 4–5. Neurosphere formation from stem cells of aged rats proceeded until passage 2 under our culture conditions, and there were no identifiable neurospheres at passage 3. In some studies, NPCs of aged rats were expanded through more passages. This study will add more information about the derivation and expansion of adult rat NSPCs of the DG.
Notes
Skulls of aged rats are thicker than those of young adult rats, so their removal is harder and takes longer.
Count cells as quickly as possible, especially in aged rat NSPCs, which are more sensitive.
It requires special attention and effort to handle rat NSPCs gently and slowly while exchanging media. Any vigorous manipulation leads to the detachment of NSPCs.
If there were large enough tissue particles on the first day of culture as to prevent precise counting, count and plate cells at this step (according to our experiments).
These cells are very sensitive, and factors such as quick pipetting, alterations of supplement concentrations, and severe movement of culture vessels prevent cells from attaching. Try not to form bubbles.
Three replicates were used for each marker.
Do not delay taking photos as it may result in fading fluorescence.
Necessary precautions:
Read safety data sheets for all materials and pieces of equipment.
Trypan blue is a carcinogenic agent. Use personal protective clothing and avoid contact with your hands, skin, and eyes. Do not breathe it.
Manipulate all products in an aseptic condition. Use gloves, masks, and protective clothing.
Acknowledgments
This work was supported by a grant (No. 3/54633) from the Ferdowsi University of Mashhad. The authors are very grateful to the Shefa Neuroscience Research Center, Khatam Alanbia Hospital, Sepideh Ghasemi, and Dr. Hadi Aligholi for their technical and scientific support. This study was a part of the Ph.D. thesis of Mina Afhami at Ferdowsi University of Mashhad.
Competing interests
The authors have no conflicts of interest.
References
Akers, K. G., Martinez-Canabal, A., Restivo, L., Yiu, A. P., De Cristofaro, A., Hsiang, H. L., Wheeler, A. L., Guskjolen, A., Niibori, Y., Shoji, H., et al. (2014). Hippocampal Neurogenesis Regulates Forgetting During Adulthood and Infancy. Science 344(6184): 598–602.
- Aligholi, H., Hassanzadeh, G., Azari, H., Rezayat, S. M., Mehr, S. E., Akbari, M., Attari, F., Khaksarian, M. and Gorji, A. (2014). A new and safe method for stereotactically harvesting neural stem/progenitor cells from the adult rat subventricular zone. J. Neurosci. Methods 225: 81–89.
- Azari, H., Rahman, M., Sharififar, S. and Reynolds, B. A. (2010). Isolation and Expansion of the Adult Mouse Neural Stem Cells Using the Neurosphere Assay. J. Vis. Exp.: e3791/2393.
- Bond, A. M., Ming, G. l. and Song, H. (2015). Adult Mammalian Neural Stem Cells and Neurogenesis: Five Decades Later. Cell Stem Cell 17(4): 385–395.
- Chen, K., Hughes, S. M. and Connor, B. (2007). Neural Progenitor Cells Derived from the Adult Rat Subventricular Zone: Characterization and Transplantation. Cell Transplant. 16(8): 799–810.
- Christian, K. M., Song, H. and Ming, G. l. (2014). Functions and Dysfunctions of Adult Hippocampal Neurogenesis. Annu. Rev. Neurosci. 37(1): 243–262.
- Gage, F. H. (2019). Adult neurogenesis in mammals. Science 364(6443): 827–828.
- Gobbel, G. T., Choi, S. J., Beier, S. and Niranjan, A. (2003). Long-term cultivation of multipotential neural stem cells from adult rat subependyma. Brain Res. 980(2): 221–232.
- Guo, W., Patzlaff, N. E., Jobe, E. M. and Zhao, X. (2012). Isolation of multipotent neural stem or progenitor cells from both the dentate gyrus and subventricular zone of a single adult mouse. Nat. Protoc. 7(11): 2005–2012.
- Kase, Y., Shimazaki, T. and Okano, H. (2020). Current understanding of adult neurogenesis in the mammalian brain: how does adult neurogenesis decrease with age? Inflamm. Regener. 40(1): e1186/s41232-020-00122-x.
- Obernier, K. and Alvarez-Buylla, A. (2019). Neural stem cells: origin, heterogeneity and regulation in the adult mammalian brain. Development 146(4): e156059.
- Ray, J. and Gage, F. H. (2006). Differential properties of adult rat and mouse brain-derived neural stem/progenitor cells. Mol. Cell. Neurosci. 31(3): 560–573.
- Reynolds, B. A. and Rietze, R. L. (2005). Neural stem cells and neurospheres—re-evaluating the relationship. Nat. Methods 2(5): 333–336.
- Sun, T., Wang, X., Xie, S., Zhang, D., Wang, X., Li, B., Ma, W. and Xin, H. (2011). A comparison of proliferative capacity and passaging potential between neural stem and progenitor cells in adherent and neurosphere cultures. Int. J. Dev. Neurosci. 29(7): 723–731.
- Takei, Y. (2019). Age-dependent decline in neurogenesis of the hippocampus and extracellular nucleotides. Hum. Cell 32(2): 88–94.
- Walker, T. L. and Kempermann, G. (2014). One Mouse, Two Cultures: Isolation and Culture of Adult Neural Stem Cells from the Two Neurogenic Zones of Individual Mice. J. Vis. Exp.: e3791/51225-v.
- Zhao, X. and Moore, D. L. (2018). Neural stem cells: developmental mechanisms and disease modeling. Cell Tissue Res. 371(1): 1–6.
Article Information
Copyright
© 2023 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Afhami, M., Behnam-Rassouli, M., Gorji, A., Karima, S. and Shahpasand, K. (2023). Isolation and Culture of Neural Stem/Progenitor Cells from the Hippocampal Dentate Gyrus of Young Adult and Aged Rats. Bio-protocol 13(19): e4843. DOI: 10.21769/BioProtoc.4843.
Category
Neuroscience > Cellular mechanisms > Cell isolation and culture
Cell Biology > Cell isolation and culture > Monolayer culture
Stem Cell > Adult stem cell > Neural stem cell
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.