- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Detection of Cytoplasmic and Nuclear Circular RNA via RT-qPCR
Published: Vol 13, Iss 17, Sep 5, 2023 DOI: 10.21769/BioProtoc.4798 Views: 3457
Reviewed by: Durai SellegounderSalim Gasmi
Original research article
The authors used this protocol in:

Jul 2021
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Circular RNA (circRNA) is an intriguing class of non-coding RNA that exists as a continuous closed loop. With the improvements in high throughput sequencing, biochemical analysis, and bioinformatic algorithms, studies on circRNA expression became abundant in recent years. However, functional studies of circRNA are still limited. Subcellular localization of circRNA may provide some clues in elucidating its biological functions by performing subcellular fractionation assay. Notably, circRNAs that are predominantly found in the cytoplasm are more likely to be involved in post-transcriptional gene regulation, e.g., acting as micoRNA sponge, whereas nuclear-retained circRNAs are predicted to play a role in transcriptional regulation. Subcellular fractionation could help researchers to narrow down and prioritize downstream experiments. The majority of the currently available protocols describe the steps for subcellular fractionation followed by western blot analysis for protein molecules. Here, we present a protocol for the subcellular fractionation of cells to detect circRNA via RT-qPCR with divergent primers. Moreover, detailed steps for the generation of specific circRNAs-enriched cDNA included in this protocol will enhance the amplification and detection of low-abundance circRNAs. This will be useful for researchers studying low-abundance circRNAs.
Key features
• This protocol builds upon the method developed by Gagnon et al. (2014) and extends its application to circRNA study.
• Protocol for amplification of low levels of circRNA expression.
• Analysis takes into consideration the ratio of cytoplasmic RNA concentration to nuclear RNA concentration.
Graphical overview
Background
Circular RNA (circRNA) is an intriguing class of non-coding RNA that is formed through a unique mechanism known as backsplicing, in which the 5′ and 3′ termini are covalently joined (Jeck and Sharpless, 2014). Due to the absence of free termini in the circular structure, circRNA can easily escape from hydrolysis by numerous cellular exonucleases. CircRNAs can be derived from exons, introns, or both, with a great diversity in length. Emerging evidence has shown that circRNAs are involved in many different biological processes and human diseases by functioning as miRNA sponges, RNA-binding protein sponges, transcriptional regulators, mRNA traps, regulators of assembly and transport of cellular proteins, as well as templates for translation.
Several computational methods, such as CIRI2, CIRCexplorer2, and find_circ, have been developed to identify circRNA by employing alignment-based strategies to recognize the back-spliced junction (BSJ), which is a unique feature of circRNAs (Szabo and Salzman, 2016; Cai et al., 2020). To confirm the presence of a circRNA identified in silico, an experimental approach is applied to validate the computationally predicted circRNAs (Zhang et al., 2016).
Reverse-transcription PCR (RT-PCR) is commonly used for the validation of circRNAs identified by RNA sequencing using divergent primers. In this approach, circRNA is converted to cDNA via reverse transcription (RT) with the presence of random hexamer or gene-specific reverse primers. Priming with random hexamers offers flexibility by ensuring RT of all RNA sequences, but gene-specific RT enhances the sensitivity of detecting the target of interest by converting the target of interest—instead of transcribing all RNA in the mix—into cDNA. Priming with gene-specific primers could enhance the detection of circRNA of interest, especially for those present in low abundance (Bustin and Nolan, 2004).
The conventional RT-PCR assay using convergent primers is unable to differentiate circRNAs from their linear transcript when the linear genome is used as template for primer design. Divergent primers are needed for the RT-PCR, as they allow direct detection of BSJ and quantification of circRNA. Divergent primers are a pair of outward-facing primers that allow amplification of circRNAs and not linear mRNA with the same sequence in cDNA and not gDNA (Figure 1). Sanger sequencing is usually performed on the PCR product amplified with the divergent primers to confirm the presence of a BSJ, as to rule out any incorrect amplification or artifact. Quantitative RT-PCR (RT-qPCR) can be used to relatively quantify the expression of circRNA across a panel of samples.
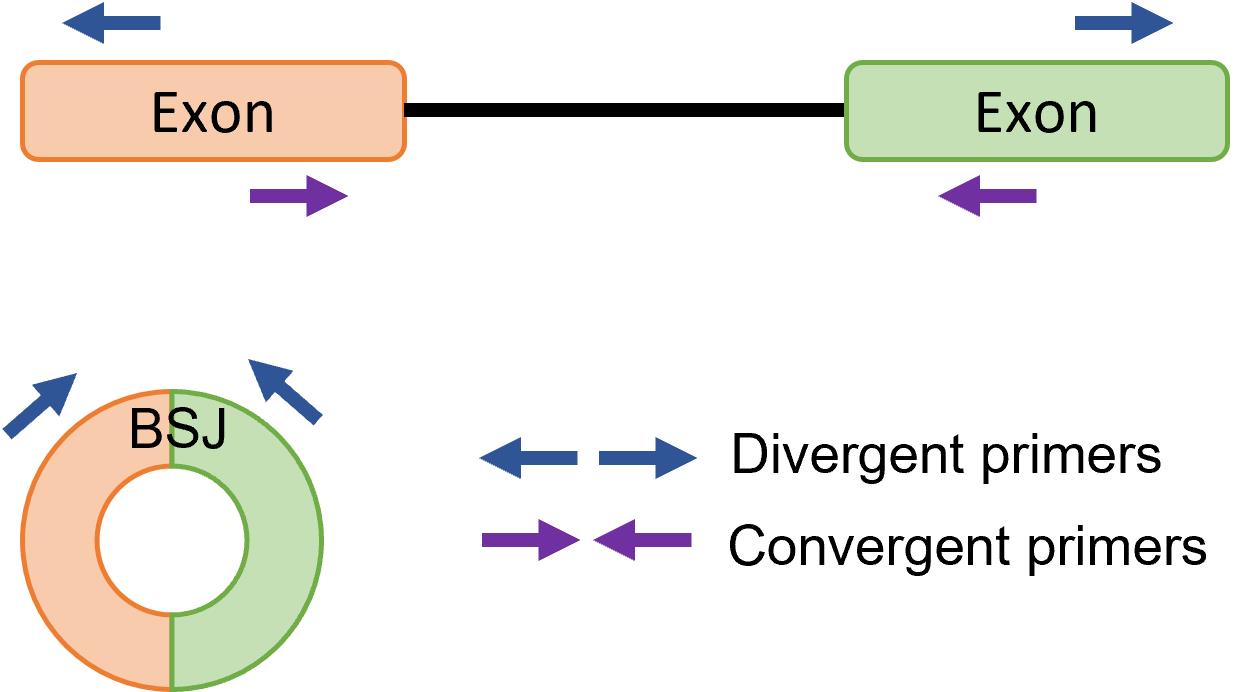
Figure 1. Primer design for circular RNA (circRNA) detection. Divergent primers are a pair of outward-facing primers that allow amplification of circRNA backsplice junction (BSJ), whereas convergent primers are a pair of inward-facing primers that allow amplification of linear mRNA from prepared cDNA.
Combining subcellular fractionation and RT-qPCR with divergent primers may provide some hints on circRNA’s biological functions. For example, circRNAs that are predominantly found in the cytoplasm are more likely to be involved in post-transcriptional gene regulation, whereas nuclear-retained circRNAs are predicted to play a role in transcription regulation. Currently available protocols are mainly for protein work, describing the steps for subcellular fractionation followed by western blot analysis, which is not entirely suitable for RNA work. Instead, this protocol allows the downstream detection of circRNA through RT-qPCR with divergent primers. A detailed protocol for the generation of specific circRNAs-enriched cDNA will also be included to better detect low-abundance circRNAs.
Materials and reagents
Biological material
GM12878 cell line
Reagents
Dulbecco’s phosphate-buffered saline (PBS) (Fisher Scientific, catalog number: BP399-1)
Trypsin-EDTA solution (0.5%) (Gibco, catalog number: 15400054)
Tissue culture media: Roswell Park Memorial Institute (RPMI) 1640 medium (Gibco, catalog number: 11875093)
Fetal bovine serum (FBS) (Gibco, catalog number: 10270106)
Penicillin-Streptomycin (Gibco, catalog number: 15140122)
Trypan blue (Alfa Aesar, catalog number: A1860)
Tris-Base (Fisher Scientific, catalog number: BP152-5)
Potassium chloride (KCl) (Fisher Scientific, catalog number: BP366-1)
Magnesium chloride (MgCl2) (Fisher Scientific, catalog number: BP214-500)
DL-Dithiothreitol (DTT) (Sigma-Aldrich, catalog number: D0632)
NP-40 (Sigma-Aldrich, catalog number: 9016-45-9)
Ribonucleoside vanadyl complexes (NEB, catalog number: S1402S)
Sodium acetate (Fisher Scientific, catalog number: BP333-500)
Molecular grade ethanol (Merck, catalog number: 64-17-5)
TRIzol (Life Technologies, catalog number: 15-596-018)
Chloroform:isoamyl alcohol (1:24) (Fisher Scientific, catalog number: BP1752I-400)
Molecular grade isopropanol (Fisher Scientific, catalog number: BP2618-1)
RNase-free water (Macherey Nigel, catalog number: 740378)
10× M-MuLV RT buffer (NEB, catalog number: #M0253)
DNase I (NEB, catalog number: #M0303)
M-MuLV reverse transcriptase (NEB, catalog number: #M0253)
RNase inhibitor (NEB, catalog number: #M0314)
dNTP mix (Thermo Scientific, catalog number: R0192)
Random hexamer (Invitrogen, catalog number: N8080127)
Primers (IDT, origin: USA)
SYBR Fast qPCR Master Mix (KAPA Biosystem, catalog number: KK4602)
Solutions
Complete media for GM12878 cell line (see Recipes)
Hypotonic buffer (see Recipes)
RNA precipitation solution (RPS) (see Recipes)
Recipes
Complete media for GM12878 cell line
Reagent Stock concentration Final concentration Volume (mL) RPMI media 395 FBS 100% 20% 100 Penicillin-Streptomycin 10,000 U/mL 100 U/mL 5 Total 500 Hypotonic buffer
Reagent Stock concentration Final concentration Volume (μL) Tris (pH 7.5) 100 mM 10 mM 500 KCl 100 mM 10 mM 500 MgCl2 150 mM 1.5 mM 50 DTT 100 mM 0.5 mM 25 NP-40 10% 0.075% 37.5 Ribonucleoside vanadyl complexes 200 mM 2 mM 50 H2O 3837.5 Total 5,000 RNA precipitation solution (RPS)
Reagent Stock concentration Final concentration Volume (mL) Sodium acetate (pH 5.5) 3 M 150 mM 0.5 Ethanol 99.5% 9.5 Total 10 Final concentration of each component is 0.15 M sodium acetate (pH 5.5) in ethanol.
Keep at -20 °C for storage.
Equipment
Refrigerated tabletop centrifuge (15 or 50 mL conical tube adaptors) (Thermo Scientific, model: Multifuge X1R)
Refrigerated benchtop centrifuge (1.5 mL tube rotor) (Eppendorf, model: 5424R)
Light microscope (Olympus, origin: USA)
Vortexer (Biosan, model: V-1 plus)
Micropipettors (Gilson, origin: USA)
NanoDrop 2000c UV-Vis Spectrophotometer (Thermo Scientific, origin: USA)
Applied Biosystems Veriti Dx 96-Well Thermal Cycler PCR Thermocycler (Applied Biosystem, origin: USA)
Bio-Rad Connect Real-Time PCR System (Bio-Rad, origin: USA)
Procedure
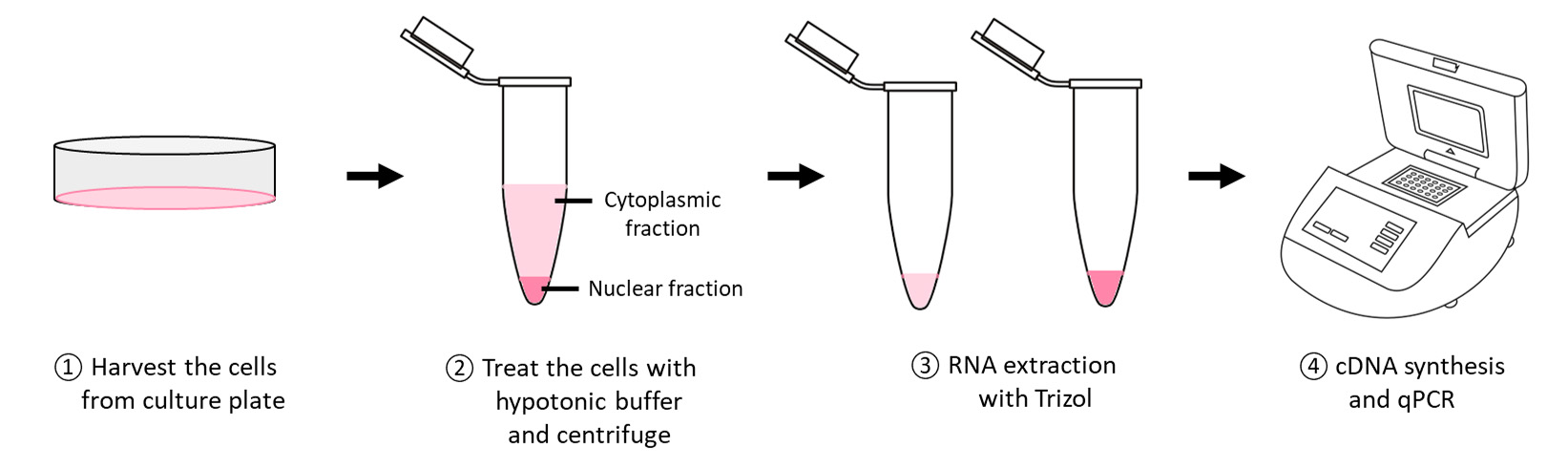
Subcellular fractionation
Cell harvesting from culture plates
Grow the cells to a confluency of ~80%. Rinse the cells with 1× PBS.
For adherent cells, detach the cells with Trypsin-EDTA. For suspension cells, proceed to step A1d.
Add an equal volume of complete media to stop the trypsin proteolysis.
Collect the cell suspension in a conical tube and centrifuge at 200× g for 5 min.
Remove the supernatant and resuspend the cell pellet with 1× PBS.
Perform cell counting and aliquot 1 × 106 cells into a 1.5 mL microcentrifuge tube.
Centrifuge the tube at 200× g for 5 min and remove the supernatant.
Keep the cell pellet on ice prior to the next step.
RNA isolation from cytoplasmic and nuclear fractions of cells
Resuspend the cell pellet gently with 100 μL of ice-cold hypotonic buffer and incubate on ice for 5 min.
Note: Volume of the hypotonic buffer can be adjusted based on the size of cell pellet.
Check for membrane lysis with trypan blue. Proceed with centrifugation at 500× g for 10 min at 4 °C until > 90% of cells are lysed. Otherwise, increase the incubation time for step A2a until > 90% of cells are lysed.
Note: Do not overlyse the cells.
Collect the supernatant (cytoplasmic fraction) and wash the pellet (nuclear fraction) with 300 μL of ice-cold hypotonic buffer three times with centrifugation at 500× g for 5 min at 4 °C.
Add 1 mL of RPS to the cytoplasmic fraction and incubate at -20 °C for at least 1 h.
Vortex the cytoplasmic fraction in RPS and centrifuge at 18,000× g for 15 min at 4 °C.
Discard the supernatant and rinse the pellet with 70% (v/v) ice-cold ethanol.
Add 1 mL of TRIzol to the semi-dry nuclear and cytoplasmic pellets followed by the addition of 10 μL of 0.5 M EDTA.
Heat both fractions at 65 °C until the pellet dissolves with vortexing.
Cool the samples to room temperature and add 200 μL of chloroform:isoamyl alcohol (1:24).
Vortex the samples and centrifuge at 18,000× g for 10 min at room temperature.
Transfer the aqueous supernatant into a clean microcentrifuge tube.
Add an equal volume of isopropanol and incubate at -20 °C for at least 1 h.
Vortex the samples and centrifuge at 18,000× g for 15 min at room temperature.
Wash the pellet with 70% (v/v) ethanol and centrifuge at 18,000× g for 5 min.
Air-dry the pellet for 5–10 min.
Dissolve the air-dried RNA pellet in 30 μL of RNase-free water.
Quantify the RNA concentration and purity with NanoDrop 2000c UV-Vis spectrophotometer.
cDNA synthesis
DNase I treatment for isolated RNA
Prepare the RNA in PCR tube for DNase I treatment as shown in Table 1.
Table 1.Components used for DNase I treatment
Components Stock concentration Final concentration Volume (μL) MuLV RT Buffer 10× 1× 1.4 DNase I 1 U 0.5 RNA 500–2,000 ng x H2O Top up to 14 Incubate the tube in a thermal cycler at 37 °C for 30 min.
Reverse transcription (RT)
Prepare the RT reaction as shown in Table 2.
Table 2. Components used for reverse transcription
Components Stock concentration Final concentration Volume (μL) Random hexamer Primer specific MuLV RT Buffer 10× 1× 0.6 0.6 dNTP mix 10 mM 0.5 mM 1 1 Random hexamer 50 mM 2.5 mM 1 − Reverse primers 10 μM 0.5 μM − 0.5 for each primer RNase inhibitor 2 U 0.5 0.5 Reverse transcriptase 10 U 0.5 0.5 H2O 2.4 Top up to 6 Subtotal 6 6 DNAse I–treated RNA 14 14 Total 20 20 Perform the cDNA conversion on a thermal cycler with the setup of 42 °C for 60 min and follow by heat inactivation at 65 °C for 20 min.
Notes:
i. If more than four pairs of primer sets (targeting linear mRNAs and/or circRNAs) are needed for primer-specific cDNA conversion, prepare 5 μM primer mix and add 1 μL into the RT reaction.
ii. One may start with random hexamer for the cDNA conversion and switch to primer-specific cDNA conversion if the amplification of the targeted circRNA BSJs is weak, absent, or unspecific.
iii. Random hexamer-generated cDNA allows amplification of any circRNA target of interest at any one time, while primer-specific-generated cDNA only allows amplification of the planned circRNA targets and does not allow any amplification of any unplanned circRNA targets.
iv. Caution is needed when generating circRNA-specific cDNA, as the remaining unused reverse primer in the RT reaction could possibly interrupt or participate in qPCR process and lead to undesired amplification from linear mRNA.
Quantitative PCR (qPCR)
Prepare the PCR reaction as shown in Table 3.
Table 3.Components used for SYBR Green RT-qPCR
Components Stock concentration Final concentration Volume (μL) SYBR Fast qPCR Master Mix 2× 1× 5 Forward primer 10 μM 0.1 μM 0.1 Reverse primer 10 μM 0.1 μM 0.1 6-fold diluted cDNA 3 RNase-free water 1.8 Total 10 Perform the qPCR on a real time thermal cycler with the cycling parameter of 95 °C for 3 min, followed by 40 cycles of 95 °C for 2 s and 60 °C for 20 s.
Calculate the relative expression of each gene based on the equation below:

RNA ratio is the ratio of cytoplasmic RNA concentration to nuclear RNA concentration eluted in a similar volume of water. Gene expression for a cytoplasmic marker is calculated using formula (a), whereas gene expression of a nuclear marker is calculated using formula (b).
Data analysis
Representative data
Subcellular fractionation was performed using GM12878 cells, which is a lymphoblastoid cell line that contains Epstein-Barr Virus (EBV). The concentration and purity of each fraction were measured using NanoDrop 2000c (Table 4). The cytoplasmic RPL30 and nuclear MALAT1 transcripts were used as positive controls to indicate the purity of cytoplasmic and nuclear fractions, respectively. As shown in Figure 2, RPL30 was enriched in the cytoplasm, whereas MALAT1 was enriched in the nuclear fraction, indicating that subcellular fractionation was successfully performed. In this representative data, the linear EBV latent membrane protein 2 (LMP-2) transcripts were found in both cytoplasmic and nuclear fractions. The subcellular localization pattern of circLMP-2_e5 is similar to that of linear LMP-2A and LMP-2B.
Note that the protocol described here requires a real-time PCR machine to generate the data, which may not be available in all laboratories. In such a case, conventional agarose gel–based RT-PCR can be conducted to obtain a semi-quantitative result.
Table 4. Concentration and purity of cytoplasmic and nuclear fraction
| Concentration (ng/μL) | 260/280 | 260/230 | |
|---|---|---|---|
| Cytoplasmic fraction 1 | 1,279 | 1.96 | 1.95 |
| Cytoplasmic fraction 2 | 1,242 | 1.93 | 1.74 |
| Nuclear fraction 1 | 566 | 1.76 | 0.71 |
| Nuclear fraction 2 | 430.3 | 1.87 | 1.46 |
1 and 2 represent biological replicates.
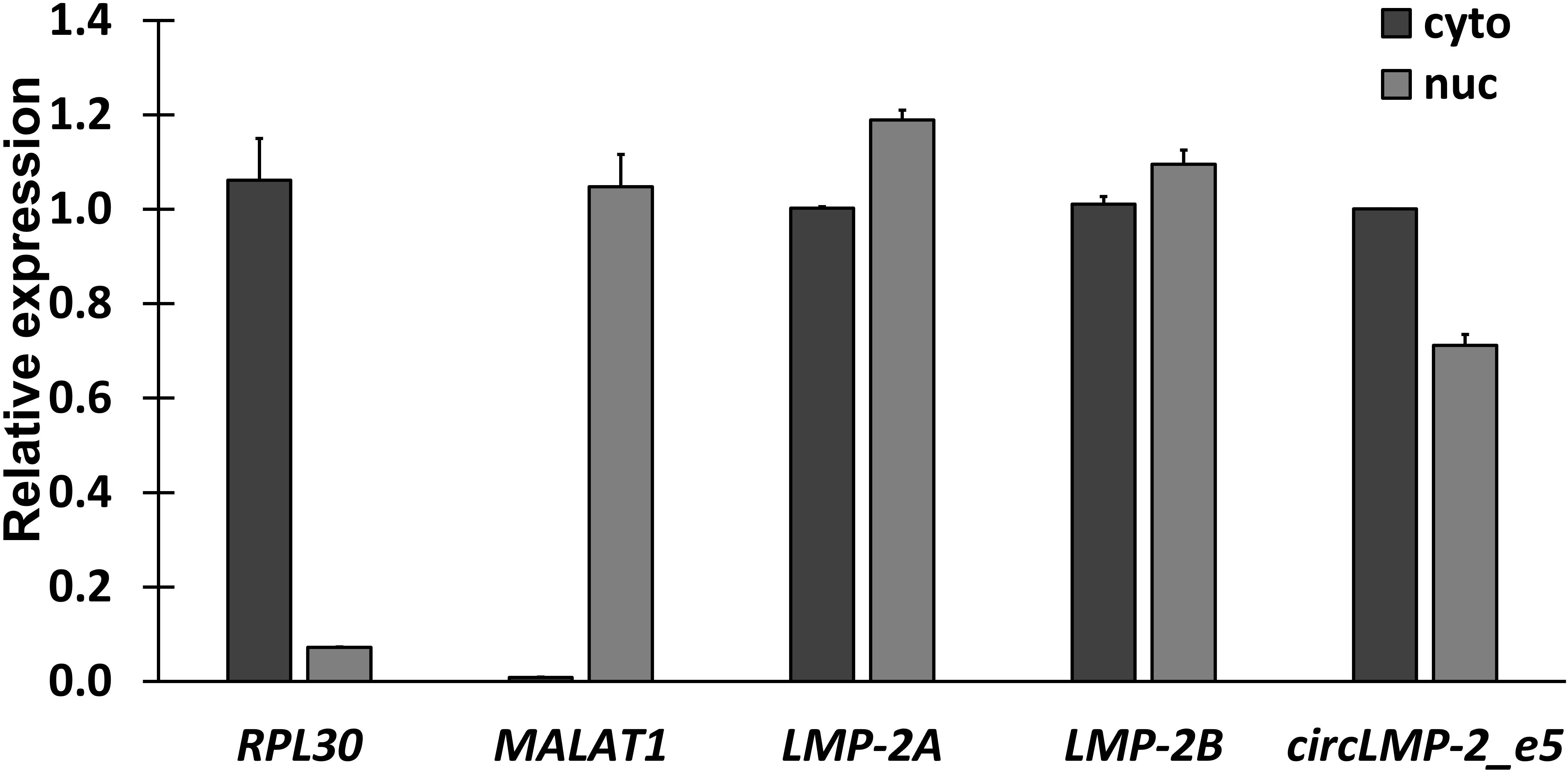
Figure 2. Subcellular localization of circLMP-2_e5. RT-qPCR analysis showed LMP-2 and circLMP-2_e5 were localized in both nucleus and cytoplasm of GM12878 cells. Data was normalized to the RNA yield ratio and represents the mean ± SD of two independent experiments. This result was published in Figure 3C in Tan et al. (2021).
Validation of protocol
This protocol or parts of it has been used and validated in the following research article:
Tan et al. (2021). Identification and characterization of a novel Epstein-Barr Virus-encoded circular RNA from LMP-2 Gene. Scientific Reports (Figure 3, panel C).
Acknowledgments
The work was supported financially by the Ministry of Higher Education Malaysia via Fundamental Research Grant Scheme (FRGS/1/2017/SKK08/UM/02/11) and University of Malaya High Impact Research Grant (UM.C/625/1/HIR/MOE/CHAN/02/07).
The original research paper in which this protocol was described and validated is Tan et al. (2021).
Competing interests
The authors declare no competing interests.
References
- Bustin S. A. and Nolan, T. (2004). Pitfalls of quantitative real-time reverse-transcription polymerase chain reaction. J. Biomol. Tech. 15(3): 155–166.
- Cai, Z., Fan, Y., Zhang, Z., Lu, C., Zhu, Z., Jiang, T., Shan, T. and Peng, Y. (2020). VirusCircBase: a database of virus circular RNAs. Briefings Bioinf. 22(2): 2182–2190.
- Gagnon, K. T., Li, L., Janowski, B. A. and Corey, D. R. (2014). Analysis of nuclear RNA interference in human cells by subcellular fractionation and Argonaute loading. Nat. Protoc. 9(9): 2045–2060.
- Jeck, W. R. and Sharpless, N. E. (2014). Detecting and characterizing circular RNAs. Nat. Biotechnol. 32(5): 453–461.
- Szabo, L. and Salzman, J. (2016). Detecting circular RNAs: bioinformatic and experimental challenges. Nat. Rev. Genet. 17(11): 679–692.
- Tan KE, Ng WL, Marinov GK, Yu KH, Tan LP, Liau ES, Goh SY, Yeo KS, Yip KY, Lo KW et al. (2021). Identification and characterization of a novel Epstein-Barr Virus-encoded circular RNA from LMP-2 Gene. Sci Rep. 2021 Jul 13;11(1):14392.
- Zhang, Y., Yang, L. and Chen, L. L. (2016). Characterization of Circular RNAs. In: Feng, Y. and Zhang, L. (Eds.). Long Non-Coding RNAs (pp. 215–227). Methods in Molecular Biology. Humana Press, New York.
Article Information
Copyright
© 2023 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Tan, K. E., Ng, W. L., Ea, C. K. and Lim, Y. Y. (2023). Detection of Cytoplasmic and Nuclear Circular RNA via RT-qPCR. Bio-protocol 13(17): e4798. DOI: 10.21769/BioProtoc.4798.
Category
Molecular Biology > RNA > qRT-PCR
Cell Biology > Organelle isolation > Fractionation
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.