- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Human Schwann Cells in vitro I. Nerve Tissue Processing, Pre-degeneration, Isolation, and Culturing of Primary Cells
Published: Vol 13, Iss 22, Nov 20, 2023 DOI: 10.21769/BioProtoc.4748 Views: 3455
Reviewed by: Vivien J. Coulson-ThomasCarmen Melendez-VasquezHeleen van 't Spijker
Original research article
The authors used this protocol in:

Aug 2018

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
This paper presents versatile protocols to prepare primary human Schwann cell (hSC) cultures from mature peripheral nervous system tissues, including fascicles from long spinal nerves, nerve roots, and ganglia. This protocol starts with a description of nerve tissue procurement, handling, and dissection to obtain tissue sections suitable for hSC isolation and culturing. A description follows on how to disintegrate the nerve tissue by delayed enzymatic dissociation, plate the initial cell suspensions on a two-dimensional substrate, and culture the primary hSCs. Each section contains detailed procedures, technical notes, and background information to aid investigators in understanding and managing all steps. Some general recommendations are made to optimize the recovery, growth, and purity of the hSC cultures irrespective of the tissue source. These recommendations include: (1) pre-culturing epineurium- and perineurium-free nerve fascicles under conditions of adherence or suspension depending on the size of the explants to facilitate the release of proliferative, in vitro–activated hSCs; (2) plating the initial cell suspensions as individual droplets on a laminin-coated substrate to expedite cell adhesion and thereby increase the recovery of viable cells; and (3) culturing the fascicles (pre-degeneration step) and the cells derived therefrom in mitogen- and serum-supplemented medium to accelerate hSC dedifferentiation and promote mitogenesis before and after tissue dissociation, respectively. The hSC cultures obtained as suggested in this protocol are suitable for assorted basic and translational research applications. With the appropriate adaptations, donor-relevant hSC cultures can be prepared using fresh or postmortem tissue biospecimens of a wide range of types and sizes.
Keywords: Human Schwann cellsBackground
Schwann cells (SCs) are a heterogeneous group of axon-ensheathing cells in the peripheral nervous system of all vertebrate species (Jessen et al., 2015). These nerve-resident neuroglial cells can be isolated and expanded in vitro using standard cell culture techniques. Human SC (hSC) cultures can be prepared using any type of nerve or ganglion from developing and adult organ donors [reviewed recently in Monje (2020)]. Culturing hSCs is a lengthier and more labor-intensive process than culturing SCs from rodents and other experimental animals. However, the hSCs obtained from an initial harvest can be amplified substantially in vitro to generate large numbers of cells for a wide range of experimental approaches (Figure 1). Once established, the hSC cultures can be managed in a manner similar to adherent cell lines, though there are limitations regarding the expandability of individual stocks.
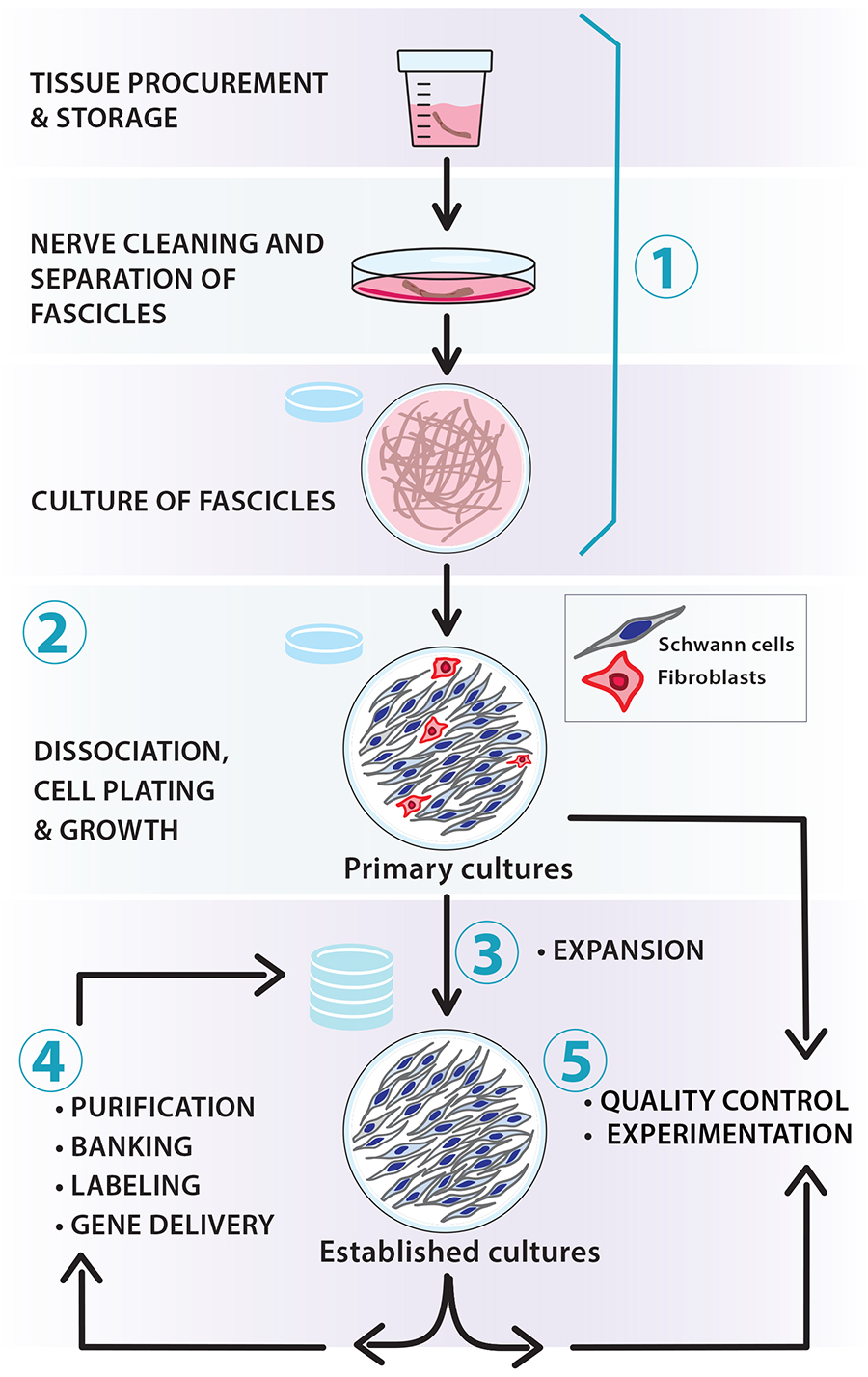
Figure 1. Scalable workflow for the preparation of nerve-derived human Schwann cell (hSC) cultures. The protocols described in this article and associated manuscripts (Monje, 2023a and 2023b) address the following basic procedures: (1) tissue procurement, dissection, and culture of fascicles (pre-degeneration); (2) enzymatic dissociation, isolation, and culture of primary hSCs; (3) derivation of established hSC cultures and amplification via serial passaging; (4) routine manipulations in vitro (e.g., purification, cryopreservation, labeling, and gene delivery); and (5) quality control of identity and bioactivity of the cells to be used in experimentation. Our protocols are adaptable; for instance, the number of hSCs can be scaled up by increasing the size of the tissue specimens used for cell isolation (primary cultures) and expanding the populations in medium containing mitogenic factors and serum (established cultures).
SC cultures from human tissues have facilitated numerous discoveries over nearly four decades [reviewed in Guest et al. (2013); Monje et al. (2021); Vallejo et al. (2022)]. These cultures are accurate in vitro models for studying neural development, differentiation, regeneration, electrophysiology, and toxicology in normal and disease states. They are also valuable for cell therapy development to repair damage caused by trauma and neurodegenerative disease. Indeed, the transplantation of cultured hSCs from a patient’s sural nerve has been implemented as a strategy to treat spinal cord and peripheral nerve injuries in USA-FDA-regulated clinical trials (Levi et al., 2016; Anderson et al., 2017; Khan et al., 2021).
Empirical data have shown that highly viable, expandable hSC cultures can be established from tissues provided by donors > 60 years of age [reviewed in Bunge et al. (2017)]. Biospecimens from live donors are not required. In fact, hSC cultures from postmortem tissues are phenotypically indistinguishable from those obtained from live donors (Boyer et al., 1994; Casella et al., 1996; Bastidas et al., 2017). Importantly, in vitro cultured SCs from adult nerves retain their ability to proliferate in response to axon contact, promote axonal growth, and form a myelin sheath (Morrissey et al., 1991), which are key functions of SCs during nerve development, maturation, and repair (Jessen et al., 2015).
Two main obstacles must be overcome to establish SC cultures from humans: (1) harvesting sufficient proliferative hSCs from the source tissue; and (2) expanding these primary hSCs sufficiently while maintaining low levels of fibroblast contamination (Morrissey et al., 1995; Peng et al., 2020). Processing adult biospecimens for cell isolation is more time demanding and challenging than doing so from developing (embryonic, neonatal) nerves. This is at least in part due to the presence of multiple layers of connective tissue (CT) and extracellular matrix (ECM), both within and around the SC-enriched fascicles. In addition, the highly elaborate cellular architecture of mature myelinating and ensheathing (non-myelinating or Remak) SCs, which are among the largest cells in the body, impose a hard-to-overcome technical barrier for disintegrating the tissue without disturbing the integrity of the cells. Although isolating hSCs immediately after nerve harvesting is feasible under certain conditions (Weiss et al., 2016), we strongly recommend pre-culturing the tissues without dissociation to achieve more consistent cell yields (Bunge et al., 2017; Chu et al., 2022). We also recommend selective immunological purification protocols to tackle the problem of fibroblast overgrowth, although other methods are available to enrich hSCs over non-glial cell populations.
In the following sections, we present generic protocols for preparing primary hSC cultures by including procedures for: (1) the dissection and harvesting of fascicles, roots, and ganglia from adult human donors; (2) the pre-degeneration of nerve segments, an intermediate step that involves the culturing of intact tissues to enrich the number of hSCs; (3) the disintegration of cultured tissues by proteolytic enzymes and the plating of initial cell suspensions on an adherent substrate; and (4) the growth and management of primary hSCs at the desired quality and quantity (Figures 1 and 2). Relevant notes on the procurement, storage, and handling of patient-derived biospecimens are included along with two different protocols for processing large and small tissue biospecimens. Various recommendations are made for managing the hSC cultures during the initial stages of growth. Supportive data on the properties of hSC cultures prepared according to these methods can be found in our published studies (Monje et al., 2006, 2008 and 2018). Our methodologies were developed using assorted tissue sources from deidentified donors. Importantly, the methods presented here are intended for non-clinical research only and differ substantially from those used in preclinical research (Bastidas et al., 2017) and clinical trials (Khan et al., 2021). However, it should be mentioned that in vitro cultured hSCs from nerves, skin, and ganglia are expected to exhibit fairly similar phenotypic and functional characteristics regardless of the tissue of origin and the mode of preparation (Stratton et al., 2017; Monje et al., 2018 and 2020; Chu et al., 2022).
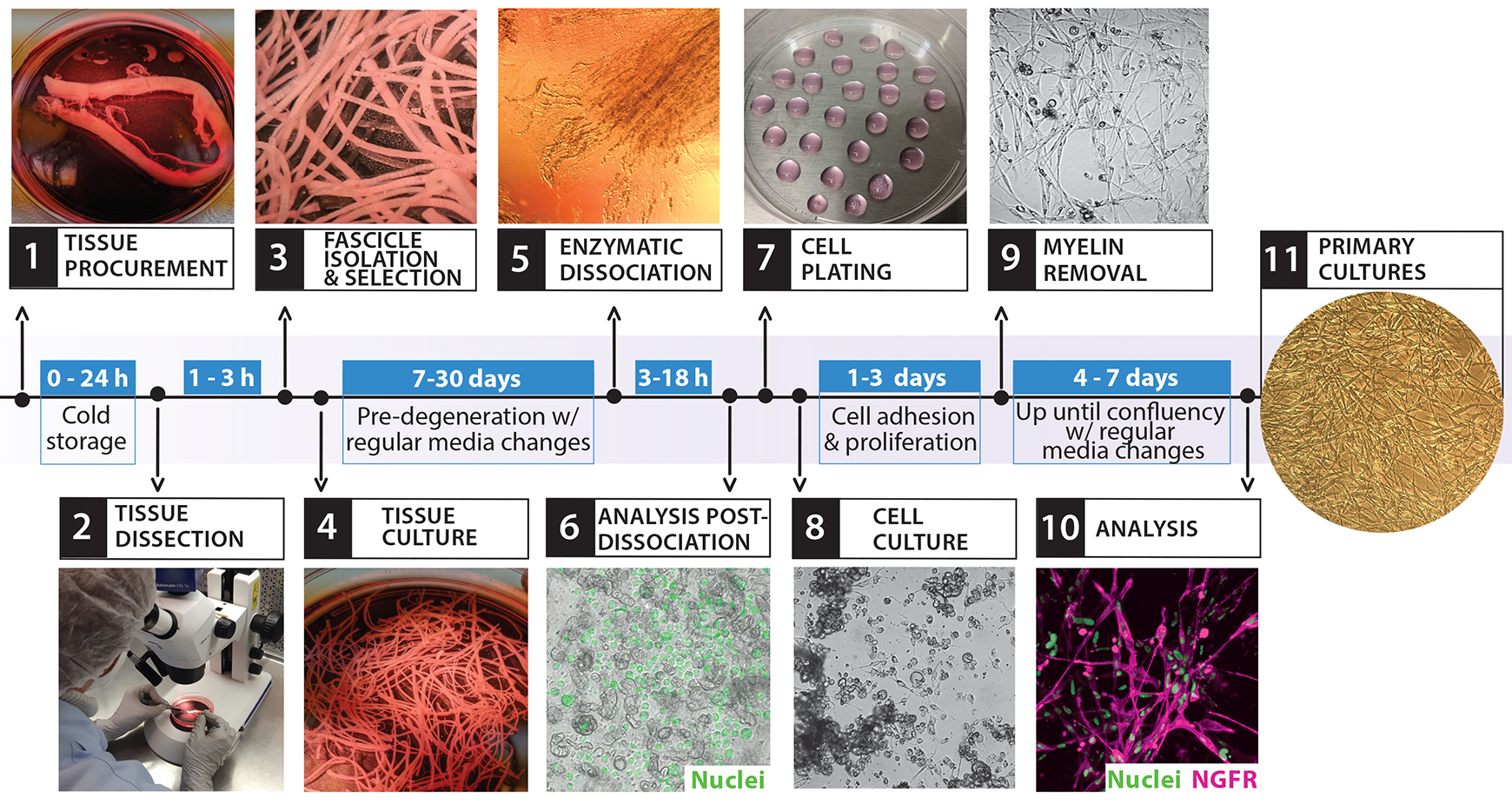
Figure 2. Isolation and culture of primary human Schwann cell (hSCs). The diagram depicts the overall cell culture strategy, highlighting the timeline for each procedure and the main steps involved, as illustrated by the representative images. This workflow starts with the bioprocessing of peripheral nerve tissues (steps 1–3) and ends with the establishment of confluent cultures of primary hSCs ready to use (step 11). The entire process can take at least three weeks, depending mostly on the length of the pre-degeneration phase (step 4, tissue culture).
Materials and reagents
All materials, reagents, and solutions should be endotoxin-free and cell culture grade. Except for dissection tools that are cleaned and sterilized for each procedure, most laboratory ware can be acquired as disposable materials for single use. It is recommended to use commercially available cell culture grade water to prepare all solutions, buffers, and culture media. In the sections below, we have added products’ information for reference only. We have mainly used disposable cell culture–treated flasks and dishes from CorningTM, but products from other brands may be equally suitable.
Supplies and consumables
Dumont forceps #3, #4, and #5; straight tip shape (Fine Science Tools, catalog numbers: 11231-30, 11242-30, and 11251-10, respectively)
Spring scissors, angled to side (Fine Science Tools, catalog number: 15006-09)
Moria Dowell spring scissors, straight tip shape (Fine Science Tools, catalog number: 15372-62)
Disposable 5 mL serological pipettes, polystyrene, sterile (VWR, catalog number: 89130-896)
Disposable 10 mL serological pipettes, polystyrene, sterile (VWR, catalog number: 19221005)
Polystyrene Pasteur pipettes, sterile and individually wrapped (VWR, Argos Technology, catalog number: 10122-560)
Borosilicate glass Pasteur (transfer) pipettes (Corning, catalog number: 7095D-9) with attached rubber bulb
100 mm Petri dishes (Corning, catalog number: 351029)
Polypropylene conical-bottom centrifuge tubes, 15 and 50 mL (Corning, catalog numbers: 430791 and 430290, respectively)
Round-bottom centrifuge tubes with a snap cap, polypropylene, 15 mL (BD, catalog number: 352059)
Polystyrene cell culture dishes, 35, 60, or 100 mm (Corning, catalog numbers: 353001, 353002, and 353003, respectively)
24-well plate, flat bottom (Corning, catalog number: 3524)
Containers with wet ice
Media, supplements, and reagents for cell culture
Distilled water, cell culture grade (Thermo Fisher Scientific, Gibco, catalog number: 15-230-147)
Dulbecco’s phosphate-buffered saline (DPBS), pH 7.2 (Thermo Fisher Scientific, Gibco, catalog number: 14190)
Hank’s balanced salt solution (HBSS), formulated without calcium or magnesium and containing phenol red, pH 7.2 (Thermo Fisher Scientific, Gibco, catalog number: 14170-112)
Leibovitz’s L15 medium (Thermo Fisher Scientific, Gibco, catalog number: 11415064)
Dulbecco’s modified Eagle’s medium (DMEM) with high glucose and phenol red, pH 7.2 (Thermo Fisher Scientific, Gibco, catalog number: 11965092)
1,000× Gentamycin (Thermo Fisher Scientific, Gibco, catalog number: 15750-060)
De-complemented, gamma-irradiated fetal bovine serum (FBS) (HyClone, catalog number: SV 30014.03)
100× GlutaMAX supplement (Thermo Fisher Scientific, Gibco, catalog number: 35050061)
Forskolin (Sigma-Aldrich, catalog number: F68861)
Heregulin-β1177-244 (referred to as heregulin) (Preprotech, catalog number: G-100-03)
Laminin stock solution, consisting of a sterile 1 mg/mL laminin from Engelbreth-Holm-Swarm murine sarcoma basement membrane (Sigma-Aldrich, catalog number: L2020). Store in aliquots at -80 °C and use as described in Andersen and Monje (2018)
Poly-L-lysine (PLL) stock solution (Sigma, catalog number: P-2636). Prepare, store in aliquots at -80 °C, and use as described in Andersen and Monje (2018)
Matrigel growth factor reduced basement membrane matrix, phenol red-free (BD Biosciences Discovery Labware, catalog number: 356231)
Dispase II or neutral protease, ≥ 0.8 units/mg protein, lyophilized powder (Roche, catalog number: 165-859)
Collagenase Type I, ≥ 125 units per milligram dry weight, dialyzed, lyophilized powder (Worthington, CLS-1, catalog number: 4196)
Dissection medium (DM) (see Recipes)
Low proliferation medium (LP) (see Recipes)
High proliferation medium (HP) (see Recipes)
Laminin coating solution (see Recipes)
10× enzymatic solution (see Recipes)
Antibodies and fluorescent dyes
Anti-NGFR mouse IgG monoclonal antibody, produced in-house from the HB-8737 hybridoma cell line (reactivity: human/primate-specific NGFR; American Type Culture Collection, ATCC). See Ravelo et al. (2018) for a step-by-step description of NGFR immunostaining in live and fixed hSC cultures. (Optional) Use the rabbit monoclonal antibody EP1039Y (Abcam, catalog number: ab52987).
Anti-O4 mouse IgM monoclonal antibody, produced in-house from the O4 hybridoma cell line (reactivity: human/rat/mouse/pig/other; kindly provided by Dr. Melitta Schachner). See Ravelo et al. (2018) for a step-by-step description of O4 immunostaining in live hSC cultures. (Optional) Use a commercially available purified O4 antibody (Novus Biologicals, catalog number: NL637)
Hoechst 33342 (Sigma, catalog number: B2261), prepared in water at 1 mg/mL
Syto-24 green, fluorescent nucleic acid stain (Invitrogen, catalog number: S75559)
Propidium iodide (PI) nucleic acid stain (Sigma, catalog number: P4170) prepared in water at 1 mg/mL. See Ravelo et al. (2018) for a step-by-step description of viability assays using PI and Hoechst 33342 or Syto-24 green
FluoroMyelin red (or green), fluorescent myelin stain (Invitrogen, catalog number: F34652)
FM 4-64FX, fixable analog of FM 4-64 membrane stain (Invitrogen, catalog number: F34653)
4’,6-Diamidino-2-Phenylindole, dilactate (DAPI) (Invitrogen, catalog number: D3571), prepared in water at 1 mg/mL
Equipment
Stereomicroscope with an attached digital camera (Zeiss, Stemi 305/Axio cam ER C52)
Double gooseneck fiber optics with intensity control for cool white light illumination (DolanJenner)
NIGHTSEA® Fluorescence Viewing System equipped with blue and green barrier filters (Electron Microscopy Sciences)
Inverted phase contrast microscope with an attached digital camera (VWR V5MP)
Inverted fluorescence microscope with an attached digital camera (Olympus IX71)
Benchtop centrifuge (Beckman Coulter, model: Allegra X-I2R)
Cell counter for automated counting of cells in suspension (Bio-Rad, TC20 Automated cell counter). (Optional) Hemocytometer for manual cell counting
CO2 cell incubator set up at 37 °C and 8%–9% CO2 (Thermo Fisher Scientific, Forma Steri-Cycle)
Biosafety cabinet, BL2 level (Thermo Fisher Scientific, 1300 Series A2)
Germinator 500, bench-top sterilizer (Cell Point, catalog number: 5517)
Procedure
Protocol 1: Procurement and dissection of human nerve tissues
This protocol relies on the availability of nerves or ganglia harvested under aseptic conditions. Freshly collected tissue remains from patients undergoing therapeutic or plastic surgeries or diagnostic biopsies are well-suited and may be preferred for hSC culturing if made available promptly from local providers. Nerves harvested postmortem are also appropriate if collected and stored properly until arrival at the laboratory.
Most tissues that become available for research are procured from adult subjects. Embryonic and fetal tissues render highly proliferative hSC cultures (Boyer et al., 1994; Scarpini et al., 1988), but they may be difficult to obtain due to ethical concerns or other restrictions. The nerve biospecimens may not be limited to a particular type or size as long as the appearance is normal, and the anatomical layers are clearly distinguishable. The best results are obtained by using nerves (or ganglia) of sufficient length (> 1 cm) and caliber (> 2–3 mm) because they can be manipulated and visualized with ease under a standard stereomicroscope. The protocols below explain how to manually isolate individual segments (fascicles, roots, and ganglia) from the surrounding connective tissue using typical long segmental nerves (Protocol 1B) and dorsal root ganglia (DRG) with attached nerve roots (Protocol 1C). Removing as much ECM as possible from the outside of the nerve or ganglia can prevent the introduction of contaminating fibroblasts into the hSC cultures. The dissection protocols will have to be adapted for the isolation of hSCs from normal tissues enriched in nerve terminals, such as the gut or the skin, and pathological tissues known to contain hSCs, such as tumors from nerves or the skin.
Tissue procurement, handling, and storage
Collect and store the human nerve in a sterile container filled with an excess volume of storage medium as appropriate for the size of the specimen, e.g., 50 mL of medium for a specimen that is 5 cm long. Use a gentamycin-supplemented balanced solution, such as University of Wisconsin (UW)® cold storage solution, high-glucose DMEM, or L15 medium, for storage. It is important to minimize the time between the surgical removal of the tissue and its immersion in the storage medium. Maintain the container in wet ice (4 °C) until beginning the dissection procedure (see Note a).
Transfer the tissue to the laboratory and start the cleaning of the nerve as soon as possible. If the procedure cannot be started immediately, the tissue can be safely stored inside a 4 °C refrigerator (for more details on storage time and conditions, please see Recommendations and Troubleshooting).
Isolation of fascicles from typical mature nerves
Transfer the nerve into a sterile 100 or 150 mm Petri dish (depending on the size of the biospecimen) containing ice-cold DM (see Recipe 1) (Figure 3A).
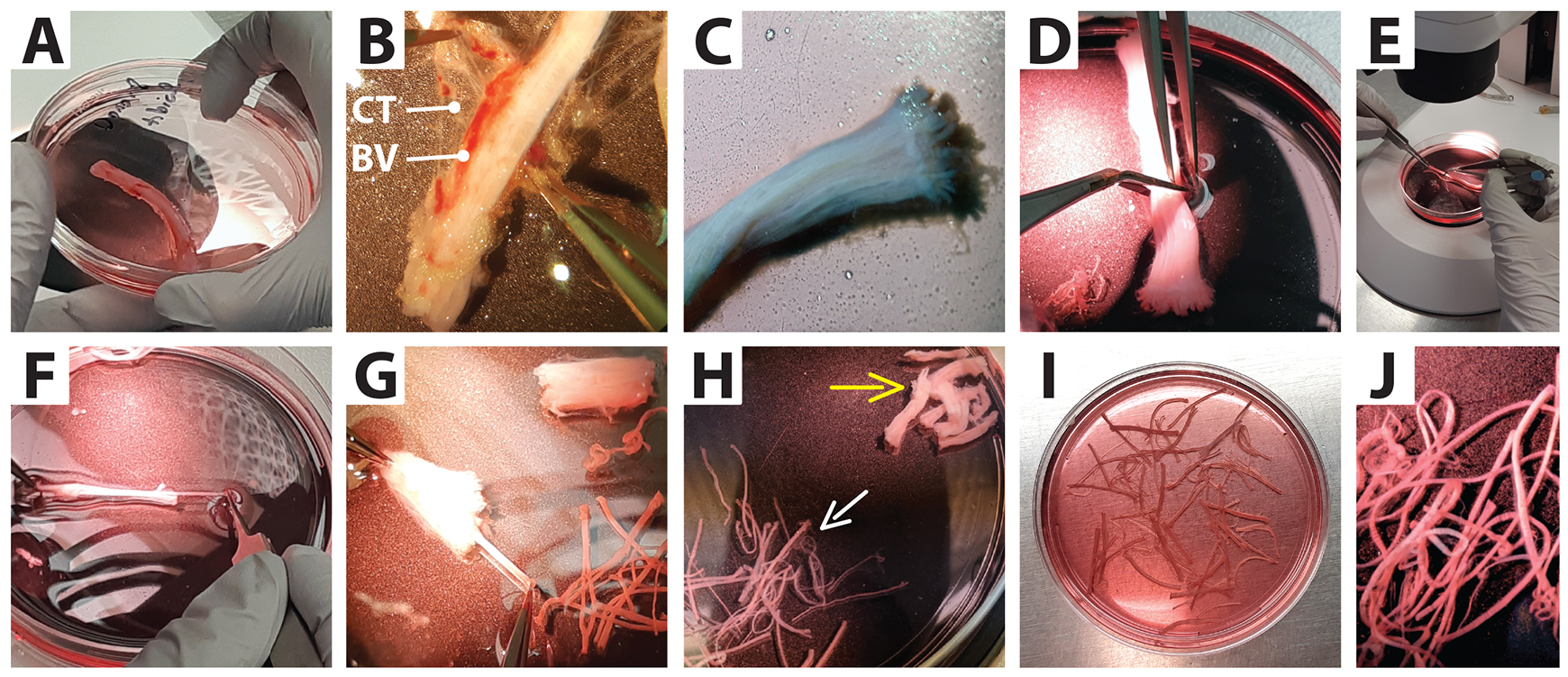
Figure 3. Separation of nerve fascicles from connective tissue. Low magnification images of a whole nerve biopsy (tibial) as received from a surgical procedure and its dissection under a stereomicroscope. (A) A nerve fragment placed in a Petri dish. (B) Removal of externally attached tissues: connective tissue (CT) and blood vessels (BV). (C) Appearance of a clean nerve. (D and E) Sectioning of the nerve into smaller segments. (F and G) Fascicle separation from the surrounding connective tissue. (H) Final harvest. White arrow: fascicles to be transferred to a culture dish; yellow arrow: connective tissue to be discarded. (I and J) Selected fascicles inside a tissue culture dish ready to be cultured.By working under the dissecting stereomicroscope illuminated with a cool light system, carefully remove the loosely attached collagen-enriched connective tissue, blood vessels (BV), fat, and muscle around the nerve using the appropriate combination of tools. For instance, for biospecimens > 2 cm in length, use blunt forceps or number 3 fine forceps to hold the nerve from one end, and spring scissors or forceps to remove the excess tissue around the nerve (Figure 3B) (see Note b).
Replace the DM (or transfer the nerves to new dishes) as frequently as needed to maintain an as-clean-as-possible solution during the whole cleaning procedure. Use ice-cold solutions and work fast to prevent an increase in temperature and consequent sample alteration. This procedure may be labor intensive depending on the length and condition of the nerve specimen. Nerves of a higher caliber, such as the adult human sciatic nerve, can contain adipose tissue surrounding and intermixing between the fascicles.
Once the nerves are cleared of loosely attached external material (Figure 3C), use sharp scissors (e.g., Metzenbaum scissors or spring scissors, depending on the caliber of the specimen) to cut the specimens transversally and generate smaller, more manageable segments. These segments can be ~1 cm and ~2–3 cm long for larger (e.g., sciatic) and lower caliber (e.g., tibial) nerves, respectively (Figure 3D and 3E). Ensure that the proximal (close to the spinal cord) and distal (close to the periphery) ends of the segments are appropriately oriented for expedited fascicle removal (see step 6).
Transfer the individual segments into a new dish containing DM and work with one segment at a time. Visually identify the individual fascicles (in cross section) protruding in between the surrounding connective tissue (whitish material) by exposing the sectioned area on the distal end of the fascicles (Figure 4A–4C).
Separate the fascicles from the collagenous connective tissue one by one by grabbing them from their distal end and pulling outward using #4 or #5 forceps while gently holding the proximal side of the nerve segment with another pair of forceps. Always pull the nerves out in the same direction while visually following their trajectory until they separate fully. If the tissue is maintained steady from the proximal end, all fascicles within one segment can be pulled out quickly and smoothly, thus leaving a carcass of collapsible connective tissue composed mostly of the epifascicular and interfascicular epineurial layers (Figure 3F–3H, 4B–4D, and 5G–5H) (see Note c).
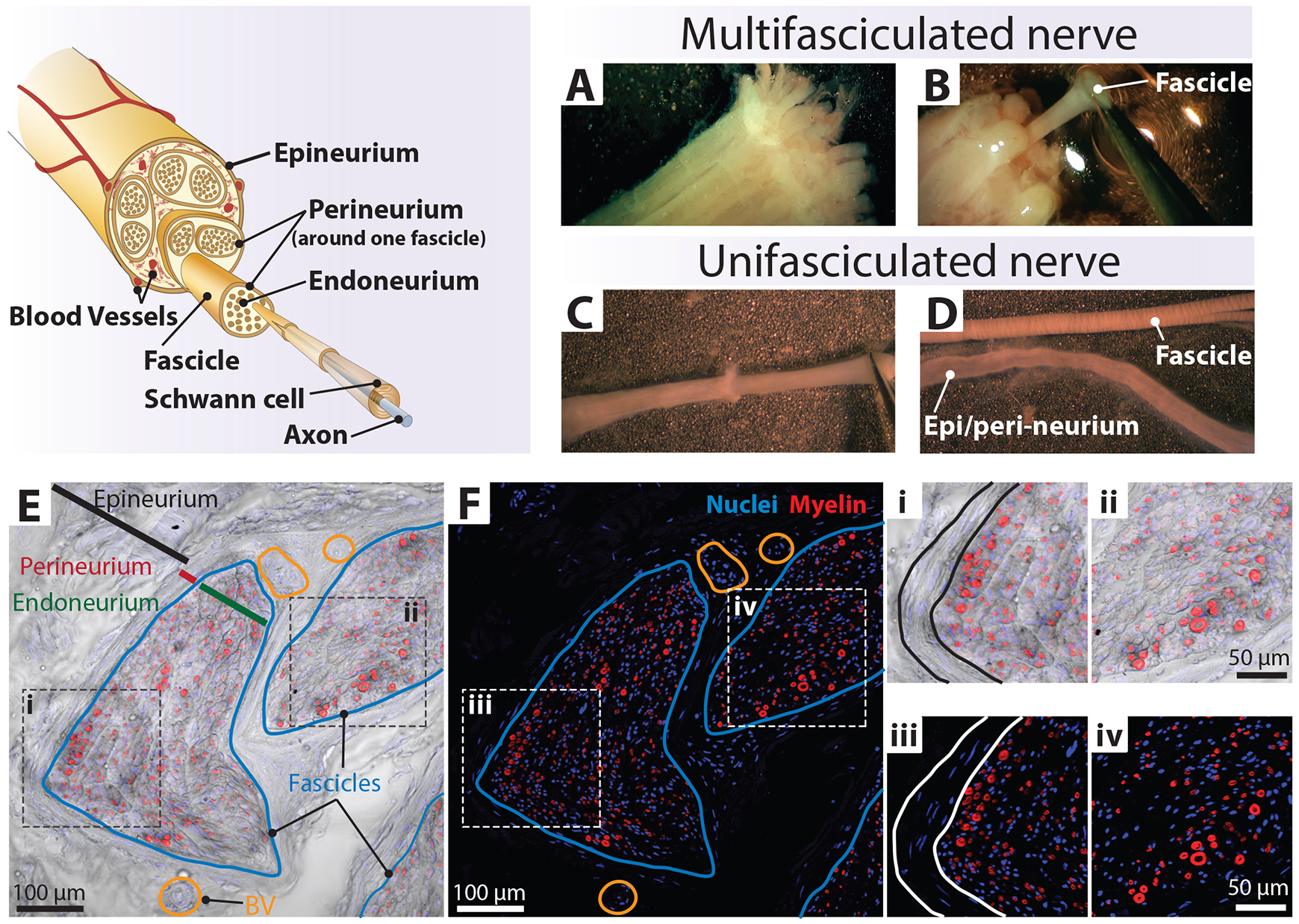
Figure 4. Technique to separate nerve fascicles. Upper-left panel: nerve fascicles are a bundle of fibers [axon–Schwann cell (SC) units] enclosed by the perineurium layer. These bundles are organized into larger bundles surrounded by substantial extracellular matrix (ECM) and enclosed within a continuous membranous layer, the epineurium. Human SC (hSC) cultures are derived from mature myelinating (shown in the diagram) and non-myelinating/Remak (not shown) SCs within the fascicles that become proliferative during the tissue culture step. Upper-right panel: higher magnification views of individual fascicles protruding from the distal end of a nerve segment before (A) and at the time of being detached from the connective tissue layers (B, C). (D) Close-up views of a nerve fascicle exhibiting clearly defined Fontana bands (top) and the corresponding collapsible layers comprising the epineurium and possibly also the perineurium (bottom). Lower panels: (E and F) Low-magnification images of a human sciatic nerve (seen in cross section) labeled with DAPI (nuclei, blue) and FM 4-64FX (myelin, red). The brightfield image was combined with the fluorescence images (F) to visualize the complex organization of the nerve consisting of endoneurium (green line, E), perineurium (red line, E), and epineurium (black line, E). The fascicles (blue line, E–F) are surrounded by multilayered perineurial and epineurial membranes enriched in blood vessels (BV, orange circles). (i–iv) Zoom-in images to highlight myelinating SCs depicting myelin sheaths of various calibers. Section thickness was 20 µm.(Optional) Discard the fascicles with indistinct boundaries, attached ECM, and/or perineurium unless they can be eliminated easily with fine forceps. These layers contain abundant non-glial cells and can introduce a higher-than-acceptable number of fibroblasts into the hSC cultures (see Note d and Figure 5G and 5H).
Proceed as described in Protocol 2A.
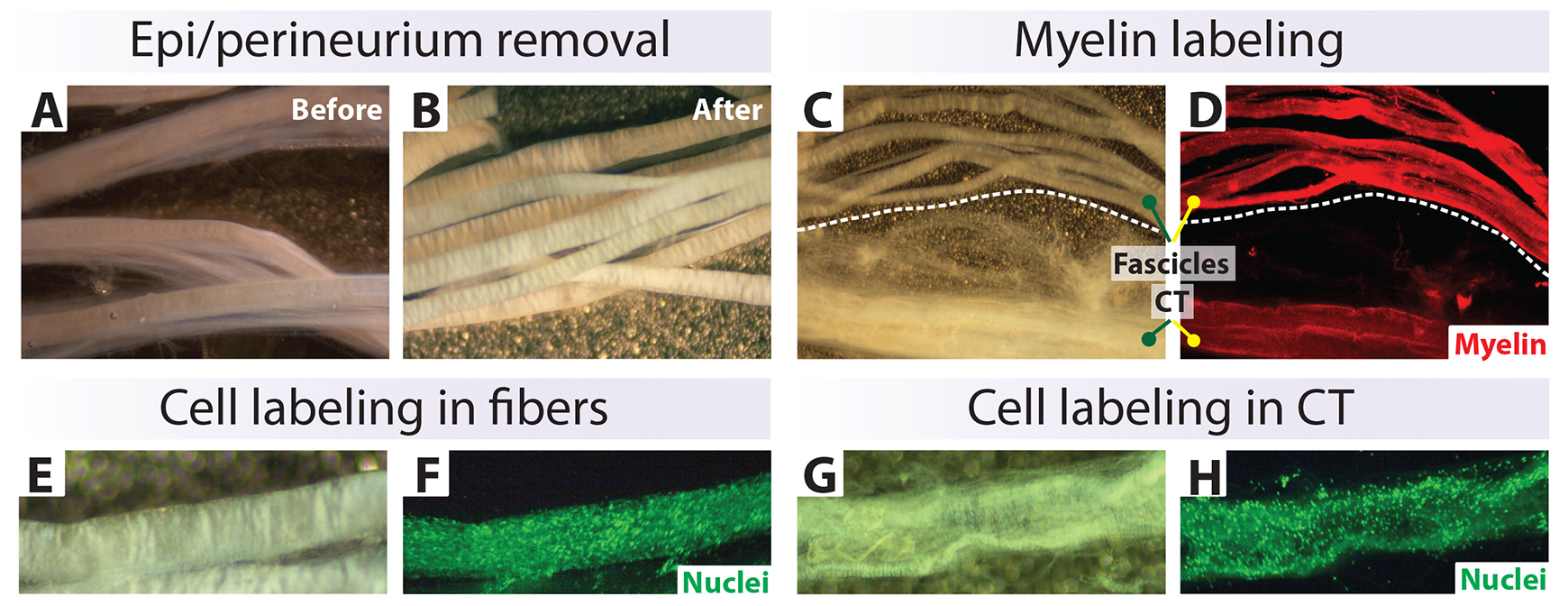
Figure 5. Characterization of nerve biospecimens suitable for cell culture. (A, B) Appearance of fascicles before and after removal of connective tissue (CT). (C–H) Fluorescent labeling of myelin (red) (C, D) and total cell nuclei (green) (F, H) in clean fibers and CT layers. The tissues were stained live using Fluoromyelin Red (D) and Syto-24 (F and H), which detect myelin and cell nuclei, respectively. Cells from the CT can contaminate the hSC cultures if not eliminated from the onset (G, H).
Isolation of nerve roots and ganglia from the adult DRG
Transfer the tissue into a 100 mm dish containing ice-cold DM. Carefully trim off the external connective tissue using a combination of spring scissors and fine forceps. Expose the body of the ganglia and the protruding nerve roots as shown in Figure 6A and 6B.
Transfer the partially cleaned DRG with its attached roots to a new 100 mm dish with ice-cold DM. Using spring scissors, cut away all remaining connective tissue capsule trying not to pinch the roots or the ganglia. Then, cut off the nerve roots by positioning the spring scissors as close as possible to the DRG body. The clean DRG has a creamy-yellowish, spongy appearance with a heart or football shape. The roots have a whiteish appearance (see Note d). If the roots or DRGs have bloody areas, they should be cut away together with any remaining roots, fat, or connective tissue (Figure 6C and 6D).
Transfer the roots and DRG bodies separately to new dishes containing DM. Use fine spring scissors or scalpels (with #11 blades, stainless steel) to slice the nerve roots into smaller 1 mm segments (Figure 6E and 6F). Use the same tools to cut the DRG bodies longitudinally into halves, then fourths, then eighths and so forth up until the segments are approximately 1 mm in diameter. Notice that the individual DRG neurons are observable at low (4×) magnification.
Proceed as described in Protocol 2B.
Notes:
The conditions of nerve harvesting, transfer, and storage for the biospecimens may not be controlled and standardized, but a record for each material should be maintained regarding all relevant variables that can affect the viability of the cells. If tissues are provided by a local surgeon, the surgical team can be provided with enough collection tubes, ready-to-use storage medium, and other materials for expedited tissue transfer.
Prepare an excess of sterile dissection tools for the fast replacement of instrumentation. It is good practice to clean and sterilize the forceps as frequently as possible using a microbead sterilizer (e.g., Germinator) positioned inside the biosafety cabinet. Confirm that the tips of the fine forceps do not become damaged while performing the procedure. Replace distorted forceps as frequently as needed.
Most fascicles inside the collagenous carcass are identifiable by visual observation under the stereomicroscope (Figure 4A and 4B). When attempting to isolate the fascicles, hold each nerve segment with fine forceps at the opposite end from where the fascicles are pulled. Then, grip and pull each fascicle up until no fascicles remain, as per visual inspection, in any of the segments. Note that the number and caliber of individual fascicles recovered from each nerve can vary substantially from donor to donor, even when the nerves are taken from comparable anatomical locations. For instance, the human sural nerve, a nerve from the lower leg, has a variable number of fascicles (usually 3–20 bundles with an average of 10 per nerve) of a range of calibers (Mizia et al., 2014).
The fascicles and nerve roots should exhibit a smooth surface with striated whitish bands (bands of Fontana) and no visibly attached connective tissue (Figures 4D and 5B). The interfascicular epineurium and/or the perineurium can be pulled away easily using #5 forceps if they remain attached to the isolated fascicles. The perineurium is a delicate semi-transparent tube (veil) that surrounds the fascicles, but its presence is not always obvious by inspection under the stereomicroscope.
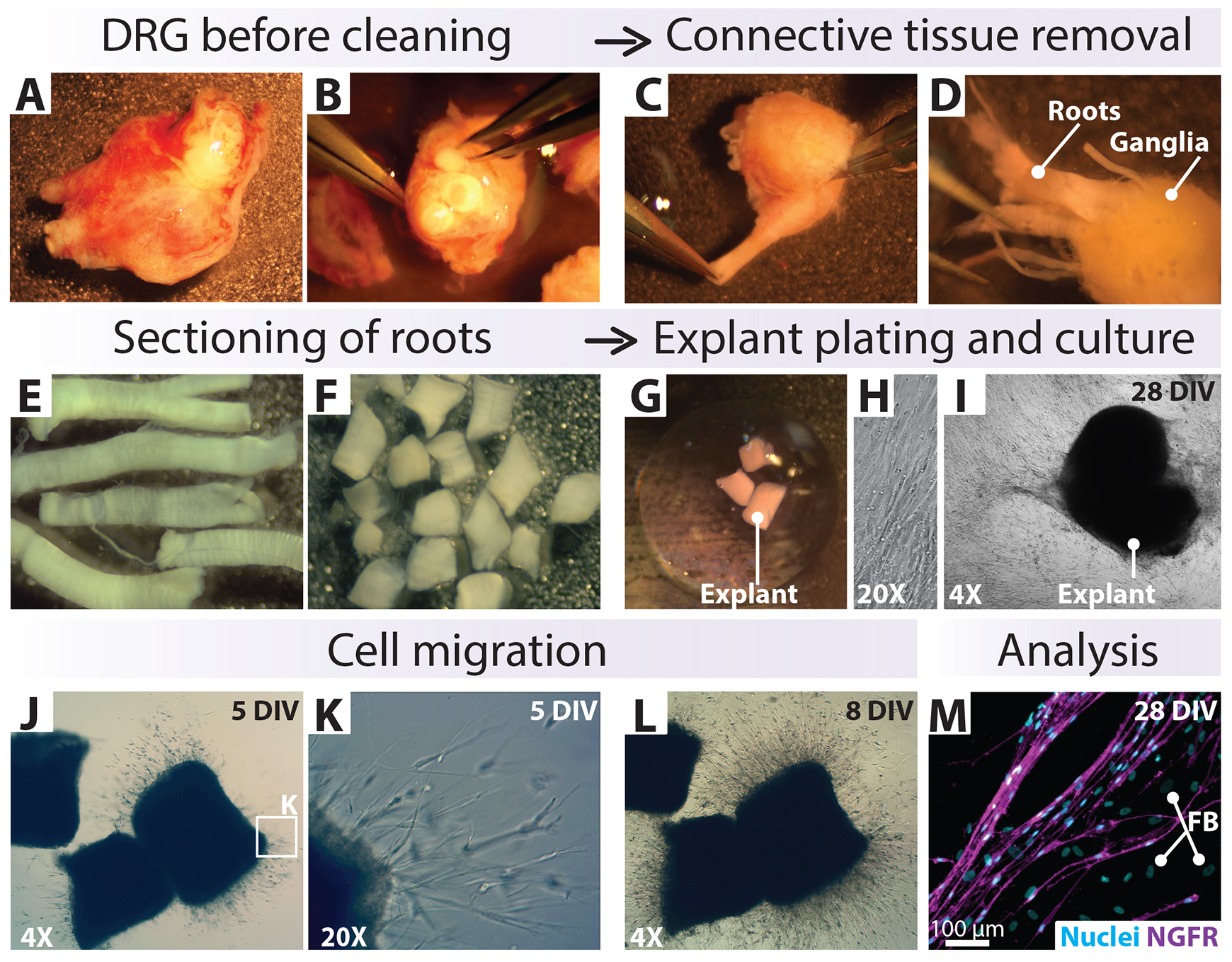
Figure 6. In vitro pre-degeneration of small tissue biospecimens: adherent explants inside a Matrigel drop. (A–D) General view of tissue specimens [dorsal root ganglion (DRG) with attached roots] as obtained from a surgical procedure (A, B), after the removal of external connective tissue (C), and during dissection of the roots (D). (E–G) Harvest of clean nerve roots, sectioning into 1 mm segments (explants), and immersion into a drop of Matrigel inside a culture well. (H–L) Cultured explants, shown at five and eight days post plating (DIV: days in vitro), depict the outgrowth of cells that continue to migrate, proliferate, and cover the available surface in the following weeks. This method can be used for the culture of hSCs from nerve roots (E–M) or ganglia (not shown), with the consideration that the culture conditions do not support the survival of the DRG neurons. (M) NGFR-immunostaining was performed to reveal the cellular constituents of the outgrowth. The cells around the explant comprise NGFR+ human Schwann cells (hSCs, magenta) and NGFR- fibroblasts (FB), as indicated. The cell nuclei were labeled with DAPI (cyan). Tissues were courtesy of Jamie Bradbury.
Protocol 2: Culturing of undissociated tissues
Tissue culturing has been implemented for over three decades to enhance the yields and viability of cell suspensions resulting from enzymatic disintegration of the nerves (Morrissey et al., 1991). Whereas immediate dissociation is best suited for SC isolation from developing nerves, delayed dissociation with an intermediate step of culturing is preferred for adult tissues (Morrissey et al., 1991). This step is often referred to as pre-degeneration due to its resemblance to the process of Wallerian degeneration associated with injury-induced axonal loss and SC activation in vivo (Jessen and Arthur-Farraj, 2019). The culture of undissociated nerves is understood to facilitate the prompt dedifferentiation of the hSCs within their basal lamina tubes while receiving trophic and nutritional support from the culture medium. Early studies revealed that the culturing of human nerve fascicles led to less cellular damage after dissociation (Casella et al., 1996). Even though adult rat nerves can be dissociated immediately after harvesting to generate enough SCs for cell culture (Andersen et al., 2016; Andersen and Monje, 2018), immediate dissociation of adult human nerves often leads to poor cell viability or inconsistent cell yields.
In the following sections, we suggest using distinct pre-degeneration methods according to the size and type of explant tissue. Whereas free-floating explants (Protocol 2A) are most suitable for large tissue specimens (nerves > 1 cm in length), adherent explants (Protocol 2B) are recommended for smaller-sized specimens (e.g., nerve roots or ganglionic tissue). In the former method, individual nerve fascicles are allowed to pre-degenerate while suspended in the culture medium. In the latter method, small pieces of tissue (1–3 mm in length) are immersed inside a droplet of Matrigel to stably attach them to the surface of a culture dish or well. Once the Matrigel matrix becomes a solid gel, culture medium can be flooded around the explant for continued culture. This method renders a population of mixed adherent cell types that migrate out from the explant, including hSCs (Figure 6H–6M).
Free-floating method
Starting with a harvest of nerve fascicles (Figure 3I–3J) and using a sterile transfer pipette, completely aspirate the dissection medium from the periphery of the dish without disturbing the fascicles.
Transfer the fascicles to a new culture dish, well, or flask (according to the volume of tissue) containing HP medium (see Recipe 3) by gently grabbing all the fascicles together using blunt forceps or aspirating them using a pre-wet, wide-bore, glass pipette.
Incubate the fascicles at 37 °C in an 8% CO2 incubator, ensuring they are immersed in medium.
Replace the HP medium on average every three days or as needed, e.g., when the medium turns yellowish (Figure 7A), for the entire pre-degeneration period. Aspirate the medium around the fascicles using a transfer pipette, avoiding the fascicles, and add an equivalent volume of new medium. Cells within the explants are highly active and acidification of the medium is expected (see Notes a and b).
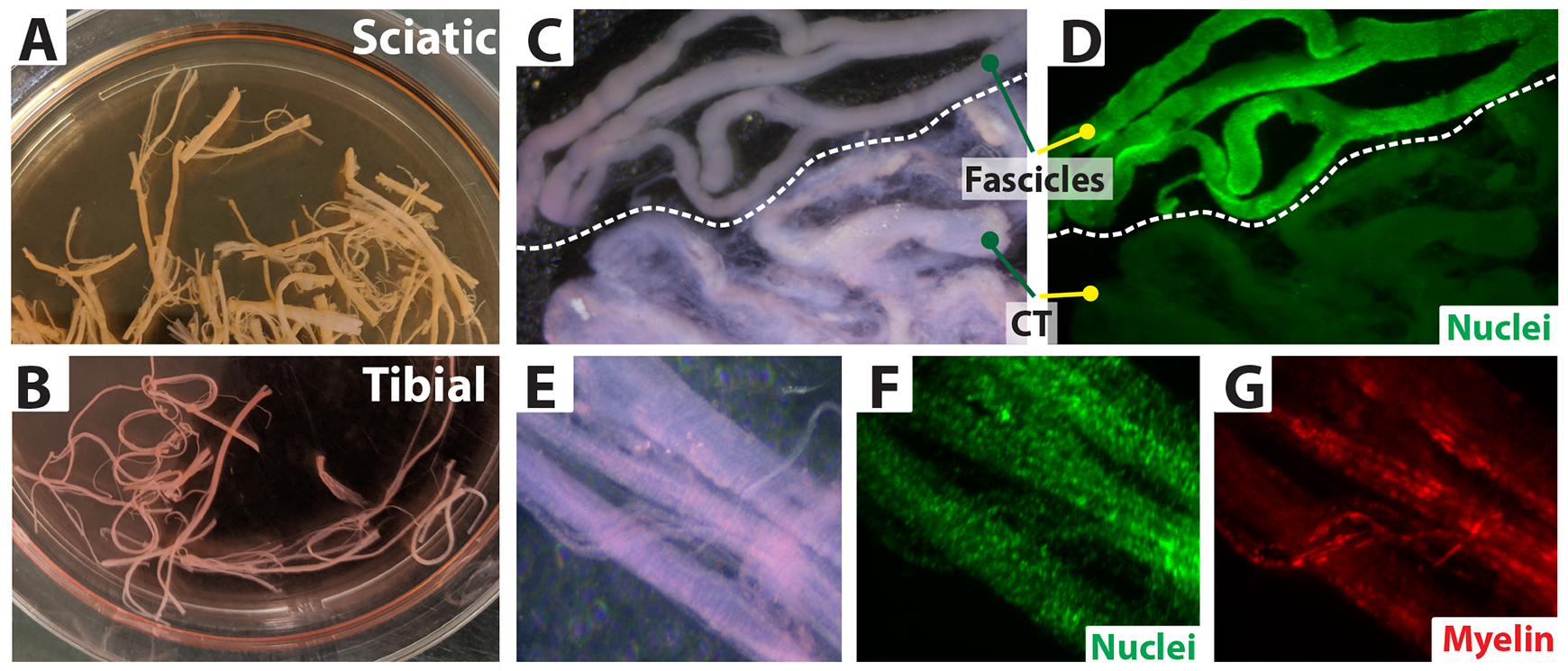
Figure 7. In vitro pre-degeneration of large nerve biospecimens: floating explants. (A–G) Floating (non-attached) spinal nerves (as indicated) are shown at low (A and B) and high magnification (C–G) under white (C, E), blue (D, F), and green light (G) illumination (NIGHTSEA® stereomicroscope). The fascicles were pre-degenerated in HP medium inside a 100 mm dish. In C and D, degenerated fibers are shown next to degenerated connective tissue (CT) from the corresponding nerve segment to reveal the cellular enrichment within the fibers after staining live tissue with Syto-24 (nuclei, green). Pre-degenerated fibers exhibit scattered myelin debris, as shown by staining with FM 4-64FX (G, red). Note that the fibers rather than the CT are filled with cells, indicating that human Schwann cells (hSCs) rather than non-glial cells survive and multiply under our pre-degeneration conditions. The tissue-resident hSCs are highly active metabolically, especially within the first two weeks of culture, leading to quick acidification of the medium, which turns yellow (A). This acidification results from secreted metabolic products and should be distinguished from microbial growth by appropriate tests. The number of dead cells (PI positive) in these samples was negligible (not shown). Tissues shown in this figure were courtesy of Michael Murphy and Kristen Wanczyk.
Adherent explant method
Place 30–40 µL of cold Matrigel matrix in the center of a culture well (e.g., 24-well plate) or a dish placed on ice (see Note c).
Immediately transfer a 1 mm long piece of nerve or ganglia (Protocol 1C) into the matrix using fine forceps, ensuring the matrix embeds the tissue thoroughly (Figure 6G).
Immediately transfer the dish to the CO2 incubator.
Once the Matrigel becomes solid and the tissue explant is secured, use a transfer pipette to gently add culture medium around the explant to fill the dish.
Observe regularly and perform media changes as explained in the free-floating method (see Note d).
Notes:
The length of the pre-degeneration phase can range from one to five weeks. However, we have found that hSCs can remain alive within undissociated fascicles for a much longer time (>3 months) (unpublished). An important control is to assess the content and viability of cells inside pre-degenerated nerves before attempting an enzymatic dissociation (Figure 7D and 7F).
Reducing the size of the fascicles to increase the surface area and more efficiently expose the cells to soluble media components is recommended. By doing so, we expect to expedite the diffusion of the growth factors contained in the medium to act directly on the hSCs to promote their survival and multiplication.
Matrigel may not be needed for secure adhesion of the explants, but it is highly recommended to prevent accidental tissue loss during media changes. Tissue explants substantially shrink during culture and detachment can be problematic when dealing with smaller samples.
After the tissue explant is attached to the Matrigel, the hSCs start to come out of the explant into the surrounding Matrigel, and then into the substrate of the dish, usually within the first week (Figure 6J and 6L). Use precoated dishes with PLL (or equivalent) and laminin to favor the growth of adherent cells in a monolayer (Figure 6H and 6I). The cultures obtained in this manner contain a substantial proportion of fibroblasts (Figure 6M). Attachment of nerve explants to tissue culture plastic was initially used to reduce fibroblast contamination (Morrissey et al., 1991). Here, we use Matrigel as physical support to retain small tissue explants and prevent them from being lost during media changes rather than to eliminate fibroblasts.
Protocol 3: Nerve tissue dissociation, cell plating, and growth
Harvesting sufficient cells from nerve tissues is the most critical step in the culture workflow. Early empirical observations showed that enzymatic dissociation, as opposed to mechanical dissociation, is more appropriate to disorganize the adult nerve tissue (Casella et al., 1996), and that prolonged incubation with a combination of collagenase and neutral protease leads to more consistent cell yields (Casella et al., 1996; Casella et al., 2000). We have confirmed these observations and suggest proceeding with nerve tissue dissociation essentially as shown in the Casella et al. (1996 and 2000) papers (Protocol 3A) with a few modifications for the seeding and growth of the cells. A simple drop plating method (Protocol 3B) where cells are seeded in the form of small-volume discrete droplets onto dishes sequentially coated with PLL and laminin is recommended to increase cell recovery and expedite cell attachment. After testing the effect of various growth factors on hSC proliferation (Monje et al., 2018), we recommend using medium supplemented with serum, heregulin, and forskolin from the onset, as this formulation induces hSC mitogenesis as soon as 1–2 days post plating and maintains the hSCs in an active proliferative state during the initial stages (Protocol 3C) and for several passages. It is expected that the cells plated under these conditions reach confluence within 5–10 days depending on the initial plating density.
Enzymatic dissociation of pre-cultured tissues
Prepare the pre-degenerated fascicles for dissociation by cutting the fibers into smaller 1 cm segments, if needed. Before proceeding with the next steps, inspect the tissue under a dissecting or phase contrast microscope to ensure no connective tissue or debris are attached (Figure 8A).
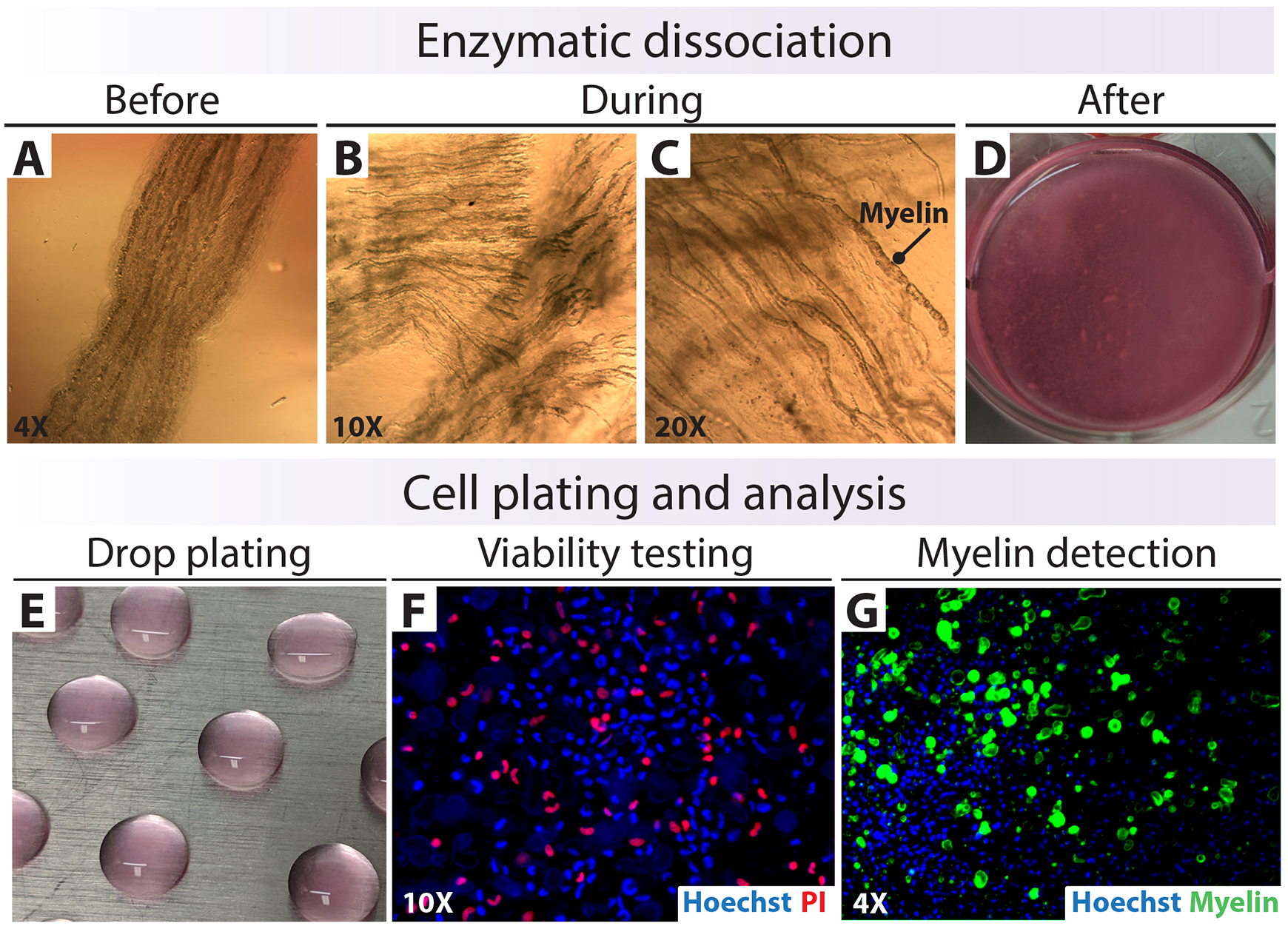
Figure 8. Isolation, plating, and analysis of human Schwann cells (hSC)-enriched suspensions. (A–D) Enzymatic dissociation of nerve fascicles. (E) Drop plating on a Poly-L-lysine (PLL)-laminin substrate. (F and G) Testing for cell viability and myelin contamination right after tissue digestion. The total cell nuclei were labeled with Hoechst (blue), dead cells were labeled with PI (red), and myelin was labeled with Fluoromyelin (FM, green).Remove the culture medium and wash the pre-degenerated fibers once with a large volume of DMEM before adding the 1× enzymatic solution (see Recipe 5). For instance, use 1.5 mL of enzymatic solution to digest the fibers harvested from a 1 cm sural nerve biopsy placed in a 3 cm culture dish. Scale up the volume of enzymatic cocktail to match the volume of tissue and the dimensions of the dish to ensure that the tissue is fully covered by enzymatic solution.
Transfer the fascicles to an incubator set at 37 °C and 8% of CO2 for overnight dissociation (15–18 h) without agitation. Use phase contrast microscopy to monitor the progression of the digestion at regular time points (Figure 8B and 8C) (see Note a).
The next day, transfer the digested explants into a 15 mL snap-cap tube (round-bottom tube) containing 5–10 mL of DMEM medium with 40% FBS to rapidly neutralize the action of the enzymes. Rinse the dish and collect the explants using LP medium (see Recipe 2) until there is nothing left in the dish.
Centrifuge the tissues at 200× g for 8 min at 4 °C to collect the particulate material. Discard the enzymatic solution.
Remove the supernatant manually using a sterile transfer pipette and add 5 mL of LP medium. Alternatively, aspiration can be used with care to avoid disturbing the pellet.
Gently resuspend the cells by pipetting up and down with a wide end bore glass pipette until the tissue disintegrates as observed by visual inspection. Then, add LP medium to a final volume of 15 mL. This suspension should look as shown in Figure 8D. Do not overdo the mechanical disintegration, as excessive passing of the cells through a pipette can increase cell death. Consider that large undigested tissues, if still present, can be removed in subsequent steps.
Repeat step 5 and 6 to create a suspension ready for plating and remove traces of the enzymatic solution. Resuspend the cell pellet gently in the desired volume of HP medium using a narrow-end glass transfer pipette. The cells are ready to be plated as described in Protocol 3B.
(Optional) Set aside a 50–100 µL aliquot of this cell suspension to determine cell counts, viability, and myelin content using nucleic acid- and myelin-specific fluorochromes (Figure 11). For live/dead nuclear labeling, use a combination of the following: (1) Syto-24 (total cells, green) and PI (dead cells, red) (not shown); or (2) Hoescht (total cells, blue under UV light) and PI (red) (Figure 8F). For myelin staining, use Fluoromyelin (red or green) or FM 4-64FX (red) alone or in combination with a nuclear stain (Figure 8G).
Plating of the initial cell suspensions
Confirm under a phase contrast microscope that a refined suspension containing single cells (or small clumps) was obtained in the prior step (Figure 8F and 8G) (see Note b).
Plate the cell suspensions in small drops directly on the surface of a PLL-laminin-coated 100 mm dish. Do so by placing discrete 30–50 µL drops in a regular pattern, as shown in Figure 2 and Figure 8E. The plating density may be difficult to estimate at this stage. For a reference, plate 106 cells in a 100 mm dish considering that one dish can accommodate 20–30 drops (see Note c).
(Optional) Set aside a 100–200 µL aliquot of this cell suspension for plating droplets in a PLL-laminin-coated chamber-slide or multi-well dish. These adherent cells are to be used for quality control analysis of cell viability, purity, and myelin content (Figures 10 and 11).
Carefully transfer the culture dishes to a CO2 incubator for at least 15–18 h (see Note d).
Culture of primary hSCs
The following day, use phase contrast microscopy to confirm that the cells have attached properly. Gently and smoothly fill each 100 mm dish with HP medium to cover the surface of the dish (8–10 mL) without disturbing the cells.
Proceed with normal medium changes by refreshing the HP medium every three days on average. Change the medium slowly without creating a flow of liquid onto the cells as they detach easily at this stage.
(Optional) Include additional washes with 37 °C DMEM or L15 to remove extracellular myelin debris and dead cells, preferably after the second day of plating (Figure 9A and 9B).
Once the cells reach a high enough density (or confluency), they can be used in quality control assessments (Figure 10) or experimentation. Confluent cultures can be considered passage-zero (P0) primary hSCs (see Note e).
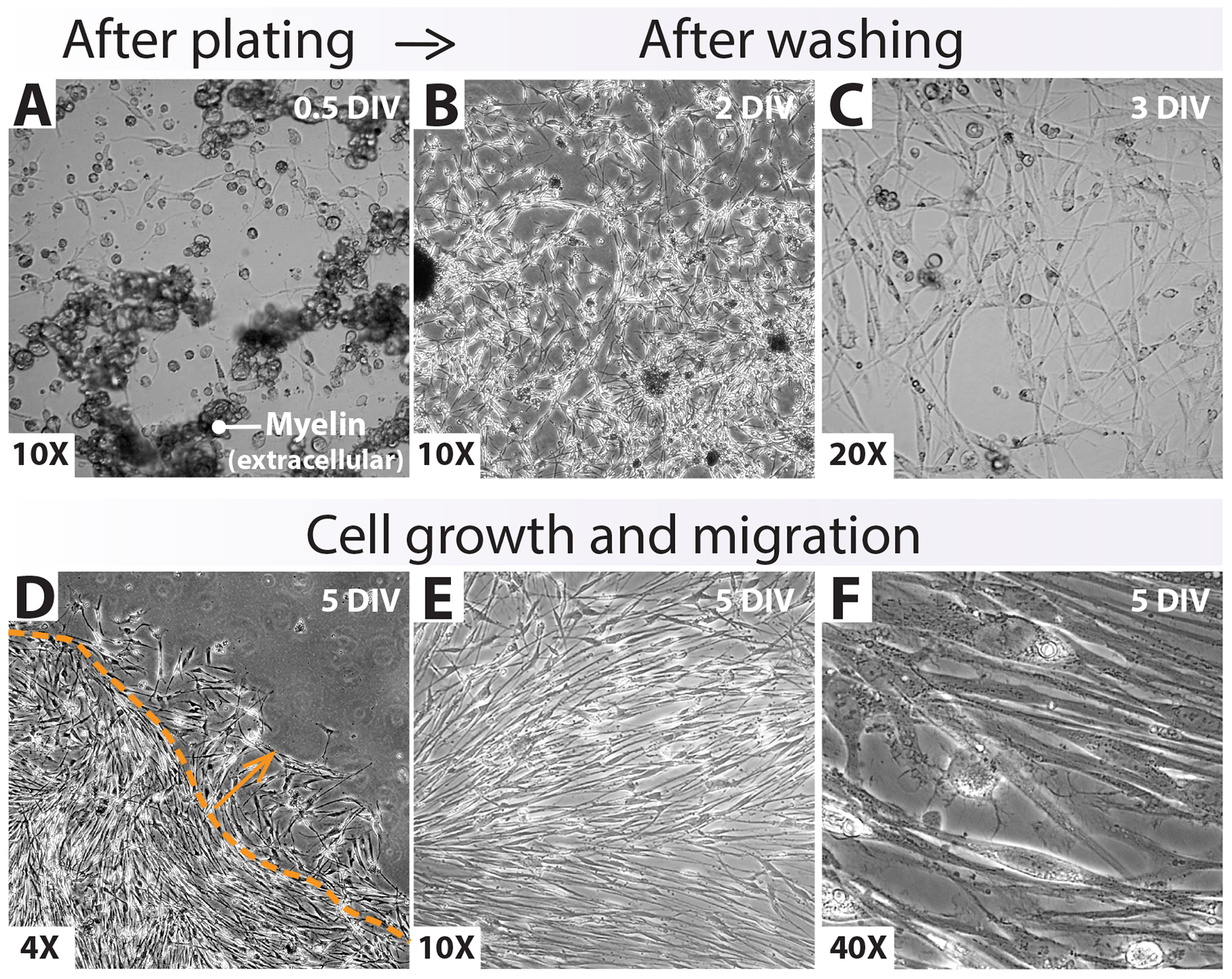
Figure 9. Growth of a drop-plated human Schwann cell (hSC) culture at passage-zero. (A–C) Cell attachment and expansion right after drop plating. Observing abundant myelin debris in the medium (A) and inside the cells (Figure 10) is a normal and expected feature of these early cultures [within three days in vitro (DIV)]. Loosely attached myelin granules (A, extracellular myelin) can be easily removed with media changes or additional washes (B). (D–F) Appearance of the monolayer outgrowth of a nearly confluent hSC culture at 5 DIV. Images were taken within the periphery (D) and the center (E, F) of an individual drop. Cells migrating out of the area initially circumscribed by the drop are denoted by the dotted line and the arrow (D). Cell alignment (E, F) may or may not be evident at this stage.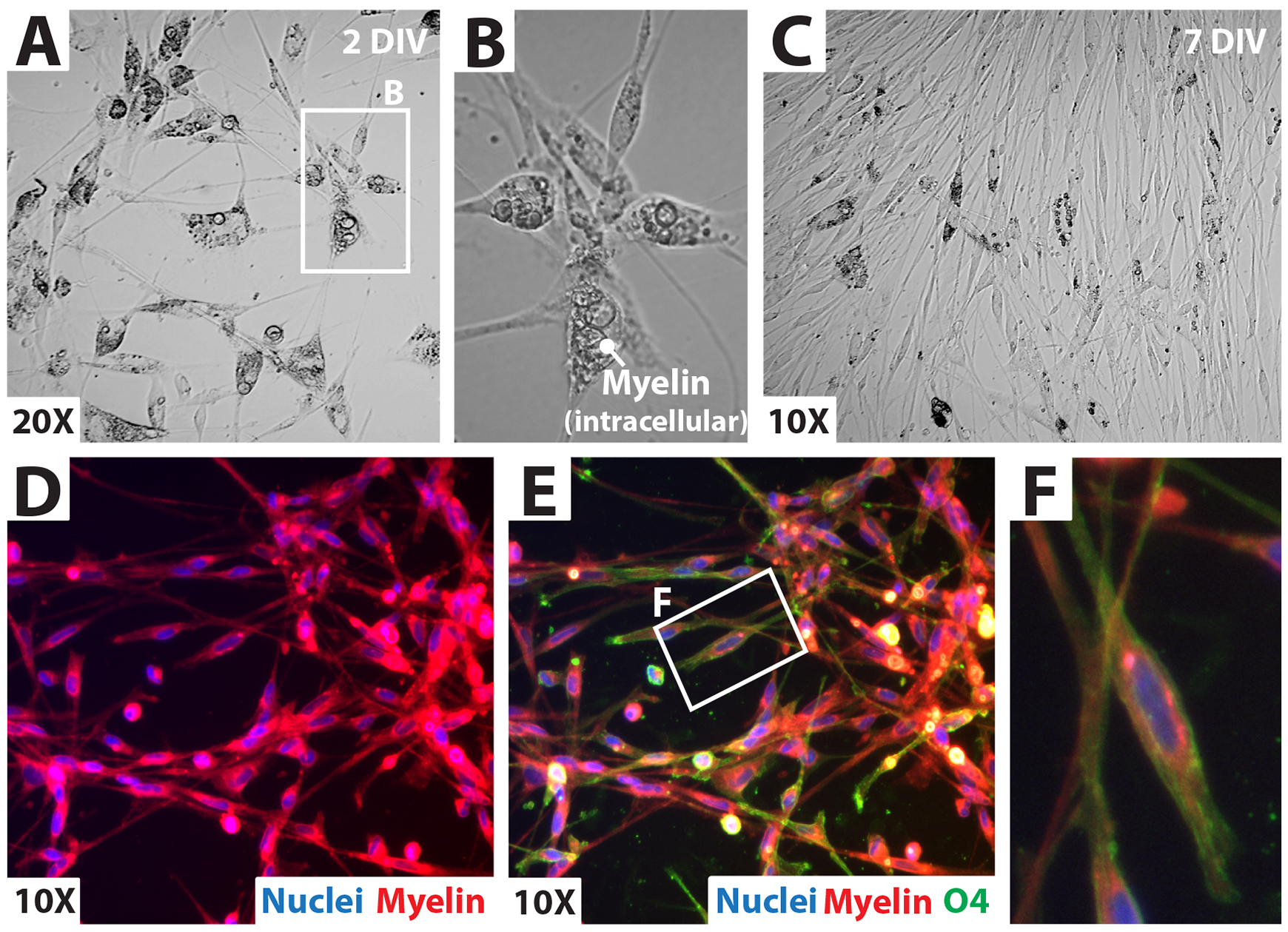
Figure 10. Analysis of a drop-plated human Schwann cell (hSC) culture at passage-zero. (A, B) Phase contrast microscopy showing cell morphology and intracellular myelin content in the cytoplasm of hSCs. Myelin persists for several days. However, granules of larger diameter (B) are no longer appreciated five to seven days post plating due to intracellular degradation (C). (F) Fluorescence microscopy revealing myelin-loaded hSCs using a myelin stain (FM 4-64FX, Myelin, red) in combination with O4 immunolabeling. The hSCs are NGFR+ (see Figure 2) and O4+ (green, E and F) at this stage [5 to 7 days in vitro (DIV)]. O4 staining declines with prolonged culture whereas NGFR persists (Monje et al., 2018). Nuclei were labeled with DAPI (blue).
Notes:
Check the progression of tissue dissociation by phase contrast microscopy 1–2 h after adding the enzymatic cocktail to appreciate the action of the enzymes (Figure 8B and 8C). It is possible to observe individual cells in suspension being released into the medium quite early after the addition of the enzymes. However, the smooth and progressive disassembly of adult nerve tissue is generally achieved after prolonged (15–18 h) incubation. Introducing mechanical steps or agitation is not recommended because it can negatively affect cell viability.
The preparations that result from enzymatic dissociation consist of single cells and cells in small clumps. A variable proportion of undigested tissues, dead cells, and myelin debris of varied granularities are also expected. To prevent further stress on the cells, proceed with the plating even if cellular aggregates and undigested fibers are observed.
Plating the suspensions as small droplets allows SCs to rapidly attach to the surface and separate easily from floating myelin debris (Andersen and Monje, 2018). This method is preferred over plating the cells directly in a large volume of medium (traditional method).
Maintain the cells inside a humidified CO2 incubator set at 37 °C with 8%–9% CO2. Monitor the cultures within 3 h post plating only to verify prompt adhesion. Do not disturb the cultures otherwise. The cells adhere and extend processes quickly onto a PLL-laminin substrate. The morphology of the cells is variable in the initial stages and detach easily if disturbed. Cell yields and purity usually differ in independent isolation experiments.
These cells can be replated and sub-cultured for several passages. Accompanying papers provide information on managing and analyzing both primary and expanded hSCs (Monje, 2023a and 2023b).
Recommendations and troubleshooting
Performing daily microscopic observations of the cultured tissues and cells and introducing proper controls are essential throughout the protocol. Primary hSCs are variable and delicate cultures until they become established. For this reason, this section includes useful conceptual and technical information to help investigators make rational use of the materials and procedures as well as trace and correct problems as they arise (Figure 11).
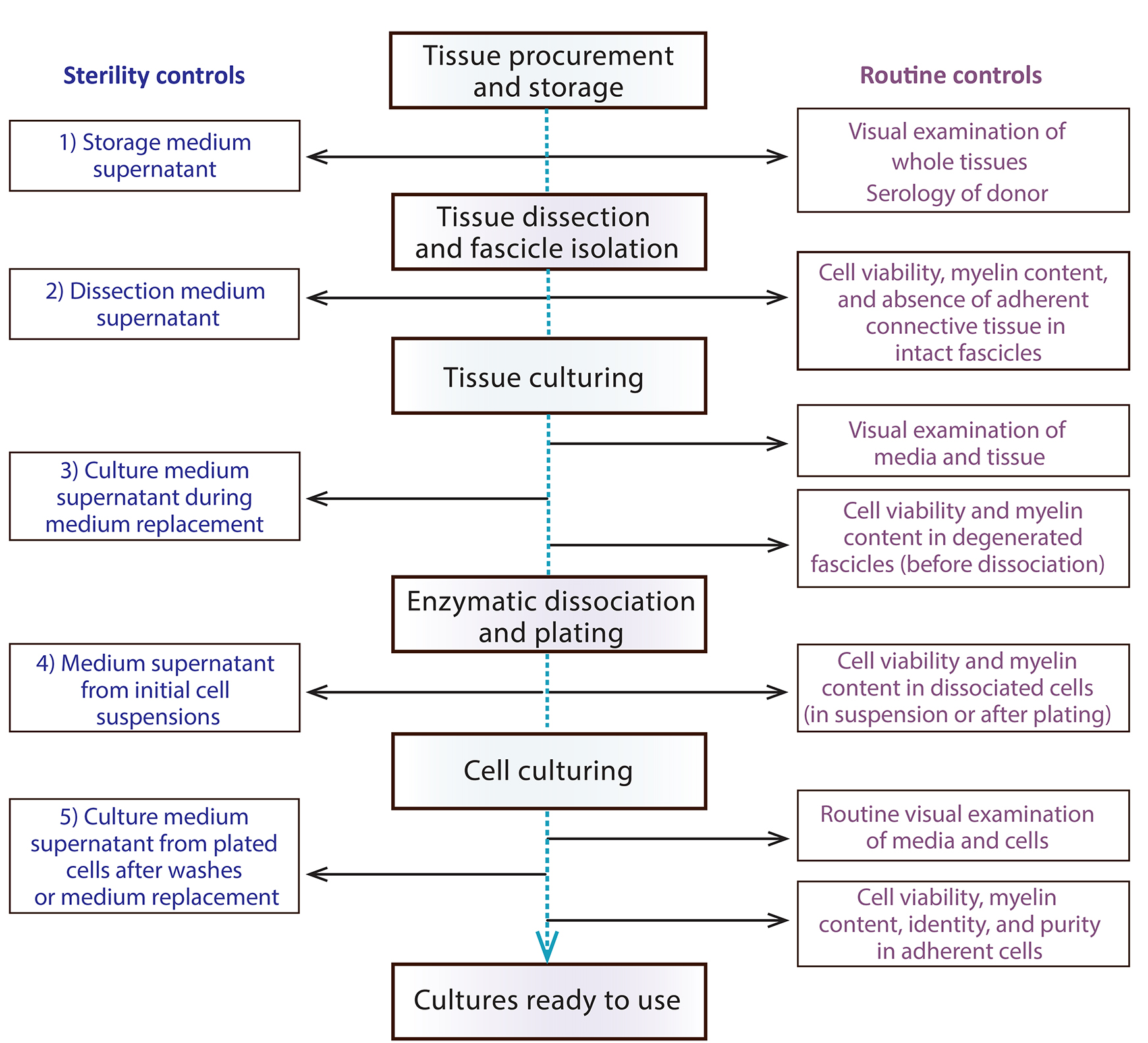
Figure 11. Suggested controls at each step during the culturing of nerve tissues and cells. The diagram depicts an action plan for performing sterility controls (left) and other assessments (right) to rule out microbial contamination and confirm the purity of the primary cell cultures, respectively.
Tissue procurement and use. The protocols for tissue harvesting, transfer, and use in laboratory research must be reviewed and approved by the appropriate institutional committees. Tissues are often furnished to researchers by local medical facilities, organ procurement centers, and tissue banks under proper authorization and transfer agreements that follow institutional guidelines for protecting donor-relevant information. Most biospecimens are deidentified before transfer. However, it is recommended that the recipient laboratory is given access to pertinent information to assess factors impacting the hSC cultures and the operators. Such information may include the date and time of tissue extraction, patients’ demographics, medical condition (or cause of death), and the presence of communicable diseases. Ideally, this information should be made available prior to unpacking the tissue in the laboratory to allow the investigative team to make an informed decision on the suitability of the tissues for cell culture, and/or the need to take additional precautions. Evidence of cancer, peripheral neuropathy, or chronic disease affecting the nerve, such as insulin-dependent diabetes, may be considered an exclusion factor for the derivation of hSC cultures unless the intention is to investigate the properties of hSCs under these conditions. The investigators must decide if they will process tissues from patients who have tested positive for blood-borne pathogens or other communicable diseases. We routinely exclude tissues if we become aware that the donors have tested positive for HIV, hepatitis B, or hepatitis C. Samples from donors that tested positive for other viruses may be processed for cell culturing after being quarantined and labeled appropriately.
Ideally, the culture of the fascicles should be initiated within 24 h following nerve harvesting or 24 h postmortem time. However, the culturing can be delayed for several days if the tissues are stored appropriately at 4 °C. A simple live/dead test (Figures 8 and 11) can suffice to assess both cell content and viability prior to tissue processing. It has been shown that short intervals (< 1 week) of cold storage do not substantially impair the viability and function of SCs obtained from adult human and rat nerves. If the cold storage period is extended to three weeks, the viability declines; however, some cells survive and can be isolated for culturing (Levi et al., 1994). In general, we suggest not to exclude tissues based on time of collection from the body (for autopsy specimens) or the cold storage period (for fresh or autopsy specimens) without empirical verification of cell viability.
Biosafety. Importantly, all human tissues and cells should be managed under the assumption that unknown pathogens are present. Universal precautions should be used, and personnel should be educated in the handling of biohazardous materials. Operators should follow institutional guidelines to work at the appropriate biosafety level and limit occupational exposure to biohazardous materials. Human cells and tissues should be managed inside a certified BL2 biosafety cabinet. Procedures that minimize the creation of aerosols and the transport of potentially hazardous materials should be implemented. Fluids and materials in contact with the human cells/tissues should be labeled, inactivated by autoclaving, and disposed of as biohazards.
Sterility. Implement best practices in the handling of the tissues and cells to maintain sterility. Examine the cultures daily by phase contrast microscopy using low (4×–10×) and high (40×) power objectives to assess the overall health and progression of the cultures and to rule out gross microbial contamination. Routine sterility testing is recommended throughout the main steps but mostly at the onset of culturing (Figure 11). Microbial contamination of the source tissue prior to arrival to the laboratory and/or during the dissection procedure is a likely cause of contamination of the cell cultures. Once the cultures become contaminated, they should be discarded. We have a simple method in place for microbial testing using liquid cultures in Luria-Bertani (LB) broth. For this, we inoculate 10 mL of LB medium with approximately 50 µL aliquots of the cell/tissue supernatants from the media or buffers used for tissue storage, dissection, pre-degeneration, and culturing along with aliquots of the unused media stocks. These LB cultures, grown at 37 °C for at least five days, can allow the source of microbial contamination to be traced before it arises in the cell/tissue cultures. This method is useful but has limitations. For more reliable testing of banked cell cultures, we outsource samples to an analytical lab that conducts testing for the presence of bacteria (aerobic, anaerobic), fungi, and mycoplasma (culture-based methods). We do not usually conduct routine mycoplasma testing in primary cultures used in basic in vitro cell research unless the cells are banked. Detection by fluorescence microscopy and PCR (commercial kits) are useful for such purposes.
Culture media formulation and use. Adding heregulin and forskolin is recommended but not essential for the growth and survival of primary hSCs. Medium containing FBS at 10%–15% can be used for plating the cell suspensions when chemical mitogens are unavailable or undesirable experimentally, with the caveat that these conditions do not lead to substantial hSC proliferation; instead, they exacerbate the propagation of fibroblasts. Researchers may optimize their culture media formulation. Serum-free or low-serum media have been used by other groups (Haastert et al., 2007; Aghayan et al., 2012; Weiss et al., 2018). We do not generally recommend the removal of serum unless necessary experimentally.
Regarding the feeding schedule, we recommend feeding twice a week on average using 10 mL of medium for a 100 mm dish, or the volume that matches the size of the dish. To feed the cultures, remove the medium carefully from the side and gently add new medium (previously warmed at a 37 °C water bath) to the side to prevent turbulence on the surface, as this can lead to cell detachment. To prevent alkalization of the media, do not leave the cells and the culture media stocks open in the biosafety cabinet for long periods. The viability of cultured hSCs is impaired when exposed to alkaline media or buffers, even for short periods. Use properly calibrated solutions and follow best methodological practices to prevent sudden pH changes. If available, use phenol red–containing media to visually monitor and correct fluctuations in pH. One consideration is that phenol red can have potential estrogenic effects (Berthois et al., 1986) and redox activity (Morgan et al., 2019) and may be avoided in cell culture. Nevertheless, our empirical observations have shown that hSCs can be cultured equally well in the presence or absence of phenol red.
Substrate requirements. Freshly plated and passage-zero hSC cultures are prone to detachment, which can be reduced by (re)plating the hSCs on PLL-laminin. Prepare PLL-laminin-coated dishes as suggested in this protocol and use them preferably within 1–3 days post coating. We cannot recommend commercial dishes that provide a good substrate for hSCs (empirical data); however, other substrates (or matrices) may be used if hSC adhesion and proliferation are confirmed empirically (Vleggeert-Lankamp et al., 2004).
Myelin and other impurities. The myelin content in intact and pre-degenerated nerves varies substantially from donor to donor and even from fascicle to fascicle within a nerve (Figure 4E and 4F). Use HP rather than LP medium for more efficient myelin clearance by hSCs within the fascicle explants (pre-degeneration) and after plating (Casella et al., 1996). While the presence of myelin can be reduced over time, it is rarely eliminated during the pre-degeneration phase, even with prolonged incubation (>1 month). This is a distinctive feature of nerves obtained from humans (Aparicio and Monje, unpublished).
Myelin interferes with the visualization of cells after enzymatic digestion of the fascicles. Notice that standard phase contrast microscopy is inaccurate for this purpose. A practical way to estimate the number of cells in freshly isolated preparations is to stain them with vital fluorophores and count fluorescent nuclei instead of whole cells using a Neubauer hemocytometer or an appropriate automated device. The discrimination between live/dead cells is best accomplished by co-staining the preparations with a combination of green (e.g., Syto-24) and red (e.g., PI) fluorophores to prevent the interference of myelin autofluorescence in the UV channel. The Hoescht/PI combination is useful in plated preparations where the myelin floats (Figure 8F and 8G).
In addition to myelin debris, the initial cell suspensions prepared directly from tissues contain abundant dead cells, membranous vesicles, and other impurities intermixed with the cells. Passage-zero hSC cultures may contain myelin debris floating in the medium or loosely attached to the surface of the cells (Figure 9). A proportion of the hSCs contain cytoplasmic myelin granules of various sizes that are degraded quickly after the hSCs are plated on a substrate (Figure 9A–9C, Figure 10A–10C). Intracellular and extracellular myelin from the original tissue is drastically reduced approximately one week post plating. However, smaller-sized myelin granules and lipid droplets may persist in the cytoplasm of hSCs for some time. The extent of myelin contamination in each hSC preparation is variable, as determined by the type of nerve and the level of pre-degeneration. Nevertheless, the presence of myelin does not impair the viability or the proliferation of the cultured hSCs. A trained experimentalist can easily discriminate myelin contamination from microbial growth. If this discrimination is doubtful, perform appropriate sterility tests.
Management of tissues and cultured cells. The enzymatic dissociation of the nerve fibers is the most important step that affects cell yields and viability. The temporal course of dissociation must be controlled for each preparation, particularly if mature nerves with fully developed connective tissue layers are used. Increasing the concentration of the enzymes and teasing the fibers with fine forceps, as described in Andersen and Monje (2018) are two distinct ways to disaggregate the tissue more expeditiously with the caveat of increased cell death. Experimenters should always optimize the dissociation step by testing the viability of the cells throughout the course of digestion (Figure 8B and 8C).
Primary hSCs can be plated in their final dishes for direct experimentation. However, these cells proliferate at a high rate in the presence of HP medium (with a duplication time of < 2 days) and the appearance of the cultures changes daily. If cells are plated as drops, we recommend using the cells in experimentation once they reach confluency within the surface area initially delimited by each individual drop (Figure 9D and 9E). Over-confluent cultures may show areas with frequent SC clusters. Use the hSC cultures as soon as signs of migration and clustering in local areas become evident. If the drops are plated initially in a 100 mm plate, these cells can be re-plated into a new 100 mm plate, or the equivalent surface area of a multi-well dish, for recovery. One limitation of our method is the variable cell yields per isolation experiment (see Donor variability). hSC purity is usually high (>80%) under these conditions.
Identification and analysis of cultured cells. Passage-zero hSCs are highly active and migratory. Even though normal hSCs tend to grow in monolayer, the primary cells overlap their processes and rarely align with one another to form fingerprint-like patterns on the surface, as is expected of established SC cultures from rodents and humans. SCs and fibroblasts are not easily distinguishable at this stage. Therefore, researchers should use immunological methods to confirm the composition of the cultures. Additional relevant controls are to estimate the proportion of live/dead cells (Figure 8) and the content of myelin debris (Figure 10) by fluorescence microscopy. We have previously reported detailed methods for purity and viability assessments (Ravelo et al., 2018; Peng et al., 2020); thus, here we have simply provided examples of typical results in Figure 2 (NGFR staining), Figure 10 (O4 staining), and Figure 8 (viability). Researchers may consider using image analysis software such as ImageJ to quantitatively assess purity, viability, and myelin contamination from adherent cells or cells in suspension. If intracellular myelin is confirmed in a proportion of the cells, it can be used as a criterion for hSC identification, as myelin engulfment is a specific property of the SCs. Fibroblasts do not normally engulf myelin fragments, and macrophages are rarely seen under these experimental conditions.
Minimal manipulation. Primary cells are considered more reliable model systems than established cultures or cell lines because they more closely reflect the phenotype present in the original tissue. However, maintaining this phenotype in vitro is a challenge. It has been shown that the characteristics of hSCs change with passaging, but the extent to which in vitro–cultured hSCs diverge from those in the nerves is unknown thus far. Therefore, limit the time of exposure to mitogenic factors and the rounds of expansion if the expectation is to generate a product with as little manipulation as possible. The protocols described herein have favored consistency in cell isolation and growth over time in vitro and exposure to mitogens. Other protocols emphasize minimal manipulation to produce hSCs from normal nerves and schwannomas (Dilwali et al., 2014).
Donor variability. One general caveat of patient-derived cultures is that the quality of the populations varies from donor to donor and batch to batch for reasons that are seldom clear to the investigators. This variability may be difficult to control but cannot be ignored when evaluating outcomes from in vitro and in vivo experiments. The heterogeneity of hSC cultures and the divergence from the cells of origin may be problematic if the cultured cells are intended to represent endogenous hSCs from patients. Factors as diverse as the donors' genetic background and medical history, the conditions of tissue procurement, and cell type selection in culture may contribute to batch variability [reviewed in Monje (2020)]. Having accurate records of the history of the tissues and the cells may help explain variation. If possible, derive cultures from different donors and use those to simultaneously compare the responses of cells from different batches before making generalizable conclusions on the effect of experimental variables.
Recipes
Dissection medium (DM)
Reagent Final concentration Amount L15 medium n/a 499.5 mL Gentamycin (1,000×) 1× 0.5 mL Total 500 mL Low proliferation medium (LP)
Reagent [Stock concentration] Final concentration Amount DMEM (or DMEM/F12) n/a 445 mL FBS (100%) 10% 50 mL GlutaMAX (100×) 1× 5 mL Gentamycin (1,000×) 1× 0.5 mL Total n/a 500 mL Note: Adjust the pH of this and other culture media to be 7 or lower (up to 6), sterilize by filtering through a 0.22 µm filter, and maintain at 4 °C.
High proliferation medium (HP)
Reagent [Stock concentration] Final concentration Amount Low proliferation medium n/a 500 mL Heregulin-β1177-244 (25 µM) 10 nM 200 µL Forskolin (15 mM) 2 µM 69.25 µL Total 500 mL Note: Use this medium preferably within a month of preparation and re-filter if needed. Do not freeze.
Laminin coating solution
Reagent [Stock concentration] Final concentration Amount Laminin (1 mg/mL) 15.6 µg/µL 78 µL DPBS n/a 5 mL Total 5 mL Note: This recipe coats one 100 mm cell culture dish (growth area = 78 cm2). We use 1 µg of laminin/cm2 of coating surface for the growth of hSCs. See our paper for the PLL-laminin coating protocol (Ravelo et al., 2018).
10× enzymatic solution
Reagent [Stock concentration] Final concentration Amount Dispase II (powder) 25 mg/mL 125 mg Type I collagenase (powder) 5 mg/mL 25 mg DMEM n/a 5 mL Total 5 mL Note: Prepare the 10× enzymatic solution as indicated above and sterilize by filtration using a 0.22 µm filter. Aliquot and store this solution at -80 °C for up to one year. To prepare the 1× working solution thaw the 10× aliquots on ice and use DMEM to bring the concentration of enzymes to 1×. Do not add antibiotics.
Acknowledgments
We recognize Ketty Bacallao and Natalia Andersen for expert technical assistance, and Patrick Wood and Mary Bunge for their valuable mentoring. Michael Murphy, Kristen Wanczyk, Jamie Bradbury, Greg Gerhardt, Craig van Horne, and George Quintero contributed tissue specimens via institutional Materials Transfer Agreements. Former colleagues from the University of Miami (UM), Yelena Pressman, Linda White, Anna Gomez, Gagani Athauda, Peggy Bates, Aisha Khan, Adriana Brooks, Risset Silvera, Maxwell Donaldson, and James Guest are kindly acknowledged for their valuable input over many years of shared work. The protocols described here were developed and optimized during a >10-year period supported by funding (to P.V.M.) from the National Institutes of Health NIH-NINDS (NS084326), the Craig H Neilsen Foundation, The Miami Project to Cure Paralysis and the Buoniconti Foundation from the University of Miami, and the Indiana State Department of Health (grants 33997 and 43547). G.I.A received support from the International Society for Neurochemistry-Committee for Aid and Education in Neurochemistry (ISN-CAEN), the Fulbright, Bunge & Born, Williams Foundations (program 2020–2021), and CONICET-Argentina. P.V.M. and G.I.A. received support from the Department of Neurosurgery at the University of Kentucky. The contents of this article are the authors' responsibility and do not necessarily represent the official views of the funding agencies. We are grateful to Lingxiao Deng for fruitful scientific discussions and lab support. We thank Beth Ansel for critical review, Valeria Nogueira for illustrations, and Louise Pay for English editing. We are greatly indebted to the generosity of the anonymous patients and their families for donating tissues for research.
Competing interests
P.V.M. is the founder of GliaBio LLC, a consulting company focusing on glial cell research. The authors declare that the research described here was conducted without commercial or financial relationships that could be construed as a potential conflict of interest.
Author’s contributions: P.V.M.: conceptualization, experimentation, data acquisition and analysis, manuscript writing, figure preparation, funding. G.I.A.: experimentation, data acquisition, manuscript writing, figure preparation.
Ethical considerations
Experimentation with human tissues and cells was deemed to constitute non-human subjects research by the Human Subjects Research Offices of the University of Miami (2003–2018) and Indiana University (2018–2022). Procedures were further reviewed and approved by the Institutional Biosafety Committee of Indiana University. Human tissues were acquired from multiple sources, as follows. Michael Murphy and Jamie Bradbury (Indiana University) contributed deidentified nerves and ganglia from surgical remains. Craig van Horne (University of Kentucky) contributed deidentified sural nerve biopsies from clinical trial participants. The National Disease Research Interchange (NDRI) provided cadaveric sural and sciatic nerves under an institutional agreement with P.V.M while affiliated to U.M. Protocols for tissue procurement and sharing by the provider scientists were approved by the respective institutional review boards. Experiments were conducted using non-pathological tissues from males and females as made available from the provider scientists or organ procurement centers without regard to the gender or age of the donors. Requests for information and materials (cells, antibodies, and other materials) should be directed to P.V.M. Investigators are encouraged to contact P.V.M. for feedback on the reported protocols.
References
- Aghayan, H. R., Arjmand, B., Norouzi-Javidan, A., Saberi, H., Soleimani, M., Tavakoli, S. A., Khodadadi, A., Tirgar, N. and Mohammadi-Jahani, F. (2012). Clinical grade cultivation of human Schwann cell, by the using of human autologous serum instead of fetal bovine serum and without growth factors. Cell Tissue Bank 13(2): 281-285.
- Andersen, N. D. and Monje, P. V. (2018). Isolation, Culture, and Cryopreservation of Adult Rodent Schwann Cells Derived from Immediately Dissociated Teased Fibers. Methods Mol Biol 1739: 49-66.
- Andersen, N., Srinivas, S., Piñero, G. and Monje, P. V. (2016). A rapid and versatile method for the isolation, purification and cryogenic storage of Schwann cells from adult rodent nerves. Sci Rep 6: 31781.
- Anderson, K. D., Guest, J. D., Dietrich, W. D., Bunge, M. B., Curiel, R., Dididze, M., Green, B. A., Khan, A., Pearse, D. D., Saraf-Lavi, E., et al. (2017). Safety of Autologous Human Schwann Cell Transplantation in Subacute Thoracic Spinal Cord Injury. J Neurotrauma 34(21): 2950-2963.
- Bastidas, J., Athauda, G., De La Cruz, G., Chan, W. M., Golshani, R., Berrocal, Y., Henao, M., Lalwani, A., Mannoji, C., Assi, M., et al. (2017). Human Schwann cells exhibit long-term cell survival, are not tumorigenic and promote repair when transplanted into the contused spinal cord. Glia 65(8): 1278-1301.
- Berthois, Y., Katzenellenbogen, J. A. and Katzenellenbogen, B. S. (1986). Phenol red in tissue culture media is a weak estrogen: implications concerning the study of estrogen-responsive cells in culture. Proc Natl Acad Sci U S A 83(8): 2496-2500.
- Boyer, P. J., Tuite, G. F., Dauser, R. C., Muraszko, K. M., Tennekoon, G. I. and Rutkowski, J. L. (1994). Sources of human Schwann cells and the influence of donor age. Exp Neurol 130(1): 53-55.
- Bunge, M. B., Monje, P. V., Khan, A. and Wood, P. M. (2017). From transplanting Schwann cells in experimental rat spinal cord injury to their transplantation into human injured spinal cord in clinical trials. Prog Brain Res 231: 107-133.
- Casella, G. T., Bunge, R. P. and Wood, P. M. (1996). Improved method for harvesting human Schwann cells from mature peripheral nerve and expansion in vitro. Glia 17(4): 327-338.
- Casella, G. T., Wieser, R., Bunge, R. P., Margitich, I. S., Katz, J., Olson, L. and Wood, P. M. (2000). Density dependent regulation of human Schwann cell proliferation. Glia 30(2): 165-177.
- Chu, T. H., Baral, K., Labit, E., Rosin, N., Sinha, S., Umansky, D., Alzahrani, S., Rancourt, D., Biernaskie, J. and Midha, R. (2022). Comparison of human skin- and nerve-derived Schwann cells reveals many similarities and subtle genomic and functional differences. Glia 70(11): 2131-2156.
- Dilwali, S., Patel, P. B., Roberts, D. S., Basinsky, G. M., Harris, G. J., Emerick, K. S. and Stankovic, K. M. (2014). Primary culture of human Schwann and schwannoma cells: improved and simplified protocol. Hear Res 315: 25-33.
- Haastert, K., Mauritz, C., Chaturvedi, S. and Grothe, C. (2007). Human and rat adult Schwann cell cultures: fast and efficient enrichment and highly effective non-viral transfection protocol. Nat Protoc 2(1): 99-104.
- Guest, J., Santamaria, A. J. and Benavides, F. D. (2013). Clinical translation of autologous Schwann cell transplantation for the treatment of spinal cord injury. Curr Opin Organ Transplant 18(6): 682-689.
- Jessen, K. R. and Arthur-Farraj, P. (2019). Repair Schwann cell update: Adaptive reprogramming, EMT, and stemness in regenerating nerves. Glia 67(3): 421-437.
- Jessen, K. R., Mirsky, R. and Lloyd, A. C. (2015). Schwann Cells: Development and Role in Nerve Repair. Cold Spring Harb Perspect Biol 7(7): a020487.
- Khan, A., Diaz, A., Brooks, A. E., Burks, S. S., Athauda, G., Wood, P., Lee, Y. S., Silvera, R., Donaldson, M., Pressman, Y., et al. (2021). Scalable culture techniques to generate large numbers of purified human Schwann cells for clinical trials in human spinal cord and peripheral nerve injuries. J Neurosurg Spine: 1-10.
- Levi, A. D., Evans, P. J., Mackinnon, S.E., Bunge, R.P. (1994). Cold storage of peripheral nerves: an in vitro assay of cell viability and function. Glia 10(2): 121-131
- Levi, A. D., Burks, S. S., Anderson, K. D., Dididze, M., Khan, A. and Dietrich, W. D. (2016). The Use of Autologous Schwann Cells to Supplement Sciatic Nerve Repair With a Large Gap: First in Human Experience. Cell Transplant 25(7): 1395-1403.
- Mizia, E., Tomaszewski, K. A., Rutowicz, B., Konopka, T., Pasternak, A. and Walocha, J. A. (2014). Computer-assisted assessment of the histological structure of the human sural nerve. Folia Morphol (Warsz) 73(3): 292-297.
- Monje, P. V. (2020). The properties of human Schwann cells: Lessons from in vitro culture and transplantation studies. Glia 68(4): 797-810.
- Monje, P. V., Athauda, G. and Wood, P. M. (2008). Protein kinase A-mediated gating of neuregulin-dependent ErbB2-ErbB3 activation underlies the synergistic action of cAMP on Schwann cell proliferation. J Biol Chem 283(49): 34087-34100.
- Monje, P. V., Bartlett Bunge, M. and Wood, P. M. (2006). Cyclic AMP synergistically enhances neuregulin-dependent ERK and Akt activation and cell cycle progression in Schwann cells. Glia 53(6): 649-659.
- Monje, P. V., Deng, L. and Xu, X. M. (2021). Human Schwann Cell Transplantation for Spinal Cord Injury: Prospects and Challenges in Translational Medicine. Front Cell Neurosci 15: 690894.
- Monje, P. V., Sant, D. and Wang, G. (2018). Phenotypic and Functional Characteristics of Human Schwann Cells as Revealed by Cell-Based Assays and RNA-SEQ. Mol Neurobiol 55(8): 6637-6660.
- Monje, P. V. (2023a). Human Schwann cells in vitro II. Passaging, purification, banking and labeling of established cultures. Bio Protoc 13(22): e4882.
- Monje, P. V. (2023b). Human Schwann cells in vitro III. Analytical Methods and a Practical Approach for Quality Control. Bio Protoc 13(22): e4840.
- Morgan, A., Babu, D., Reiz, B., Whittal, R., Suh, L. Y. K. and Siraki, A. G. (2019). Caution for the routine use of phenol red - It is more than just a pH indicator. Chem Biol Interact 310: 108739.
- Morrissey, T. K., Kleitman, N. and Bunge, R. P. (1991). Isolation and functional characterization of Schwann cells derived from adult peripheral nerve. J Neurosci 11(8): 2433-2442.
- Morrissey, T. K., Kleitman, N. and Bunge, R. P. (1995). Human Schwann cells in vitro. II. Myelination of sensory axons following extensive purification and heregulin-induced expansion. J Neurobiol 28(2): 190-201.
- Peng, K., Sant, D., Andersen, N., Silvera, R., Camarena, V., Pinero, G., Graham, R., Khan, A., Xu, X. M., Wang, G. and Monje, P. V. (2020). Magnetic separation of peripheral nerve-resident cells underscores key molecular features of human Schwann cells and fibroblasts: an immunochemical and transcriptomics approach. Sci Rep 10(1): 18433.
- Ravelo, K. M., Andersen, N. D. and Monje, P. V. (2018). Magnetic-Activated Cell Sorting for the Fast and Efficient Separation of Human and Rodent Schwann Cells from Mixed Cell Populations. Methods Mol Biol 1739: 87-109.
- Scarpini, E., Kreider, B. Q., Lisak, R. P. (1988). Cultures of human Schwann cells isolated from fetal nerves. Brain Res. 440(2): 261-266.
- Stratton, J. A., Kumar, R., Sinha, S., Shah, P., Stykel, M., Shapira, Y., Midha, R. and Biernaskie, J. (2017). Purification and Characterization of Schwann Cells from Adult Human Skin and Nerve. eNeuro 4(3): ENEURO.0307-16.2017.
- Vallejo, F. A., Diaz, A., Errante, E. L., Smartz, T., Khan, A., Silvera, R., Brooks, A. E., Lee, Y. S., Burks, S. S. and Levi, A. D. (2022). Systematic review of the therapeutic use of Schwann cells in the repair of peripheral nerve injuries: Advancements from animal studies to clinical trials. Front Cell Neurosci 16: 929593.
- Vleggeert-Lankamp, C. L., Pego, A. P., Lakke, E. A., Deenen, M., Marani, E. and Thomeer, R. T. (2004). Adhesion and proliferation of human Schwann cells on adhesive coatings. Biomaterials 25(14): 2741-2751.
- Weiss, T., Taschner-Mandl, S., Ambros, P. F. and Ambros, I. M. (2018). Detailed Protocols for the Isolation, Culture, Enrichment and Immunostaining of Primary Human Schwann Cells. Methods Mol Biol 1739: 67-86.
- Weiss, T., Taschner-Mandl, S., Bileck, A., Slany, A., Kromp, F., Rifatbegovic, F., Frech, C., Windhager, R., Kitzinger, H., Tzou, C. H., et al. (2016). Proteomics and transcriptomics of peripheral nerve tissue and cells unravel new aspects of the human Schwann cell repair phenotype. Glia 64(12): 2133-2153.
Article Information
Copyright
© 2023 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Aparicio, G. I. and Monje, P. V. (2023). Human Schwann Cells in vitro I. Nerve Tissue Processing, Pre-degeneration, Isolation, and Culturing of Primary Cells. Bio-protocol 13(22): e4748. DOI: 10.21769/BioProtoc.4748.
Category
Neuroscience > Peripheral nervous system > Schwann cell
Cell Biology > Cell isolation and culture > Cell isolation
Cell Biology > Cell isolation and culture > Monolayer culture
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.