- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Development of a Gene Replacement Method for the Filamentous Bacterium Leptothrix cholodnii SP-6

Published: Vol 13, Iss 8, Apr 20, 2023 DOI: 10.21769/BioProtoc.4652 Views: 2639
Reviewed by: Alberto J. Martin-RodriguezGonzalo Durante-RodríguezAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Dec 2022
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Genetic strategies such as gene disruption and fluorescent protein tagging largely contribute to understanding the molecular mechanisms of biological functions in bacteria. However, the methods for gene replacement remain underdeveloped for the filamentous bacteria Leptothrix cholodnii SP-6. Their cell chains are encased in sheath composed of entangled nanofibrils, which may prevent the conjugation for gene transfer. Here, we describe a protocol optimized for gene disruption through gene transfer mediated by conjugation with Escherichia coli S17-1 with details on cell ratio, sheath removal, and loci validation. The obtained deletion mutants for specific genes can be used to clarify the biological functions of the proteins encoded by the target genes.
Graphical overview

Background
Leptothrix species are microorganisms with a long history as research targets. The type strain, L. ochracea, was reported in the late 18th century (Spring, 2006) and, since then, has attracted much attention from researchers in the fields of microbiology and environmental science. Leptothrix ubiquitously inhabits seeps, streams of iron-rich groundwater, and activated sludge. Leptothrix divides to produce cell chains encased with entangled nanofibrils termed sheaths and eventually generates mats composed of porous networks of cell chains (Spring, 2006). Over the time of mat aging, sheaths are encrusted with inorganic particles, especially iron oxide, resulting in the production of rigid microtubes (Furutani et al., 2011). Focusing on this innate characteristic, Leptothrix has been utilized for the removal of metal contaminations such as iron and manganese from groundwater in water purification plants (Kunoh et al., 2015a). However, overgrowth of filamentous bacteria, including Leptothrix, often results in clogging of water distribution pipes and bulking of activated sludge in wastewater treatment plants (van der Waarde et al., 2002). Therefore, understanding the functions of key proteins responsible for nanofibril formation and entanglement is significant for the artificial control of their filamentous growth.
The next-generation sequencing technologies that have been developed recently enable us to freely access significant data of the whole genome sequences existing in a wide range of organisms, from prokaryotes to eukaryotes. For Leptothrix species, the complete genome sequence of L. cholodnii SP-6 (hereafter SP-6) was published by the US DOE Joint Genome Institute (Copeland et al., 2008) [GenBank (accession: GCA_000019785.1)], which opened new doors for understanding the gene functions and interactions of this strain.
To date, a gene replacement system for Leptothrix species remains unpublished, except for the sheath-less strain L. discophora SS1 (Bocioaga et al., 2014). Although we attempted to perform gene transfer of SP-6 mediated by conjugation with Escherichia coli S17-1 by following their report, the efficiency of gene replacement was quite low relative to that of SS1. We therefore considered the possibility that the sheath structure of SP-6 might interfere with the conjugation with E. coli cells, and eventually the incorporation of extracellular DNA into SP-6 cells.
We previously reported that sheath structure plays a critical role in holding aligned cell chains of SP-6 (Kunoh et al., 2020), and that calcium depletion frequently results in breakage of cell filaments of SP-6 due to failure in the production of sheath structures encasing the cell chains (Kunoh et al., 2021). From these findings, we hypothesized that preculturing SP-6 cells in a calcium-lacking medium might enhance the accessibility of E. coli cells to SP-6 cells for conjugation, which results in efficient gene transfer and subsequent gene replacement of SP-6 cells.
Here, we describe a protocol for gene replacement in SP-6 covered with sheaths (Graphic abstract), which was modified from that reported previously (Yoshihara, 2002). This protocol is expected to contribute to efficient systematic genetic analyses of SP-6, including genes responsible for sheath formation (Kunoh et al., 2022).
Materials and Reagents
Chemical compounds
Kanamycin sulfate (FUJIFILM Wako Pure Chemical Co., catalog number: 117-00341)
Ampicillin sodium salt (FUJIFILM Wako Pure Chemical Co., catalog number: 012-23303)
Rifampicin (Nacalai Tesque Inc., catalog number: 30259-94)
Ammonium sulfate [(NH4)2SO4] (Nacalai Tesque Inc., catalog number: 02633-15)
Magnesium sulfate heptahydrate (MgSO4·7H2O) (FUJIFILM Wako Pure Chemical Co., catalog number: 131-00405)
Calcium chloride dihydrate (CaCl2·2H2O) (Nacalai Tesque Inc., catalog number: 031-00435)
Potassium dihydrogen phosphate (KH2PO4) (Nacalai Tesque Inc., catalog number: 28721-55)
Sodium phosphate dibasic heptahydrate (Na2HPO4·7H2O) (Nacalai Tesque Inc., catalog number: 31725-15)
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) (Nacalai Tesque Inc., catalog number: 17514-86)
Iron(II) sulfate (FeSO4) (FUJIFILM Wako Pure Chemical Co., catalog number: 094-02942)
Sodium pyruvate (FUJIFILM Wako Pure Chemical Co., catalog number: 191-03061)
Sodium hydroxide (NaOH) (Nacalai Tesque Inc., catalog number: 31511-05)
Sodium chloride (NaCl) (Nacalai Tesque Inc., catalog number: 31319-45)
Hexadecyltrimethylammonium bromide (CTAB) (Nacalai Tesque Inc., catalog number: 07906-82)
Sodium dodecyl sulfate (SDS) (Nacalai Tesque Inc., catalog number: 30400-85)
Phenol/chloroform/isoamyl alcohol (25:24:1) (FUJIFILM Wako Pure Chemical Co., catalog number: 311-90151)
Chloroform (Nacalai Tesque Inc., catalog number: 08401-65)
Isopropanol (Nacalai Tesque Inc., catalog number: 29113-95)
Ethanol (Nacalai Tesque Inc., catalog number: 14713-95)
Sodium acetate trihydrate (CH3COONa·3H2O) (Nacalai Tesque Inc., catalog number: 31115-05)
Tris(hydroxymethyl)aminomethane hydrochloride (Tris-HCl) (Nacalai Tesque Inc., catalog number: 35434-21)
Ethylenediaminetetraacetic acid tetrasodium salt (EDTA-4Na) (FUJIFILM Wako Pure Chemical Co., catalog number: 349-01885)
35% hydrochloric acid (HCl) (Nacalai Tesque Inc., catalog number: 18321-05)
Acetic acid (Nacalai Tesque Inc., catalog number: 00212-85)
Polyethylene glycol 8000 (PEG8000) (MP Biomedicals LLC., catalog number: 195445)
Glycerol (Nacalai Tesque Inc., catalog number: 17017-35)
Dimethyl sulfoxide (DMSO) (Nacalai Tesque Inc., catalog number: 13407-45)
Potassium chloride (KCl) (Nacalai Tesque Inc., catalog number: 28514-75)
Magnesium chloride (MgCl2) (Nacalai Tesque Inc., catalog number: 20935-05)
d-glucose (Nacalai Tesque Inc., catalog number: 16805-35)
Agarose S (FUJIFILM Wako Pure Chemical Co., catalog number: 312-01193)
1 kb Plus DNA ladder (Thermo Fisher Scientific, catalog number: 10787018)
Ethidium bromide solution (10 mg/mL) (Nacalai Tesque Inc., catalog number: 14631-94)
Green nucleic acid stain (Midori Green Advance, Nippon Genetics Co. Ltd., catalog number: NE-MG04)
Liquid nitrogen (Air Water Inc., YJ code number: 799070CX1020)
Culture media
BD Difco LB broth, Lennox (Thermo Fisher Scientific, catalog number: 240230)
BD Bacto agar (Thermo Fisher Scientific, catalog number: 214010)
BD Bacto yeast extract (Thermo Fisher Scientific, catalog number: 212750)
BD Bacto tryptone (Thermo Fisher Scientific, catalog number: 211705)
BD Difco nutrient broth (Thermo Fisher Scientific, catalog number: 234000)
Vitamins
d-biotin (Nacalai Tesque Inc., catalog number: 04822-04)
Folic acid (Nacalai Tesque Inc., catalog number: 16221-91)
Thiamine hydrochloride (HCl) (Nacalai Tesque Inc., catalog number: 36319-24)
d-calcium pantothenate (Nacalai Tesque Inc., catalog number: 26004-44)
Vitamin B12 (Nacalai Tesque Inc., catalog number: 36323-96)
Riboflavin (Nacalai Tesque Inc., catalog number: 36322-51)
Nicotinic acid (Nacalai Tesque Inc., catalog number: 24326-52)
Pyridoxine hydrochloride (Nacalai Tesque Inc., catalog number: 29611-81)
p-aminobenzoic acid (Nacalai Tesque Inc., catalog number: 01930-32)
Enzymes
Proteinase K (FUJIFILM Wako Pure Chemical Co., catalog number: 166-28913)
Ribonuclease A (FUJIFILM Wako Pure Chemical Co., catalog number: 182-01493)
Lysozyme from egg white (FUJIFILM Wako Pure Chemical Co., catalog number: 122-02673)
Tks Gflex DNA polymerase (Takara Bio Inc., catalog number: R060A)
PrimeStar Max DNA polymerase (Takara Bio Inc., catalog number: R045A)
FastDigest restriction enzymes BamHI, HindIII, and SmaI (Thermo Fisher Scientific, catalog numbers: FD0054, FD0504, FD0663)
Kits
Gel Loading Buffer (10×) (Thermo Fisher Scientific, catalog number: 10816015)
Gel Extraction kit (Promega Co., Wizard® SV Gel and PCR Clean-Up System, catalog number: A9282)
Plasmid Extraction kit (Merck KGaA, GenEluteTM Plasmid Miniprep Kit, catalog number: PLN350-1KT)
DNA Ligation kit <Mighty Mix> (Takara Bio Inc., catalog number: 6023)
Materials
50 mL polypropylene tube (IWAKI AGC Techno Glass Co., Ltd., catalog number: 2345-050)
15 mL polypropylene tube (IWAKI AGC Techno Glass Co., Ltd., catalog number: 2425-015)
1.5 mL centrifuge tube (Midland Scientific, catalog number: 311-08-052)
PCR microtube (Bio Regenerations Co., Ltd., catalog number: BRG-DC-01)
ϕ 90 × 15 mm sterilized dish (RIKAKEN Holdings Co., Ltd, catalog number: RSU-SP915-2)
10 and 30 mL syringes (TERUMO Co., Ltd., catalog numbers: SS-10ELZ, SS-30ESZ)
25 G × 1 inch injection needle (TERUMO Co., Ltd., catalog number: NN-2525R)
Stericup quick release-HV sterile vacuum filtration system (Merck Millipore, catalog number: S2HVU05RE)
MF-Millipore membrane filter, 0.22 µm pore size (Merck Millipore, catalog number: GSWP04700)
Toothpicks (e.g., Yanagi Products Co., Ltd., DN Tsumayouji L)
0.22 μm syringe filter (Sartorius AG, catalog number: S7597-FXOSK)
Vacuum filtration device (Merck Millipore, catalog number: SCGPS02RE)
2.0 mL stock tube (Sarstedt AG, catalog number: 72.694.107.03)
IWAKI 20 mL glass test tube (ϕ 16.5 × 165 mm) (AGC Techno Glass Co., Ltd., catalog number: TEST16.5NP)
Test-tube closures (ϕ 17–18 mm) (LÜDI SWISS AG, catalog number: 08418)
Erlenmeyer flask (200 mL) (AGC Techno Glass Co., Ltd., catalog number: 4980FK200)
Silicon stopper (Shin-Etsu Polymer Co., Ltd., catalog number: T-38)
100–1,000 mL medium bottles (AGC Techno Glass Co., Ltd., catalog numbers: 1516/04D, 1516/08D, 1516/10D)
Cuvette for electroporation (2 mm gap) (NEPA GENE Co. Ltd., catalog number: EC-002S)
Bacterial strains
Leptothrix cholodnii SP-6 (ATCC strain 51168)
Bacillus subtilis 168 (Lab stock)
Escherichia coli S17-1 (Lab stock)
Escherichia coli DH5α (Lab stock)
Escherichia coli SN1187 [National BioResource Project – E. coli Strain (National Institute of Genetics, Mishima, Japan)]
Plasmids
pSUP5011::Tn5-mob (Lab stock, DSMZ collection 5167)
pUC18 (Takara Bio Inc., catalog number: 3218)
Prepared recipes (see Recipes below)
MSVP (ATCC 1917) medium
LB medium
SOC medium
NB medium
1 M MgSO4 and 1 M MgCl2 solutions
1 M glucose solution
NaCl solutions (0.9%, 5 M)
TE buffer
TEN buffer
10% SDS solution
CTAB/NaCl solution
3 M sodium acetate (CH3COONa) solution
50× TAE buffer
2× TSS solution
TSS solution
50% glycerol solution
Rifampicin solution
Kanamycin solution
Ampicillin solution
Equipment
Reciprocating shakers (TAITEC Co., Ltd., models: personal-11 SM, BR-41FM)
Centrifuges (Eppendorf AG., models: 5430R, 5425R)
Thermostat (Yamato Scientific Co., Ltd., model: DG400)
Spectrophotometer (ERMA INC., model: MP-1200)
Water purification systems (ELGA LabWater, model: PURELAB Chorus 1 Complete, PURELAB Chorus 2+)
pH meter (HORIBA, Ltd., model: F-74)
Electronic scale (A&D Co., Ltd., model: HR-202i)
Magnetic stirrer with hot plate (Kenis, model: MI10 33220962)
Autoclave (TOMY SEIKO Co., Ltd., model: LBS-245)
Safety cabinet (DALTON Co., Ltd., model: Class 2 A1)
Thermal cycler (BIO-RAD, model: T-100)
Microwave (for example, Sharp Co., Ltd., model: RE-21G)
Submarine electrophoresis system (Mupid.com, model: Mupid-2plus)
Gel documentation system (ATTO Co., model: WSE-5400 Printgraph Classic)
Heat block incubator (Anatech Co., Ltd., model: 5520a)
NanoDrop UVVis spectrophotometer (Thermo Fisher Scientific, model: 2000c)
Mixer (Vortex Co., Ltd., model: VORTEX-GENIE 2)
Adjustable air displacement pipette (Gilson Inc. model: PIPETMAN P series)
Electroporation system (Bio-Rad Laboratories Inc., model: Genepluser Xcell)
-30 freezer (for example Nihon Freezer Co., Ltd., model: GS-5210HC)
-80 freezer (for example Thermo Fisher Scientific, model: TSX600G)
LED illuminator (Optocode Co., Ltd., model: LED-EXTA/GFP+RFP)
Ceramic tweezer (ASONE, catalog number: 7-166-03)
Microscope (for example Carl Zeiss, model: Axio observer Z1)
Software
ApE (Davis and Jorgensen, 2022) used as a DNA plasmid sequence editor
R software version 4.2.2 [R Core Team (2022). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. (https://www.R-project.org/)] used for statistics
Procedure
Preparation of genomic DNA from SP-6 cells
This protocol was essentially described in Kunoh et al. (2015b).
Pick SP-6 cells from a glycerol stock with a sterilized toothpick, spread on MSVP agar plate, and incubate for 3–5 days at room temperature (RT).
Pick several colonies of SP-6 cells on an MSVP plate with a sterilized toothpick, inoculate into 25 mL of MSVP liquid medium in a 50 mL polypropylene tube, and incubate for 2–3 days at RT by shaking at 70 rpm.
Collect exponentially growing SP-6 cells (total 50–100 mL) by centrifugation at 7,000 × g for 5 min.
Wash the resultant cell pellet twice in 25 mL of TE buffer, resuspend in 5 mL of TE buffer containing 20 μg/mL proteinase K and 0.6% SDS, and transfer to a 15 mL polypropylene tube.
After incubation at 65 for 1 h, add 1 mL of 5 M NaCl and 800 μL of CTAB/NaCl solution to the cell suspension and incubate at 37 °C for one additional hour by shaking at 190 rpm.
To eliminate proteins, add an equal volume (~7 mL) of phenol/chloroform/isoamyl alcohol (25:24:1) to the cell suspension, mix by inversion, and centrifuge at 7,000 × g for 15 min.
Transfer the resultant aqueous phase to a new 15 mL polypropylene tube and perform chloroform extraction as described in step A5.
For precipitation of nucleic acids, transfer the resultant supernatant to a new 15 mL polypropylene tube, add an equal volume (~5 mL) of isopropanol, and incubate at -20 °C for 1 h.
Then, collect the pellet by centrifugation (7,000 × g, 30 min) and discard the supernatant, followed by rinsing the pellet in ~5 mL of 70% ethanol.
After drying up the pellet at RT for 15 min, resuspend it in 500 μL of TE buffer containing 20 μg/mL ribonuclease A by pipetting, transfer it to a 1.5 mL centrifuge tube, and incubate the suspension at 37 °C for 30 min.
To remove ribonuclease A from the suspension, add an equal volume (~500 μL) of phenol/chloroform/isoamyl alcohol (25:24:1), mix by inversion, and centrifuge (15,000 × g, 30 min). Then, transfer the resultant aqueous phase to a new 1.5 mL centrifuge tube. Repeat this step once more.
For precipitation of nucleic acids, add an equal volume (~500 μL) of ethanol and 1/10 volume (~50 μL) of 3 M CH3COONa solution to the resultant aqueous phase.
After incubation at -20 °C for ~30 min, centrifuge the mixture (15,000 × g, 30 min) and remove the supernatant, followed by rinsing the pellet in 70% ethanol.
After drying at RT for 5 min, suspend the resultant pellet in ~100 μL of TE buffer. Then, determine DNA concentration of the suspension by using a NanoDrop 2000c.
Preparation of genomic DNA from B. subtilis 168 cells
Pick B. subtilis 168 cells from a glycerol stock with a sterilized toothpick, spread on LB agar plate, and incubate at 37 °C overnight.
Inoculate a single colony into 4 mL of LB medium in a test tube and incubate by shaking at 37 °C overnight.
Transfer cell culture to 1.5 mL centrifuge tubes and harvest the cells by centrifugation (20,000 × g, 1 min).
Resuspend the resultant cell pellet in 300 μL of TEN buffer containing 10 μg/mL ribonuclease A and 30 μg/mL lysozyme and incubate at RT for 20 min.
To lyse the cells, add 9 μL of 10% SDS solution to the cell suspension, mix gently by inversion, and incubate at 50 °C for 10 min.
Add 200 μL of TEN buffer and 500 μL of phenol/chloroform/isoamyl alcohol (25:24:1) to the cell lysate, mix gently by inversion, and centrifuge at 20,000 × g for 10 min.
Transfer the upper aqueous phase to a new centrifuge tube, add 1 mL of ethanol, and mix by inversion, followed by centrifugation at 20,000 × g for 10 min.
Discard the supernatant, add 1 mL of 70% ethanol for rinsing, and centrifuge at 20,000 × g for 5 min.
Discard the supernatant, dry the DNA pellet, and resuspend it in 200 μL of TE buffer. Dilute approximately 10-fold by TE buffer and use the resultant DNA solution as a template for PCR.
Construction of plasmids pUC18-Kmr and pUC18-mob
To construct pUC18-Kmr, digest 20–50 ng of pSUP5011 and pUC18 with SmaI and HindIII at 37 °C for 30 min. To construct pUC18-mob, digest these plasmids with BamHI.
Purify the insert DNAs from pSUP5011 (1.3 kb HindIII-SmaI and 1.8 kb BamHI fragments, respectively) and digest pUC18 by agarose gel extraction.
Clone the purified insert DNA into pUC18 by using DNA Ligation kit according to the manufacturer’s instructions.
Transform E. coli DH5α cells with the ligation mixtures by electroporation, spread the cells on LB plates containing 50 μg/mL ampicillin sodium salt, and incubate at 37 °C overnight.
Pick the resultant single colonies, re-streak them on LB agar with 50 μg/mL ampicillin sodium salt, and incubate at 37 °C overnight.
Inoculate the resultant colonies into 4 mL of LB medium containing 50 μg/mL ampicillin sodium salt in test tubes and incubate by shaking at 37 °C overnight.
Harvest the cells by centrifugation (20,000 × g, 1 min) and extract the plasmid DNA (pUC18-mob and pUC18-Kmr) using the GenEluteTM Plasmid Miniprep kit according to the manufacturer’s instructions.
Confirm the plasmid DNAs by digesting the restriction enzymes.
Construction of the plasmid pUC18-mob-ΔLcho_0008::Kmr for Lcho_0008 gene deletion (see Figures 1 and 2)
Standard overlap extension PCR, DNA extraction from agarose gel, and iVEC3 system are described in Sections H, G, and I, respectively.
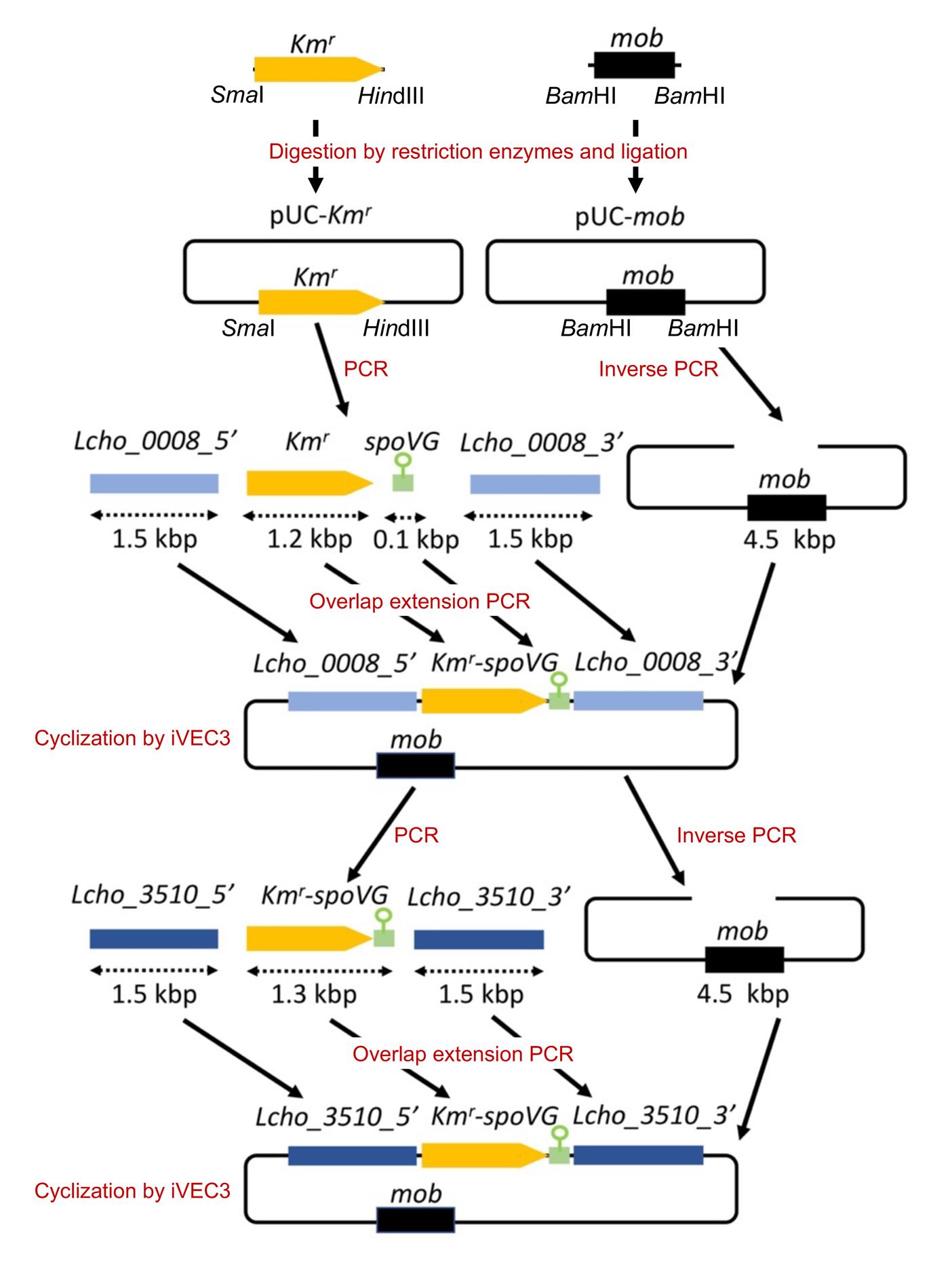
Figure 1. An overview of plasmid construction (see Kunoh et al. 2022). To construct a plasmid for the gene replacement of Lcho_0008 with the kanamycin resistance gene (Kmr), we first inserted fragments containing Kmr and mob from pSUP5011 into pUC18, respectively. Then, the upstream and downstream of Lcho_0008 (approximately 1.5 kb) and the spoVG terminator (approximately 0.1 kb) were amplified using genomic DNAs isolated from L. cholodnii SP-6 and B. subtilis 168, respectively. In parallel, Kmr was amplified using the pUC-Kmr plasmid as a template. These four fragments were stepwise joined by overlap extension PCRs (also see Figure 2) and cloned into the pUC-mob plasmid by the iVEC3 system, resulting in construction of the plasmid pUC18-mob-ΔLcho_0008::Kmr. Now, the resultant plasmid can be used not only for the gene replacement of Lcho_0008 but for the amplification of the Kmr-spoVG terminator cassette as the template to construct other plasmids, such as pUC18-mob-ΔLcho_3510::Kmr. Lcho_0008_5′ and Lcho_0008_3′, and Lcho_3510_5′ and Lcho_3510_3′ represent upstream and downstream fragments of these genes, respectively. spoVG represents the spoVG terminator.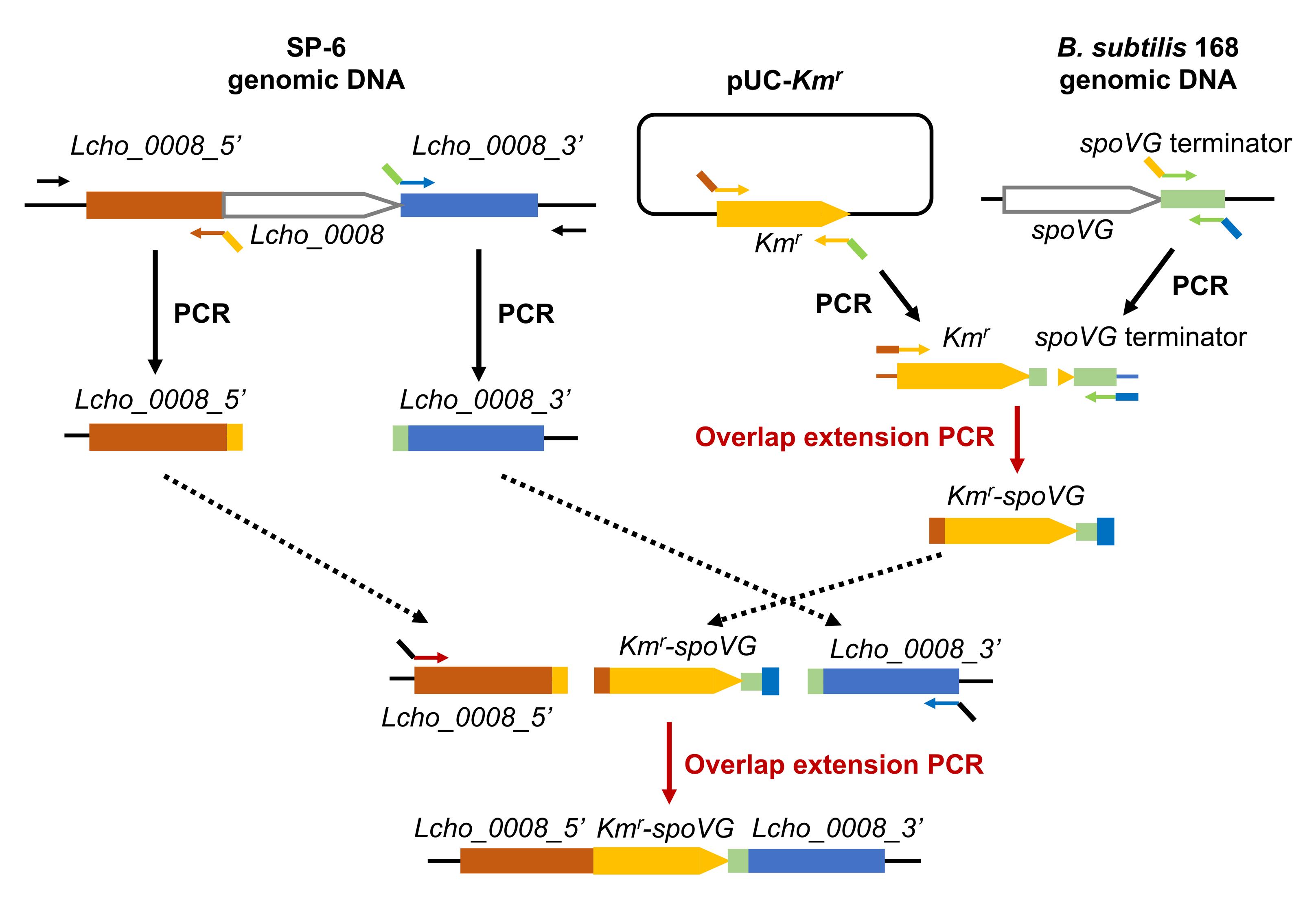
Figure 2. An overview of overlap extension PCR. The amplified PCR products contain overlapping 40 bp regions at both ends, whose Tm values were designed to be 65–76 °C (Ave. 70). These overlapping 40 bp regions are important for the overlap extension PCRs. Lcho_0008_5′ and Lcho_0008_3′ represent upstream and downstream fragments of Lcho_0008, respectively. spoVG represents the spoVG terminator.Amplify the upstream (approximately 1.5 kb) and downstream (approximately 1.5 kb) regions of Lcho_0008 gene by PCR using SP-6 genomic DNA as a template and Lc0008_F_out/Lc0008_Km_2 and term-Lc0008-1/Lc0008-R-out primer sets, respectively (see Table 1).
Table 1. Primers for the construction of pUC18-mob-ΔLcho_0008::Kmr
Name Primer sequence Lc0008-F-out 5'-AGAAGTAGTGCAGGTGCTTGAAATTG-3' Lc0008-R-out 5'-TGGTCGACGAGCTGAACCGCAAATG-3' pUC-Lc0008-1 5'-ACTCATTAGGCACCCCAGGCACAGCTGCAGCCCTTCGACTG-3' pUC-Lc0008-2 5'-AGTCGAAGGGCTGCAGCTGTGCCTGGGGTGCCTAATGAGTGAG-3' Lc0008-Km-1 5'-GCATGAACATCCTGCCCTGAGCCGCAAGCACTCAGGGCGCAAG-3' Lc0008-Km-2 5'- CCCTGAGTGCTTGCGGCTCAGGGCAGGATGTTCATGCGTCGGCAG-3' Km-term-1 5'-CGTTTTCCGGGACGCCGGCTAAAATAACCAAAAAGCAAGGAC-3' Km-term-2 5'-CCTTGCTTTTTGGTTATTTTAGCCGGCGTCCCGGAAAACG-3' term-Lc0008-1 5'-TTTCAAACTTAGTTGCACTCCCCGAGCGTGGCGGGCGGGTAC-3' term-Lc0008-2 5'-ACCCGCCCGCCACGCTCGGGGAGTGCAACTAAGTTTGAAAAATCAG-3' Lc0008-pUC-1 5'-TCGCAGCTTCAGCTCCGTGTTTTCACACAGGAAACAGCTATGACCATG-3' Lc0008-pUC-2 5'-TAGCTGTTTCCTGTGTGAAAACACGGAGCTGAAGCTGCGATGAC-3' Amplify the kanamycin resistance gene (Kmr) (approximately 1.2 kb) and the transcriptional terminator of B. subtilis spoVG gene (approximately 0.1 kb) by PCR using the primers Lc0008-Km-1/Km-term-2 and Km-term-1/term-LC0008-2 from pUC18-Kmr and the genomic DNA of B. subtilis 168, respectively.
Purify the four resultant PCR products (upstream and downstream regions of Lcho_0008, Kmr, and spoVG terminator) by agarose gel extraction.
Ligate these purified products of the Kmr and spoVG terminator by overlap extension PCR with Lc0008-Km-1 and term-Lc0008-2 primers, resulting in the production of Kmr-spoVG fragment (see Figure 2).
Purify the resultant PCR product by agarose gel extraction and ligate Kmr-spoVG fragment with upstream and downstream regions of Lcho_0008 (steps D3, D4) by overlap extension PCR with pUC-Lc0008-1 and Lc0008-pUC-2 primers, resulting in the production of the upstream Lcho_0008 + Kmr-spoVG + downstream Lcho_0008 fragment.
Purify the resultant PCR product by agarose gel extraction and use as an insert DNA in the subsequent iVEC3 procedure in step D9.
Amplify the plasmid backbone by inverse PCR with pUC-Lc0008-2 and Lc0008-pUC-1 primers using pUC18-mob plasmid as a template.
Purify the resultant PCR product by agarose gel extraction and use it as a vector DNA for the iVEC3 method in step D9.
Clone the insert DNA (upstream Lcho_0008 + Kmr-spoVG + downstream Lcho_0008) into the pUC18-mob vector by the iVEC3 system.
Pick the resultant single colonies, re-streak them on LB agar with 50 μg/mL ampicillin sodium salt, and incubate at 37 °C overnight.
Inoculate the resultant colonies into 4 mL of LB medium with 50 μg/mL ampicillin sodium salt in test tubes and incubate by shaking at 37 °C overnight.
Harvest the cells by centrifugation (20,000 × g, 1 min) and extract the plasmid DNA (pUC18-mob-ΔLcho_0008::Kmr) using the GenEluteTM Plasmid Miniprep kit according to the manufacturer’s instructions.
Confirm the construction by PCR using the extracted plasmid DNA and primer sets listed in Table 1. Sequencing especially the insert DNA of the plasmid is recommended.
Now, we are ready to use the resultant plasmid pUC18-mob-ΔLcho_0008::Kmr for the gene replacement of the Lcho_0008 gene. In addition, we can employ this plasmid as a template for the construction of other plasmid(s). For example, the plasmid pUC18-mob-ΔLcho_3510::Kmr is constructed as follows.
Construction of the plasmid for Lcho_3510 gene deletion (see Figure 1)
Detailed protocols of standard overlap extension PCR, DNA extraction from agarose gel, and iVEC3 system are described in Sections H, G, and J, respectively.
Amplify the upstream and downstream regions of Lcho_3510 gene by PCR using L. cholodnii SP-6 genomic DNA as a template and the Lc3510-F-out/Lc3510-Km-2 and Km-Lc3510-1/Lc3510-R-out primer sets, respectively (see Table 2).
Amplify the Kmr gene and the transcriptional terminator of B. subtilis spoVG gene region by PCR using the pUC18-mob-ΔLcho_0008::Kmr as a template and Lc3510-Km-1 and Km-Lc3510-2 primers.
Purify the three resultant PCR products (upstream and downstream regions of Lcho_3510 and the Kmr-terminator) by agarose gel extraction.
Ligate these purified products by overlap extension PCR with the pUC18-Lc3510-1 and Lc3510-pUC18-2 primers.
Purify the resultant PCR product by agarose gel extraction and use it as an insert DNA for the iVEC3 method in step E8.
Amplify the plasmid backbone by inverse PCR with the pUC18-Lc3510-2 and Lc3510-pUC18-1 primers using pUC18-mob plasmid as a template.
Purify the resultant PCR product by agarose gel extraction and use it as a vector DNA for the iVEC3 method in step E8.
Clone the insert DNA (upstream Lcho_3510-Kmr-spoVG terminator-downstream Lcho_3510) into the pUC18-mob vector by the iVEC3 method.
Pick the resultant single colonies, re-streak them on LB agar with 50 μg/mL ampicillin sodium salt, and incubate at 37 °C overnight.
Inoculate the resultant colonies into 4 mL of LB medium with 50 μg/mL ampicillin sodium salt in test tubes and incubate with shaking at 37 °C overnight.
Harvest the cells by centrifugation (20,000 × g, 1 min) and extract the plasmid DNA (pUC18-mob-ΔLcho_3510::Kmr) using a GenEluteTM Plasmid Miniprep kit according to the manufacturer’s instructions.
Confirm the construction by PCR using the extracted plasmid DNA and primer sets listed in Table 2. Sequencing especially the insert DNA of plasmid is recommended.
Table 2. Primers for the construction of pUC18-mob-ΔLcho_3510::Kmr
Name Primer sequence pUC18-Lc3510-1 5'-ACTCATTAGGCACCCCAGGCTCGCACGCATCCGTCCCACCGCCTTG-3' pUC18-Lc3510-2 5'-GGTGGGACGGATGCGTGCGAGCCTGGGGTGCCTAATGAGTGAG-3' Lc3510-Km-1 5'-CACGGATGTTTCGAGTTTTTTGAGCCGCAAGCACTCAGGGCGCAAG-3' Lc3510-Km-2 5'-GCGCCCTGAGTGCTTGCGGCTCAAAAAACTCGAAACATCCGTGATCCCCCTC-3' Km-Lc3510-1 5'-TTTCAAACTTAGTTGCACTCCCGCGCGTTTTCCATCCTCGGTCG-3' Km-Lc3510-2 5'-CGAGGATGGAAAACGCGCGGGAGTGCAACTAAGTTTGAAAAATCAG-3' Lc3510-pUC18-1 5'-GTTGAATCGGAGAACCGCCTTTTCACACAGGAAACAGCTATGACCATG-3' Lc3510-pUC18-2 5'-TAGCTGTTTCCTGTGTGAAAAGGCGGTTCTCCGATTCAACCAGTTTC-3' Lc3510-F-out 5'-TTCGATCGACCGACCCTTCAGATGAG-3' Lc3510-R-out 5'-TCCTGATTGGAGGTTGACGCAACGGTC-3'
DNA amplification by PCR
Mix 1 μL of template DNA, 0.15 μL of 100 μM forward primer, 0.15 μL of 100 μM reverse primer, and 25 μL of PrimeStar Max Premix (x2) and fill up to 50 μL with ultrapure water.
Perform the PCR with the initial denaturation at 94 °C for 1 min, followed by 30 cycles of 98 °C for 10 s, 55 °C for 15 s, and 72 °C for 30 s/kb.
DNA extraction from agarose gel
Separate 10–50 μL of plasmid DNA or PCR products by agarose gel electrophoresis on 0.8%–1.5% agarose gel in 1× TAE buffer.
Stain the DNA in the agarose gel with Midori Green Advance for ~15 min to visualize it with a 470 nm LED illuminator.
Excise the appropriate DNA band and extract the DNA from the agarose gel using the Wizard® SV Gel and PCR Clean-Up System according to the manufacturer’s instructions.
Overlap extension PCR
Mix 50–100 ng of gel-extracted PCR products with homologous ends and 20 μL of PrimeStar Max Premix and adjust the volume to 40 μL with ultrapure water.
Perform the PCR with an initial denaturation at 94 °C for 1 min, followed by 15 cycles of 98 °C for 10 s, 55 °C for 15 s, and 72 °C for 30 s/kb.
Mix 1 μL of the resultant PCR solution, 0.15 μL of 100 μM forward primer, 0.15 μL of 100 μM reverse primer, and 25 μL of PrimeStar Max Premix and adjust the volume to 50 μL with ultrapure water.
Perform the PCR with an initial denaturation at 94 °C for 1 min, followed by 30 cycles of 98 °C for 10 s, 55 °C for 15 s, and 72 °C for 30 s/kb.
Preparation of competent cells of E. coli SN1187 for the iVEC3 (in vivo E. coli cloning) system
Inoculate colonies of E. coli SN1187 on an LB plate into 4 mL of LB liquid medium and incubate at 37 °C by shaking at 190 rpm overnight.
Transfer 1 mL of the cell culture to 60 mL of LB medium in a 200 mL Erlenmeyer flask and further incubate for ~90 min.
Place the Erlenmeyer flask containing the cell culture on ice for ~10 min at the time of OD600 = 0.4–0.5.
Then, transfer the cell culture to 50 mL polypropylene tubes and centrifuge at 5,000 × g for 5 min.
Discard the medium and suspend the resultant cell pellets in 2 mL of ice-cold LB medium.
Add 1.6 mL of ice-cold 2× TSS solution to the cell suspension and mix gently by pipetting.
Add 0.4 mL of DMSO to the resultant suspension and mix gently at RT by pipetting.
Dispense the cell suspension (~4 mL) in 0.1 mL aliquots in centrifuge tubes.
Then, freeze the cell suspensions immediately in liquid nitrogen and keep at -80 °C.
Circularization of PCR products by the iVEC3 system
The iVEC3 system was essentially reported in Nozaki and Niki (2019). Incorporated PCR products of the vector and insert are circularized in vivo using the overlapping 40 bp regions at both ends.
Thaw the frozen competent E. coli SN1187 cells in a 1.5 mL centrifuge tube on ice.
Add 300 fmol of the insert DNA and 60 fmol of the vector DNA to the competent cells, mix gently by pipetting, and incubate for 20 min on ice.
Add the suspension to 1 mL of LB medium, mix by inversion, and incubate at 37 °C for 45 min.
Then, centrifuge the cell suspension at 20,000 × g for 1 min, discard 1 mL of the supernatant, resuspend the cell pellet with the remaining supernatant, and spread it on an LB plate containing 50 μg/mL of ampicillin sodium salt.
Preparation of competent cells of E. coli S17-1 and DH5α and transformation
Streak E. coli S17-1 or DH5α cells on an LB plate from a glycerol stock using a sterilized toothpick and incubate at 37 °C for 12 h.
Pick a single colony, inoculate into 5 mL of LB medium, and incubate at 37 °C for 12 h by shaking at 190 rpm.
Transfer 200 μL of the cell culture to 80 mL of fresh LB medium in a 200 mL Erlenmeyer flask and incubate at 37 °C for an additional ~3 h by shaking at 190 rpm.
Place the Erlenmeyer flask containing the cell culture on ice for ~10 min at the time of OD600 = 0.6–0.7.
Then, transfer the cell culture to 50 mL polypropylene tubes and centrifuge at 3,000 × g for 10 min.
For washing, suspend the resultant cell pellet in 80 mL of ice-cold sterile ultrapure water by pipetting and centrifuge at 3,000 × g for 10 min.
Repeat step K6 once more.
Suspend the resultant cell pellet in 1.4 mL of ice-cold 10% glycerol solution and transfer the suspension to a 1.5 mL centrifuge.
Then, centrifuge the cell suspension at 7,000 × g for 10 min, discard the supernatant, and suspend the resultant cell pellet in 220 μL of ice-cold 10% glycerol solution.
Dispense the cell suspension in aliquots of 42 μL in centrifuge tubes and keep them at -80 °C until use.
Thaw the competent cells in a centrifuge tube on ice.
Add 1 μL of plasmid DNA to suspension of the competent cell and mix gently by pipetting.
Transfer the resultant mixture to an electroporation cuvette.
Perform electroporation using a GenePulser X cell at 2.5 kV.
Add ~1.0 mL of SOC medium, transfer the cell suspension to a centrifuge tube, and incubate at 37 °C for 1 h.
Spread the resultant cell suspension on a LB plate containing 50 μg/mL of ampicillin sodium salt (the Kmr on the constructed plasmid optionally allows us to use kanamycin for E. coli colony selection).
Isolation of rifampicin-resistant mutant of SP-6 (hereafter WT*)
Pick several colonies of SP-6 cells from an MSVP plate with a sterilized toothpick, inoculate into 25 mL MSVP liquid medium containing 5 μg/mL rifampicin in a 50 mL polypropylene tube, and incubate for 2–3 days at RT by shaking at 70 rpm.
Spread 100 μL of the cell culture on MSVP plates containing 5 μg/mL rifampicin and incubate for 5–7 days.
Pick a single colony with a sterilized toothpick, inoculate into 25 mL of MSVP medium containing 10 μg/mL rifampicin in a 50 mL polypropylene tube, and incubate for 2–3 days at RT by shaking at 70 rpm.
Repeat steps L2 and L3 three times using MSVP medium containing 10, 15, and 20 μg/mL rifampicin in sequence.
Isolate a single colony on an MSVP plate containing 20 μg/mL rifampicin and define it as WT*.
Mix a 1-day culture of WT* with an equal volume of 50% glycerol solution, dispense the resultant cell suspension to stock tubes, and store them at -80 °C.
Gene transfer to WT* cells by conjugation with E. coli S17-1 (see Figure 3)
Preparation of WT* cells
Streak WT* cells on a MSVP plate from a glycerol stock with a sterilized toothpick and incubate at 25 °C for 3–5 days.
Pick several WT* colonies from an MSVP plate with a sterilized toothpick, inoculate into 25 mL of MSVP medium in 50 mL polypropylene tubes (25 mL × four tubes), and incubate for 2–3 days at RT by shaking at 70 rpm.
Collect exponentially growing WT* in 2-days MSVP culture by centrifugation at 7,000 × g for 5 min, wash twice in MSVP lacking Ca (MSVP-Ca), and culture in 25 mL of MSVP-Ca for additional 6 h (see Figures 7 and 8A).
Collect exponentially growing WT* cells by centrifugation at 7,000 × g for 5 min. Discard the medium and resuspend the cell pellet in 1 mL of 0.9% NaCl.
Pass the cell suspension through a needle syringe (23 G × 1 inch) using a relatively high pressure to break apart cell aggregates composed of sheathed cell chains into single cells.
Adjust the volume of the resultant cell suspension to 2 mL with 0.9% NaCl and measure the optical density at 600 nm (OD600) to determine the cell density.
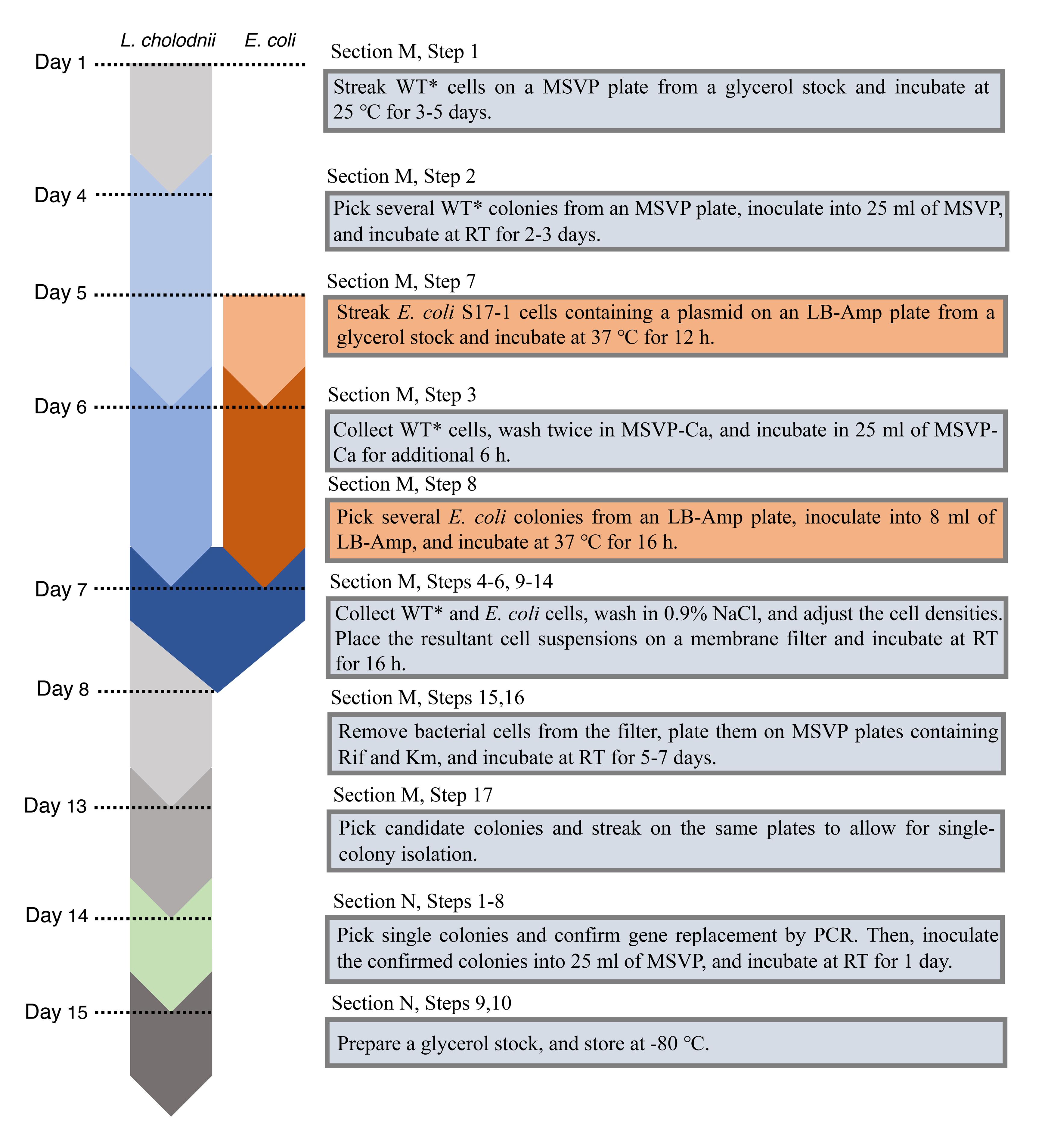
Figure 3. A detailed step-by-step chart of the culture and maintenance of L. cholodnii and E. coli
Preparation of E. coli S17-1 cells containing a plasmidStreak E. coli S17-1 cells containing a plasmid such as pUC18-mob-ΔLcho_3510::Kmr on an LB plate containing 50 μg/mL ampicillin sodium salt from a glycerol stock with a sterilized toothpick and incubate at 37 °C for 12 h.
Pick a single colony, inoculate into 4 mL of LB medium containing 50 μg/mL ampicillin sodium salt (4 mL × two tubes), and incubate at 37 °C for 16 h by shaking at 190 rpm.
Collect exponentially growing E. coli cells by centrifugation at 3,000 × g for 1 min.
Discard the medium and wash the collected cells three times with 0.9% NaCl.
Resuspend the washed cells in 2 mL of 0.9% NaCl and measure the OD600 to determine cell density.
Note: To prevent damage of pilus excreted from the cell surfaces of E. coli cells, which are important for conjugation, handle the E. coli cells gently (steps M9–M11).
Conjugation and colony selection
Drop 500 μL of the WT* suspension (from step M6) onto a membrane filter placed on an NB plate and incubate for 1 h until there is no liquid on the membrane surface (see Figure 4).

Figure 4. Image of mixing WT* and E. coli cells on a membraneDrop 500 μL of the suspension of E. coli cells (from step M11) onto the same membrane filter incubated with SP-6 and incubate for 16 h.
Transfer the membrane filter into a 15 mL polypropylene tube with a tweezer.
Remove bacterial cells from the nitrocellulose membrane filter by pipetting with 10 mL of 0.9% NaCl and collect them by centrifugation at 7,000 × g for 5 min.
Resuspend the bacterial cells in 1 mL of 0.9% NaCl, plate 100 µL each onto MSVP plates containing 20 μg/mL rifampicin and 50 μg/mL kanamycin, and incubate at RT for 5–7 days.
Pick single colonies and streak them on the same plate to allow for single-colony isolation.
Confirmation of gene replacement and preparation of glycerol stock
Colony PCR (see Figure 5)
Mix 0.15 μL of 100 μM forward primer, 0.15 μL of 100 μM reverse primer, 10 μL of 2× Gflex PCR Buffer (Mg2+, dNTP plus), and 0.4 μL of Tks Gflex DNA Polymerase (1.25 units/μL) and adjust the volume to 20 μL with ultrapure water in PCR tubes.
Pick single colonies (~0.5 mm in diameter) from MSVP plates containing 20 μg/mL rifampicin and 50 μg/mL kanamycin (Recipes 17, 18) with sterilized toothpicks and suspend them in the colony PCR mixture.
Perform the PCR with the following steps: initial denaturation at 94 °C for 1 min, followed by 30 cycles of 98 °C for 10 s, 60 °C for 15 s, and 68 °C for 1 min/kb.
Apply and electrophorese 10 mL of PCR products into an 0.8% agarose gel.
After staining the agarose gel with the ethidium bromide solution, image amplified bands using a gel documentation system.
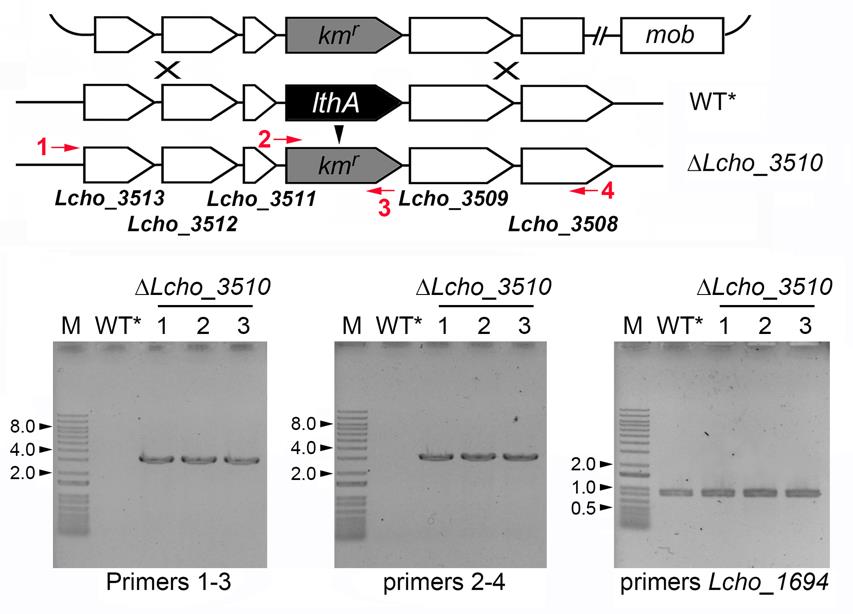
Figure 5. Confirmation of gene replacement of the Lcho_3510 gene with Kmr. Lc3510-F-out/Km-Lc3510-2 (left), Lc3510-Km-1/Lc3510-R-out (middle), and Lcho1694-F-BamHI/Lcho1694-R-BamHI (right) primer sets were employed for colony PCR (see Tables 2, 3 and the reference Kunoh et al., 2022). Size markers (kb) determined by 1 kb Plus DNA ladder (indicated as M) are indicated by numbers and arrowheads on the left.Table 3. Primers for colony PCR
Name Primer sequence Lc1694-F-BamHI 5'-CGGGATCCATGTCGAGATCGACCGCCAGCG-3' Lc1694-R-BamHI 5'-CGGGATCCTTACTTGCGTGACGAGCGCAGC-3'
Sequencing
Perform colony PCR using primers located outside the replaced gene as described in steps N1–N3.
Purify the resultant PCR products by agarose gel extraction.
Sequence the PCR products using sequencing primers listed in Table 2 (outsourced to Eurofins).
Pick the confirmed colonies (from step M17), inoculate into 25 mL of MSVP, and incubate at RT for one day.
Transfer 0.75 mL of the cell culture to a 1.5 mL tube, mix with an equal volume of 50% glycerol solution, and store at -80 °C.
The genomic DNA from B. subtilis 168 cells can be prepared using kits such as Wizard® Genomic DNA Purification kit (Promega).
To fully stop the transcription of the kmr gene at downstream of the stop codon, we employed the Bacillus subtilis spoVG transcriptional terminator because it was shown to form the stable configuration (Hudspeth et al., 1992). However, other terminators can be used for this purpose.
For in vivo cloning (the iVEC3 system), E. coli strain (SN1187) is engineered to employ the exonuclease III activity of XthA for exposure of ~40 bp of the homologous region at each end that enables annealing of DNAs, while DNA polymerase I activity of PolA is used for the gap repair of annealed DNAs (Nozaki and Niki, 2019). Although we recommend this system due to its cost-effectiveness, other cloning kits such as In-Fusion® HD Cloning kit (Clontech) can be used.
The plasmid pUC18-mob-∆Lcho_3510::Kmr is available from the RIKEN BRC Japan collection (RDB No.19817 pUC18_delLcho3510). Please contact us if the L. cholodnii strain WT* is needed.
As far as we examined by colony PCR and the following agarose gel electrophoresis, no long band corresponding to the size when the whole plasmid was integrated by single crossover recombination was detected (see Figure 5), indicating most of obtained colonies received double crossover recombination.
We employed E. coli S17-1 as a donor in this protocol. However, the S17-1 strain is known to transfer bacteriophage Mu silently. Therefore, we encourage using Mu-free donor strains such as MFDpir (Ferrières et al., 2010).
We examined the conjugation efficiencies with different donor-recipient ratios. The cell suspensions of WT* (step M6) and S17-1 (step M11) were diluted prior to mixing on a membrane filter. After conjugation, the washed bacterial suspension (step M16) was serially diluted and plated on MSVP containing rifampicin and rifampicin/kanamycin to count the total WT* cells and their transconjugates, respectively. Figure 6 indicates that the efficiency tends to be highest when the WT*:S17-1 ratio is 1:1.
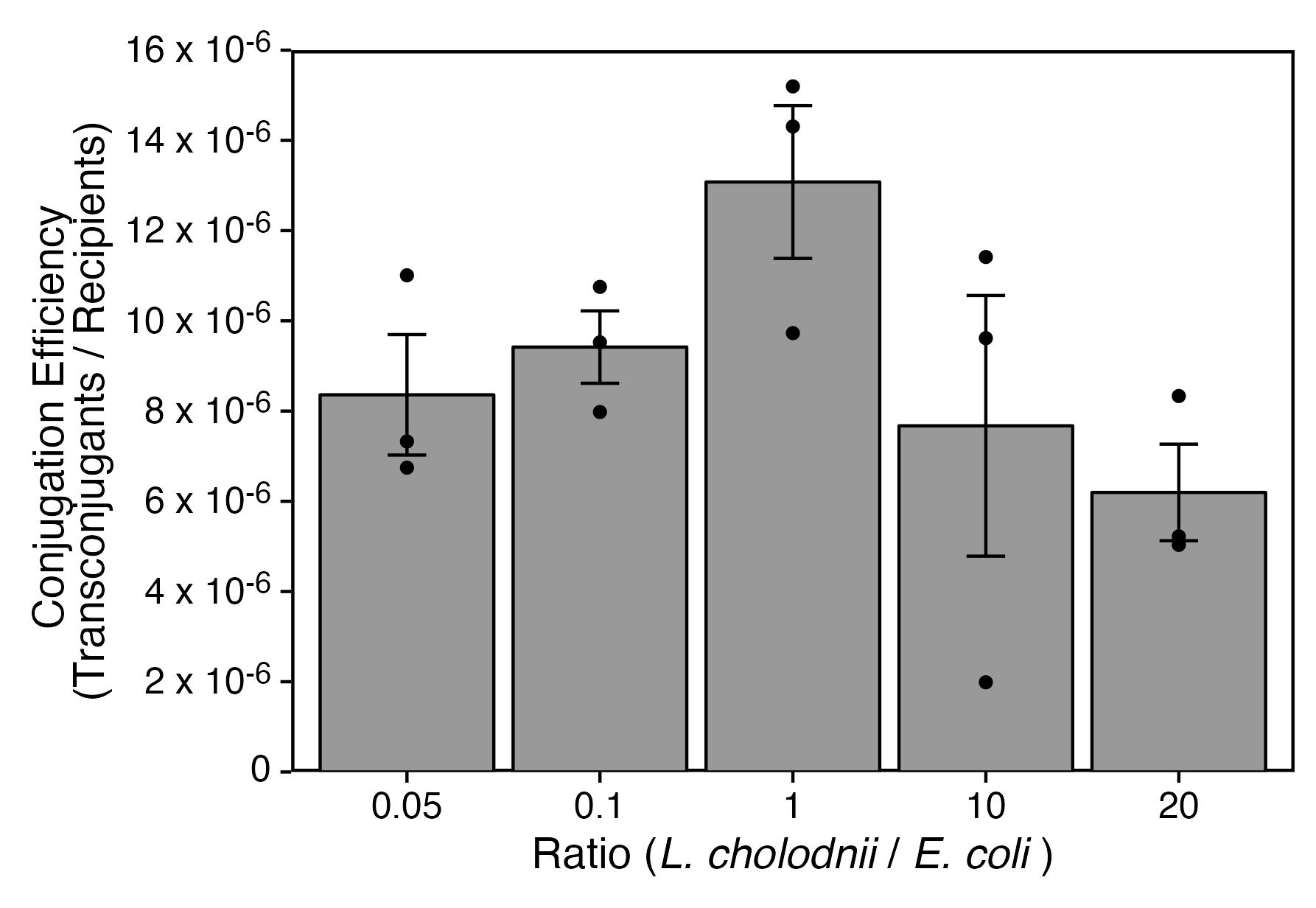
Figure 6. Conjugation efficiency with different donor-recipient ratios. Statistical analysis was performed by unpaired Welch's t-tests and Bonferroni-Holm method, suggesting no significant differences.We considered the conjugation between WT* and S17-1 to be efficient when the cell surfaces of WT* cells were free from sheaths (Figure 7). Since we previously reported that SP-6 cells become sheath-less when cultured in calcium-free conditions (Kunoh et al., 2021), WT* cells cultured in routine MSVP were harvested and further cultured in MSVP lacking Ca (MSVP-Ca) for an additional 6 h (step M3) (Figure 8A). As expected, Figure 8B indicates that the conjugation efficiency with additional culturing in MSVP-Ca is approximately 2-fold higher than that without additional culturing (MSVP+Ca).
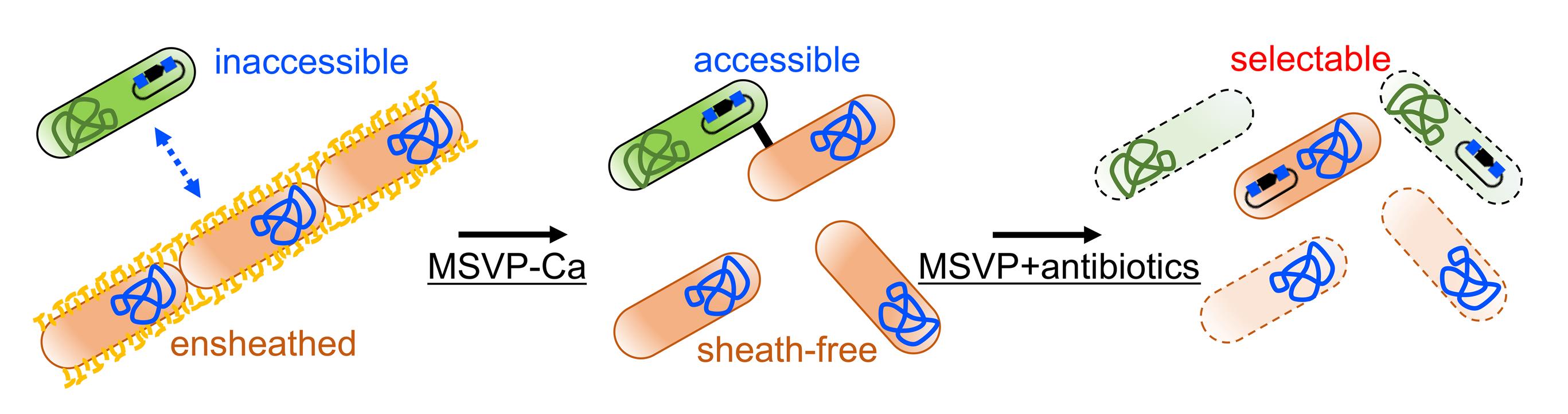
Figure 7. A strategy for enhancement of conjugation efficiency by suppressing sheath formation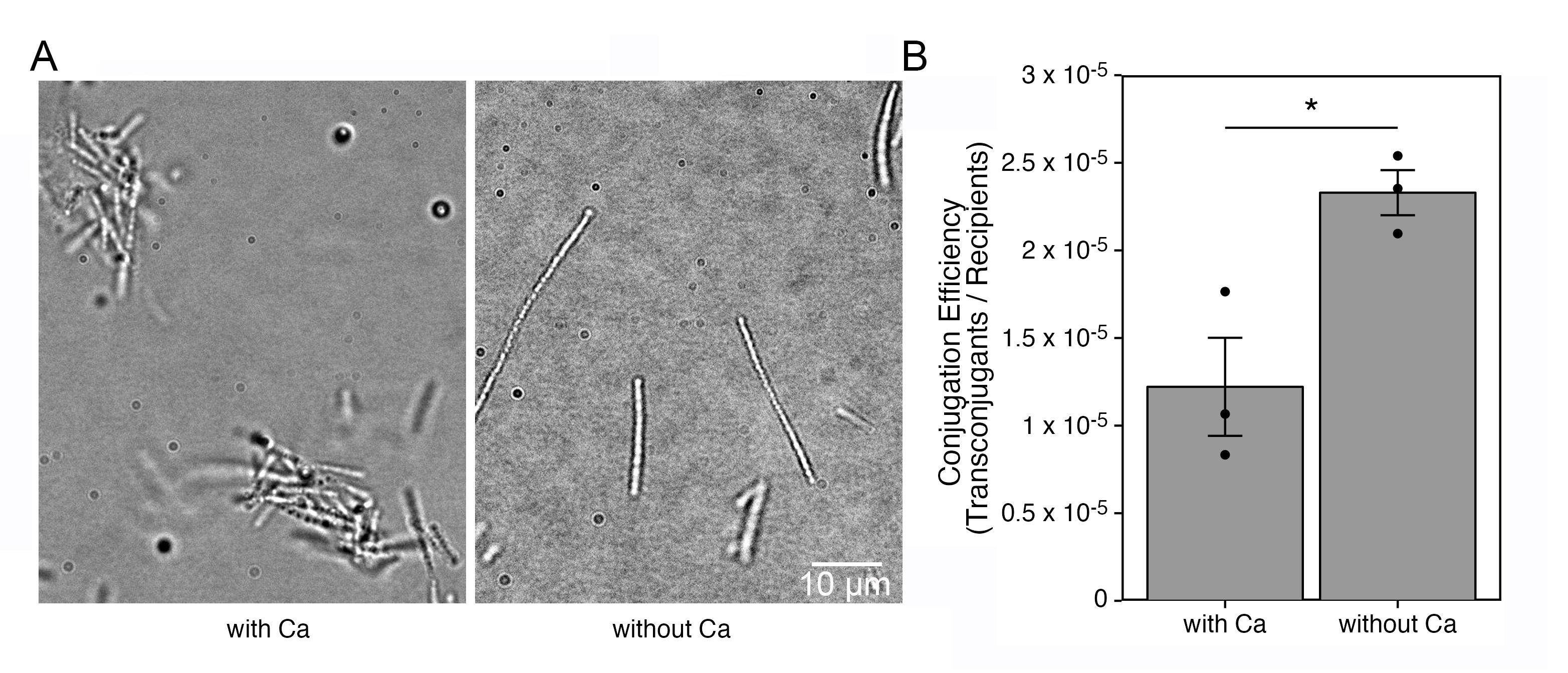
Figure 8. Conjugation efficiency with suppressing sheath formation. The brightfield images indicate that aggregate formation is reduced when cultured in the Ca-deficient condition (A). Conjugation efficiency with or without culturing cells in calcium-free medium (B). Statistical analysis was performed by unpaired Welch's t-tests. *p < 0.04.To validate whether the described method is universal for gene transfer, we employed three additional plasmids designed to target the Lcho_1694, Lcho_2352, and Lcho_2734 loci. As Figure 9 shows that the conjugation efficiencies are comparable among the representative plasmids, we consider our method to be useful for constructions targeting different loci.
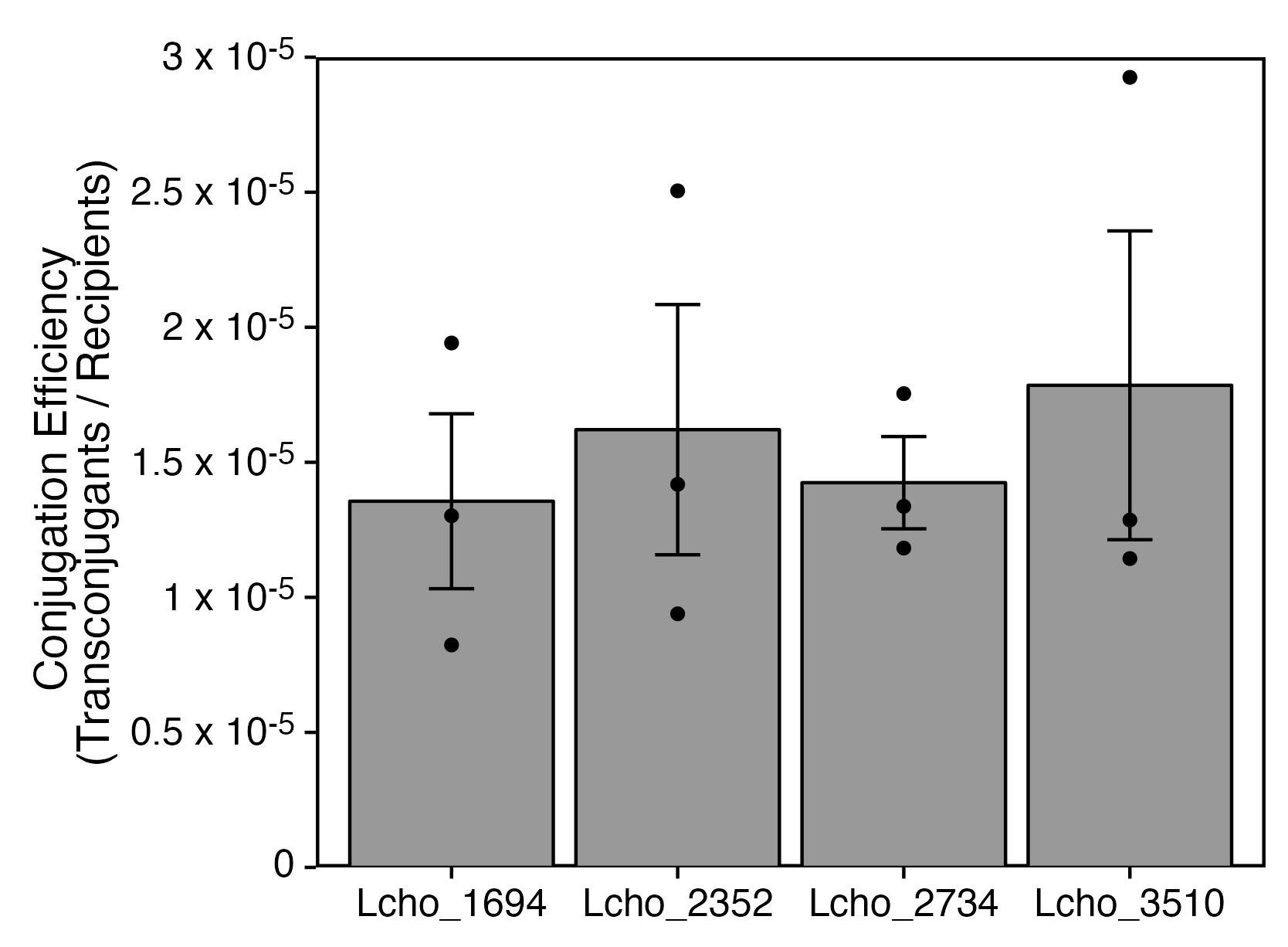
Figure 9. Conjugation efficiencies of plasmids targeting different loci. Statistical analysis was performed by Welch's t-tests and Bonferroni-Holm method, suggesting no significant differences.In experiments shown in Figure 9, approximately 250 colonies on average were obtained when 25 × 4 and 8 × 2 mL cultures of L. cholodnii and E. coli, respectively, were employed for conjugation. As far as confirmed by colony PCR, the gene replacement occurred correctly in most candidate colonies. These results implicate that we can set up the mutagenesis experiments as described in this protocol.
MSVP (ATCC 1917) medium
Prepare MVSP medium (Emerson and Ghiorse, 1993) for culturing SP-6 cells (see Tables 4, 5). Omit CaCl2·2H2O from the original recipe of MSVP for a calcium-lacking medium (MSVP-Ca). To isolate spontaneous rifampicin-resistant mutant (WT*), sequentially spread the cell suspension on MSVP plates containing 5, 10, and 20 μg/mL rifampicin. MSVP plates containing 20 μg/mL rifampicin and 50 μg/mL kanamycin sulfate can be used for the selection of recombinants of WT* containing the Kmr gene, because E. coli S17-1 cells are sensitive to rifampicin, while non-recombinant WT* cells are sensitive to kanamycin but not to rifampicin.
Table 4. Composition of MSVP medium
Component Amount (g/L) Concentration (mM) (NH4)2SO4 0.24 1.81 MgSO4·7H2O 0.06 0.24 CaCl2·2H2O 0.06 0.41 KH2PO4 0.02 0.15 Na2HPO4 0.03 0.14 HEPES 2.383 10.00 Adjust to pH 7.0 with 0.1 N NaOH, fill up to 1 L with ultrapure water, and autoclave Component Amount (mL/L) Concentration (mM) 10 mM FeSO4 1.00 0.01 20% sodium pyruvate 5.00 9.09 Vitamin stock solution (see Table 5) 1.00 Sterilize these solutions by filtration and add prior to use Table 5. Composition of vitamin stock solution
Component Amount (mg/L) Concentration (mM) Working concentration (nM) Biotin 20.00 0.04 0.04 Folic acid 20.00 0.05 0.05 Thiamine·HCl 50.00 0.15 0.15 D-(+)-Calcium pantothenate 50.00 0.10 0.10 Vitamin B12 1.00 0.74 (nM) 0.74 (pM) Riboflavin 50.00 0.13 0.13 Nicotinic acid 50.00 0.41 0.41 Pyridoxine hydrochloride 100.00 0.49 0.49 p-Aminobenzoic acid 50.00 0.36 0.36 Sterilize by filtration and add prior to use LB medium
Dissolve 20 g of LB broth in 1,000 mL of distilled water and autoclave at 121 °C for 15 min. For the LB plate, add agar at 1.5%. After autoclaving, add 50 μg/mL of kanamycin sulfate and ampicillin sodium salt as needed.
SOC medium
Dissolve 20 g of tryptone, 5 g of yeast extract, 0.5 g of NaCl, and 0.186 g of KCl in distilled water, adjust the pH to 7.0 by 5 N NaOH solution, and fill up to 1,000 mL with distilled water. After autoclaving at 121 °C for 15 min, add 10 mL of 1 M MgSO4 and 1 M MgCl2 solutions (see Recipe 5), and 20 mL of 1 M glucose solution (see Recipe 6).
NB medium
Dissolve 8 g of nutrient broth in 1,000 mL of distilled water and autoclave at 121 °C for 15 min. For the NB plate, add agar at 1.5%.
1 M MgSO4 and 1 M MgCl2 solutions
Dissolve 24.65 g of MgSO4·7H2O and 9.52 g of MgCl2 in 100 mL of distilled water and autoclave at 121 °C for 15 min.
1 M glucose solution
Dissolve 18.0 g of d-glucose in 100 mL of distilled water and sterilize using a 0.22 μm syringe filter.
NaCl solutions (0.9%, 5 M)
Dissolve 0.052 or 29.22 g of NaCl in ultrapure water, adjust the volume to 100 mL, and autoclave at 121 °C for 15 min, respectively.
TE buffer
Add 0.61 g of Tris-HCl (10 mM) and 0.23 g EDTA-4Na (1 mM) in ultrapure water, adjust pH to 8.0 with hydrochloric acid (HCl), adjust the volume to 500 mL with ultrapure water, and autoclave at 121 °C for 15 min.
TEN buffer
Mix 5 mL of 1 M Tris-HCl buffer (pH 8.0), 1 mL of 0.5 M EDTA-4Na (pH 8.0), 10 ml of 5 M NaCl, and 484 mL of ultrapure water, followed by autoclaving at 121 °C for 15 min.
10% SDS solution
Dissolve 10 g of SDS in ultrapure water and adjust the volume to 100 mL.
CTAB/NaCl solution
Dissolve 10 g of CTAB and 4.09 g of NaCl (0.7 M) in ultrapure water, adjust the volume to 100 mL, and autoclave at 121 °C for 15 min.
3 M sodium acetate (CH3COONa) solution
Dissolve 40.82 g of CH3COONa·3H2O in ultrapure water, adjust the volume to 100 mL, and autoclave at 121 °C for 15 min.
50× TAE buffer
Dissolve 242 g of Tris and 22.6 g of EDTA-4Na in ultrapure water containing 57.1 mL of acetic acid and adjust the volume to 1,000 mL.
2× TSS solution
Dissolve 4 g of PEG8000, 0.49 g of MgSO4·7H2O, and 4 mL of glycerol in LB medium (see Recipe 2), adjust the volume to 20 mL, and autoclave at 121 °C for 15 min.
TSS solution
Mix 25 mL of sterilized LB medium (see Recipe 2) and 20 mL of 2× TSS solution (see Recipe 14) with 5 mL of DMSO.
50% glycerol solution
Mix 50 mL of glycerol with 50 mL ultrapure water and autoclave at 121 °C for 15 min.
Rifampicin solution
Mix 50 mg of rifampicin with 1 mL of DMSO and stock at 4 °C.
Kanamycin solution
Mix 50 mg of kanamycin sulfate with 1 mL of ultrapure water and stock at 4 °C.
Ampicillin solution
Mix 50 mg of ampicillin sodium salt with 1 mL of ultrapure water and stock at 4 °C.
Bocioaga, D., El Gheriany, I. A., Lion, L. W., Ghiorse, W. C., Shuler, M. L. and Hay, A. G. (2014). Development of a genetic system for a model manganese-oxidizing proteobacterium, Leptothrix discophora SS1. Microbiology (Reading) 160(Pt 11): 2396-2405.
- Copeland, A., Lucas, S., Lapidus, A., Glavina del Rio, T., Dalin, E., Tice, H., Bruce, D., Goodwin, L., Pitluck, S., Chertkov, O., et al. (2008). Complete sequence of Leptothrix cholodnii SP-6. Submitted to the EMBL/GenBank/DDBJ databases.
- Davis, M. W. and Jorgensen, E. M. (2022). ApE, A Plasmid Editor: A Freely Available DNA Manipulation and Visualization Program. Front Bioinform 2: 818619.
- Emerson, D. and Ghiorse, W. C. (1992). Isolation, Cultural Maintenance, and Taxonomy of a Sheath-Forming Strain of Leptothrix discophora and Characterization of Manganese-Oxidizing Activity Associated with the Sheath. Appl Environ Microbiol 58(12): 4001-4010.
- Ferrières, L., Hémery, G., Nham, T., Guérout, A.-M., Mazel, D., Beloin, C., Ghigo, J.-M. (2010). Silent mischief: bacteriophage Mu insertions contaminate products of Escherichia coli random mutagenesis performed using suicidal transposon delivery plasmids mobilized by broad-host-range RP4 conjugative machinery. J Bacteriol 192(24): 6418-6427.
- Furutani, M., Suzuki, T., Ishihara, H., Hashimoto, H., Kunoh, H., and Takada, J. (2011). Initial assemblage of bacterial saccharic fibrils and element deposition to form an immature sheath in cultured Leptothrix sp. strain OUMS1.Minerals 1(1): 157-166.
- Hudspeth, D. S. and Vary, P. S. (1992). spoVG sequence of Bacillus megaterium and Bacillus subtilis. Biochim Biophys Acta 1130(2): 229-231.
- Kunoh, T., Kunoh, H., and Takada, J. (2015a). Perspectives on the biogenesis of iron oxide complexes produced by Leptothrix, an iron-oxidizing bacterium and promising industrial applications for their functions. J Microb Biochem Technol 7(6): 419-426.
- Kunoh, T., Suzuki, T., Shiraishi, T., Kunoh, H., and Takada, J. (2015b). Treatment of Leptothrix cells with ultrapure water poses a threat to their viability. Biology (Basel) 4(1): 50-66.
- Kunoh, T., Morinaga, K., Sugimoto, S., Miyazaki, S., Toyofuku, M., Iwasaki, K., Nomura, N. and Utada, A. S. (2020). Polyfunctional Nanofibril Appendages Mediate Attachment, Filamentation, and Filament Adaptability in Leptothrix cholodnii. ACS Nano 14(5): 5288-5297.
- Kunoh, T., Yamamoto, T., Sugimoto, S., Ono, E., Nomura, N., and Utada, A. S. (2021). Responses of Leptothrix cholodnii cells to nutrient limitation.Front Microbiol 12, 691563.
- Kunoh, T., Yamamoto, T., Prasad, M., Ono, E., Li, X., Sugimoto, S., Iida, E., Obana, N., Takeda, M., Nomura, N. and Utada, A. S. (2022). Porous Pellicle Formation of a Filamentous Bacterium, Leptothrix. Appl Environ Microbiol 88(23): e0134122.
- Nozaki, S. and Niki, H. (2019). Exonuclease III (XthA) Enforces In Vivo DNA Cloning of Escherichia coli To Create Cohesive Ends. J Bacteriol 201(5): e00660-18.
- Spring, S. (2006). The genera Leptothrix and Sphaerotilus. In: Dworkin, M., Falkow, S., Rosenberg, E., Schleifer, K. H. and Stackebrandt, E. (Eds.). The Prokaryotes (pp. 758–777). New York, Springer.
- van der Waarde, J., Krooneman, J., Geurkink, B., van der Werf, A., Eikelboom, D., Beimfohr, C., Snaidr, J., Levantesi, C. and Tandoi, V. (2002). Molecular monitoring of bulking sludge in industrial wastewater treatment plants. Water Sci Technol 46(1-2): 551-558.
- Yoshihara, H. (2002). Identification of genes essential for sheath formation in Leptothrix cholodnii. Master thesis, Graduate School of Engineering, Yokohama National University.
Data analysis
Carry out statistical analyses by unpaired Welch's t-tests and Bonferroni-Holm method using the R software version 4.1.2 [R Core Team (2022). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. (https://www.R-project.org/)] (see Figures 6, 8B, and 9).
Notes
Recipes
Acknowledgments
We acknowledge NBRP-E. coli at NIG for the E. coli strain SN1187. We also thank Drs. Tamaki H. and Morinaga K. (AIST) for providing the pSUP5011 plasmid. This study was financially supported by the Japan Science and Technology Agency (JPMJER1502, JPMJMI21G8) and a scholarship donation from Bridgestone Corporation.
Competing interests
The authors declare that they have no known competing financial interests or personal relationships that could have influenced the work reported in this paper.
Ethics
This study was approved by the committee for the safe handling of living modified organisms at the University of Tsukuba and was carried out according to the guidelines of the committee.
References
Article Information
Copyright
© 2023 The Author(s); This is an open access article under the CC BY license (https://creativecommons.org/licenses/by/4.0/).
How to cite
Kunoh, T., Ono, E., Yamamoto, T., Suzuki, I., Takeda, M. and Nomura, N. (2023). Development of a Gene Replacement Method for the Filamentous Bacterium Leptothrix cholodnii SP-6. Bio-protocol 13(8): e4652. DOI: 10.21769/BioProtoc.4652.
Category
Microbiology > Microbial genetics > Genome editing
Cell Biology > Cell engineering
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.