- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

A Novel Method to Isolate RNase MRP Using RNA Streptavidin Aptamer Tags
(*contributed equally to this work) Published: Vol 13, Iss 4, Feb 20, 2023 DOI: 10.21769/BioProtoc.4615 Views: 3216
Reviewed by: Alessandro DidonnaKirsten A. CoprenAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Feb 2022
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Interactions between RNA-binding proteins and RNA molecules are at the center of multiple biological processes. Therefore, accurate characterization of the composition of ribonucleoprotein complexes (RNPs) is crucial. Ribonuclease (RNase) for mitochondrial RNA processing (MRP) and RNase P are highly similar RNPs that play distinct roles at the cellular level; as a consequence, the specific isolation of either of these complexes is essential to study their biochemical function. Since their protein components are nearly identical, purification of these endoribonucleases using protein-centric methods is not feasible. Here, we describe a procedure employing an optimized high-affinity streptavidin-binding RNA aptamer, termed S1m, to purify RNase MRP free of RNase P. This report details all steps from the RNA tagging to the characterization of the purified material. We show that using the S1m tag allows efficient isolation of active RNase MRP.
Keywords: RibonucleoproteinBackground
In order to maintain RNA functions or defend the organism, most RNAs can be the substrate of ribonucleases (RNases), enzymes specialized in RNA cleavage and degradation. Ribonucleases are generally composed of a catalytic RNA-binding protein, but the catalyst can also be an associated RNA molecule, which is called a ribozyme. In the latter, the catalytic RNA is generally associated with one or more proteins, together forming a ribonucleoprotein particle (RNP). Two RNP-type ribonucleases in humans are RNase for mitochondrial RNA processing (MRP) and RNase P (Guerrier-Takada et al., 1983), complexes with a highly similar structure. These endoribonucleases, essential to cell survival, are ubiquitously expressed RNPs and are mostly located in the nucleolus of eukaryotic cells. RNase MRP and RNase P are evolutionarily related; RNase P is present in all domains of life, while RNase MRP is only found in eukaryotes (Randau et al., 2008).
RNase MRP and RNase P both contain a unique long non-coding RNA, but share most of their protein subunits. Ten protein subunits (hPop1, hPop4, hPop5, Rpp14, Rpp20, Rpp21, Rpp25, Rpp30, Rpp38, and Rpp40) have been demonstrated to interact with both particles, although hPop4, Rpp14, and Rpp21 might be more strongly associated with RNase P (Welting et al., 2006). The RNA components of RNase MRP and RNase P differ in size and sequence, but they share the same conserved domains and fold into highly similar secondary structures. Despite their similarity, RNase MRP and RNase P have very distinct functions. While RNase MRP, named after its first discovered role in Mitochondrial RNA Processing (Chang and Clayton, 1987), is also involved in ribosomal RNA (rRNA) (Lygerou et al., 1996; Lan et al., 2020) and messenger RNA (mRNA) maturation (Gill et al., 2004; Mattijssen et al., 2011), RNase P is best known for its function as a processor of transfer RNA (tRNA) precursors (Robertson et al., 1972). In addition to the maturation of pre-tRNA, RNase P has been reported to be involved in transcription by RNA polymerase I and III (Reiner et al., 2006; 2008), as well as in the maturation of small nucleolar RNAs (snoRNAs) in yeast (Coughlin et al., 2008).
Mutations in the RNase MRP RNA (RMRP) cause a spectrum of disorders characterized by similar symptoms, the major condition being the autosomal recessive disease cartilage-hair hypoplasia (CHH, OMIM #250250). This pathology is mostly characterized by severe skeletal dysplasia and multiple pleiotropic symptoms (Hermanns et al., 2006; Mattijssen et al., 2010). CHH-causing mutations, mainly single-nucleotide substitutions, are hypothesized to affect RMRP function by altering its secondary structure or its interaction with other molecules (Welting et al., 2008). Today, the molecular mechanisms of CHH pathology remain elusive. In order to better comprehend the cellular function of RNase MRP and the disorders linked to mutations in its RNA subunit, it is necessary to gain knowledge about the composition of this RNP and its interactome.
A ribonucleoprotein complex is commonly purified by either a protein-centric or an RNA-centric method, depending on which molecule is being targeted (Ramanathan et al., 2019). Protein-centric approaches, such as immunoprecipitation or the use of protein affinity tags, allow specific purification of some major RNA–protein cellular complexes. However, this is only possible if they contain a unique protein component, e.g., a protein subunit absent from any other RNP complex. No protein component unique to the human RNase MRP has been discovered to date and, as a consequence, any protein-centric method employed to purify the RNase MRP complex will also co-purify RNase P. This will result in a mixture of both enzymes in the purified samples, and the inability to differentiate between the two RNPs leads to uncertainties on their biochemical function. For instance, it was demonstrated that RNase MRP or RNase P is responsible for the endoribonucleolytic cleavage of m6A-modified RNAs (Park et al., 2019), but differentiation between these two endoribonucleases was not possible, since the purification was based on a shared protein subunit.
When RNP complexes cannot be isolated by techniques targeting their protein components with sufficient specificity, RNA-centric methods can be employed. However, the non-coding RNA subunits of both RNase MRP and RNase P are relatively short, highly structured, and shielded with proteins. This makes it quite challenging to purify them with RNA-specific probes, such as biotin-labeled antisense oligonucleotides (Hou et al., 2016).
As an alternative to antisense oligonucleotides, an RNA aptamer can be inserted in the RNA molecule of interest to serve as a tag for purification. Several RNA aptamers that bind ligands with high affinity have been developed in the last decades, making them powerful tools for RNA-specific purification (Walker et al., 2008). Examples include the StrepTag (Bachler et al., 1999), the MS2-TRAP (MS2-tagged RNA affinity purification) procedure (Yoon et al., 2012), and streptavidin aptamer (S1) tagging. Insertion of MS2 hairpins has been proven useful for the characterization of complexes containing a non-coding or messenger RNA of interest, but it requires co-expression of a tagged MS2-binding protein (Yoon et al., 2012; Yoon and Gorospe, 2016).
The S1 aptamer was identified by in vitro selection as a streptavidin-binding short sequence with a high affinity and appeared to be effectively eluted with biotin for the successful recovery of the tagged molecule (Srisawat and Engelke, 2001). Widely available streptavidin-agarose beads can be used, and this tag has been shown to allow efficient purification of RNase P from yeast (Srisawat and Engelke, 2002) and human cells (Li and Altman, 2002).
In this report, we describe a novel method to specifically isolate RNase MRP using the S1m RNA aptamer (Derksen et al., 2022), an optimized version of the original S1 aptamer (Leppek and Stoecklin, 2014). In addition to the increase in purification efficiency compared to the S1 tag, the S1m aptamer has a relatively short size. This lowers the risk of undesirable effects on the secondary and tertiary structure of the RNA of interest, known to be crucial for the function of many non-coding RNAs, including ribozymes.
Briefly, the S1m aptamer is inserted in the RNA of interest by cloning and the purified plasmid is used to transfect human cells. The complex containing the tagged RNA is purified from the lysate using streptavidin-coated beads and all proteins and RNAs are extracted from the purified material. The composition of the bound material is then identified by northern and western blotting methods (Figure 1).
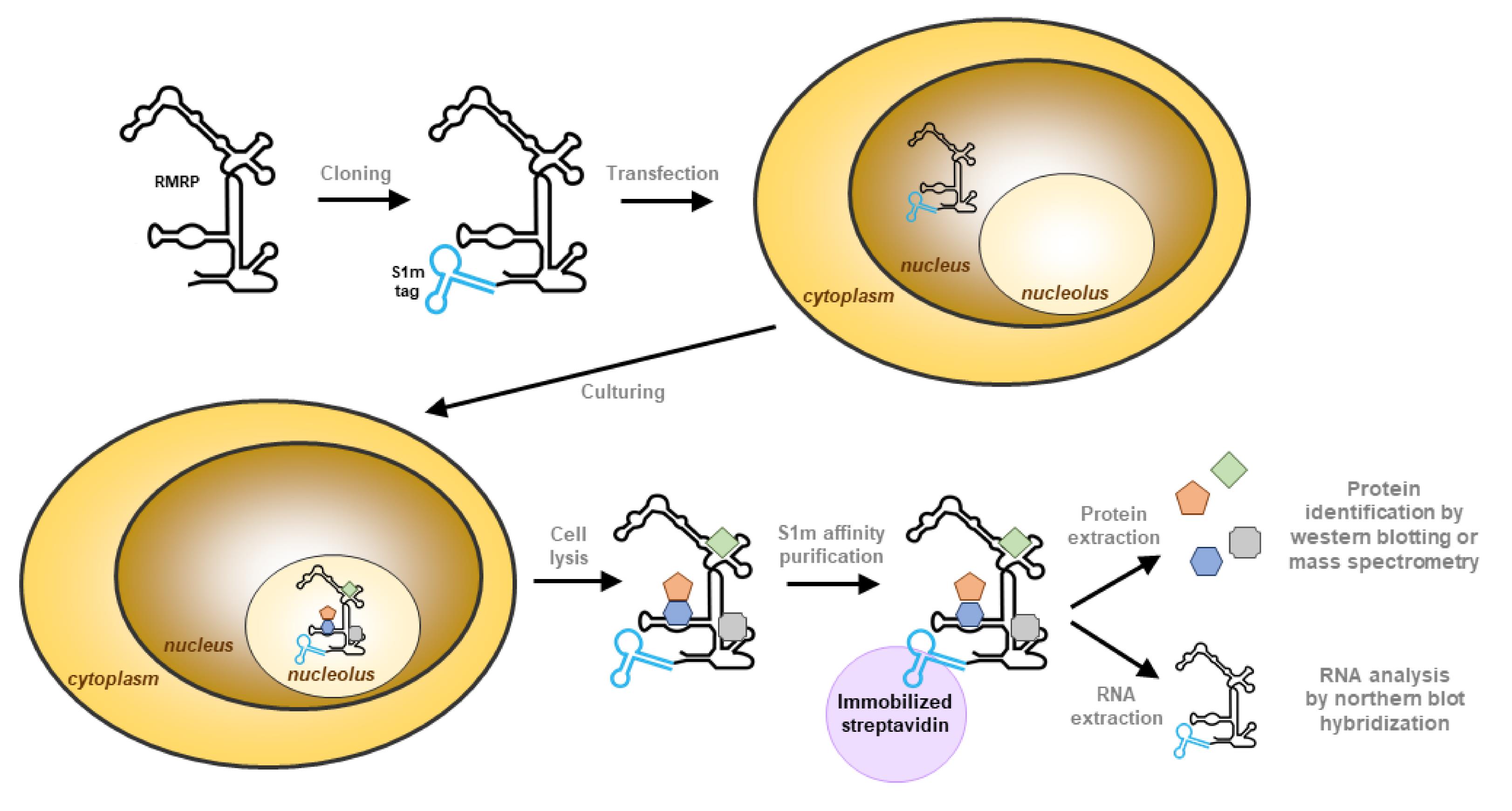
Figure 1. Schematic illustration of the isolation of RNase MRP using the S1m tag. The S1m aptamer is introduced at the desired location of the gene of interest (5′, 3′, or internal) and human cells are transfected with this construct. The tagged RNA is transcribed in the nucleus and will assemble into ribonucleoprotein particles, leading to accumulation in the nucleoli. The cells expressing the tagged RNA are lysed, and the RNA–protein complex is purified with immobilized streptavidin, which binds to the S1m tag. The purified complex is eluted from the beads to extract the proteins and RNA separately. The RNA sample can be used to analyze the specificity of purification by northern blot hybridization and the protein sample can be used to analyze the associated proteins.
Materials and Reagents
Cell lines
Human embryonic kidney HEK293T cells
Disposables
For all RNA work, use only filter tips, RNA- and RNase-free tubes, and prepare all solutions RNase-free.
1.5 mL microcentrifuge tubes (autoclaved) (any vendor)
0.2 mL PCR tubes (any vendor)
Sterile 15 mL polypropylene centrifuge tubes (any vendor)
Sterile 50 mL polypropylene centrifuge tubes (any vendor)
Sterile nuclease-free filter tips (10 µL, 200 µL, 1,000 µL) (any vendor)
Sephadex G50 column (Cytiva, catalog number: 27534001)
Cell culture 12-well plate (Greiner Bio-one, catalog number: 665180)
Cell culture dishes, 150 cm2 (Greiner, catalog number: 639160)
Serological pipette, 25 mL (any vendor)
Serological pipette, 10 mL (any vendor)
Serological pipette, 5 mL (any vendor)
Petri dishes (Sarstedt, catalog number: 82.1473.001)
Sterile surgical blades for the excision of DNA bands from gel (Swann-Morton, catalog number: 0208)
QIAquick gel extraction kit protocol (Qiagen, catalog number: 28706)
Plasmid Midi kit (Qiagen, catalog number: 12145)
Hybond-N membrane (GE Healthcare, catalog number: RPN303B)
Protran BA 85 nitrocellulose membrane, 0.45 µm pore size (VWR, catalog number: 10063-173)
Filter paper (any vendor)
Whatman paper 0.34 mm (Cytiva, catalog number: 3030-917)
0.22 µm filter (VWR, catalog number: 514-0061)
Reagents (order of use)
Sterile double distilled water (DDW), referred to as ultrapure or RNase-free water
Tryptone (MP Biomedicals, catalog number: 091010817)
Sodium chloride (NaCl) (Sigma-Aldrich, catalog number: S3014)
Yeast extract (MP Biomedicals, catalog number: 0210330391)
Agar (Thermo Scientific, catalog number: 30391023)
Sodium hydroxide (NaOH) EMSURE® (VWR, catalog number: 1.06498.1000)
Agarose (Eurogentec, catalog number: EP-0010-05)
DNA loading dye, 6× (Thermo Scientific, catalog number: R0611)
Dulbecco’s modified Eagle’s medium (DMEM) (Thermo Scientific, catalog number: 41966)
Penicillin-streptomycin solution (Thermo Scientific, catalog number: 151401)
Fetal calf serum (FCS) (Sigma-Aldrich, catalog number: F2442)
Polyethyleneimine (PEI), branched (Sigma-Aldrich, catalog number: 108727)
Opti-minimal essential medium (Opti-MEM) (Gibco, catalog number: 51985026)
Potassium chloride (KCl) (Thermo Fisher Scientific, catalog number: 15496249)
Sodium phosphate monobasic monohydrate (NaH2PO4·H2O) (VWR, catalog number: MERC1.06346)
Sodium hydrogen carbonate (NaHCO3) EMSURE® (VWR, catalog number: 1.06329)
Glucose (Invitrogen, catalog number: 15023021)
Trypsin (Sigma-Aldrich, catalog number: T4799)
Ethylenediaminetetraacetic acid (EDTA) (VWR, catalog number: 1.08418)
Dulbecco’s phosphate buffered saline (PBS) (DPBS) (Life Technologies, catalog number: 14190)
Tris (Sigma-Aldrich, catalog number: T1378)
Hydrochloric acid (HCl), 37% (VWR, catalog number: 1.00317)
Glycerol (Bio-Connect, catalog number: 4800688)
NP-40 (IGEPAL®) (Sigma-Aldrich, catalog number: 56741)
cOmplete protease inhibitor cocktail (Roche, catalog number: 1697498001)
RNasin (Promega, catalog number: N2111)
Streptavidin-conjugated SepharoseTM beads (GE Healthcare, catalog number: 17511301)
Sodium dodecyl sulfate (SDS) (Sigma-Aldrich, catalog number: L4390)
β-Mercaptoethanol (β-ME) (Sigma-Aldrich, catalog number: M3148)
Bromophenol blue (BFB) (Sigma-Aldrich, catalog number: B5525)
Trizol reagent (Invitrogen, catalog number: 15596018)
Chloroform, EMSURE® (Sigma-Aldrich, catalog number: 1.02445)
Isopropanol, EMSURE® (Sigma-Aldrich, catalog number: 1.09634)
GlycoBlue (Thermo Scientific, catalog number: AM9515)
25:24:1 phenol:chloroform:isoamyl alcohol (Invitrogen, catalog number: 15593049)
Ethanol absolute, EMSURE® (Sigma-Aldrich, catalog number: 1.00986)
Dithiothreitol (DTT) (Sigma-Aldrich, catalog number: D9779)
5× transcription optimized buffer (Promega, catalog number: P1181)
100 mM NTPs (Affymetrix, catalog number: 77245)
T7 RNA polymerase (Thermo Fisher Scientific, catalog number: EP0111)
32P-α-UTP (PerkinElmer, 3000 Ci/mmol, NEG502Z)
Boric acid (Boom, catalog number: 61000501.1000)
40% (w/v) acrylamide:bisacrylamide (19:1) solution for RNA (Serva, catalog number: 10.679.02)
Urea (Invitrogen, catalog number: 15505027)
N,N,N’,N’-Tetramethylethylenediamine (TEMED) (Sigma-Aldrich, catalog number: T7024)
Ammonium persulfate (APS) (VWR, catalog number: 97064-594)
Xylene cyanol FF (XCFF) (Sigma-Aldrich, catalog number: X4126)
Monosodium phosphate (NaH2PO4) (Sigma-Aldrich, catalog number: L2887)
Disodium hydrogen phosphate (Na2HPO4) (VWR, catalog number: 1.06580.1000)
Sodium citrate (VWR, catalog number: 1.06448)
Bovine serum albumin (BSA) (Sigma-Aldrich, catalog number: A4503)
Ficoll® 400 (VWR, catalog number: 17-0300)
Polyvinylpyrrolidone (Sigma-Aldrich, catalog number: PVP40)
Herring sperm DNA (Sigma-Aldrich, catalog number: D6898)
30% (w/v) acrylamide:bisacrylamide (37.5:1) solution (Serva, catalog number: 10.688.02)
Glycine (Merck, catalog number: 50046)
Methanol (Thermo Scientific, catalog number: 11976961)
Ponceau-S solution (Sigma-Aldrich, catalog number: P7170)
Tween-20 (Merck, catalog number: 822184)
Non-fat dry milk (any vendor)
IRDye-conjugated secondary antibodies (LiCOR Biosciences)
Luria-Bertani (LB) medium and agar (see Recipes)
Complete DMEM medium (see Recipes)
10× tyrode solution (see Recipes)
Trypsin-EDTA solution (see Recipes)
Polyethyleneimine (PEI) (see Recipes)
S1m lysis buffer (see Recipes)
S1m incubation buffer (see Recipes)
S1m wash buffer (see Recipes)
4× protein sample buffer (see Recipes)
In vitro transcription (IVT) NTP mix (see Recipes)
1× TBE (see Recipes)
RNA denaturing gel (see Recipes)
2× RNA sample buffer (see Recipes)
Blotting buffer (see Recipes)
20× SSC (see Recipes)
100× Denhardt’s (see Recipes)
10 mg/mL sheared herring sperm DNA (see Recipes)
Pre-hybridization buffer (see Recipes)
SDS-PAGE running gel (see Recipes)
SDS-PAGE stacking gel (see Recipes)
SDS-PAGE running buffer (see Recipes)
Western blotting buffer (see Recipes)
Blot blocking solution (see Recipes)
PBST (see Recipes)
Optional: BioLux® Gaussia Luciferase assay kit (New England BioLabs, catalog number: E3300)
Optional: Luminometer-compatible 96-well plate, opaque white or black (any vendor)
Plasmids
pGEM-3Zf(+)-RMRP (Pluk et al., 1999; available by request to the authors)
pcDNA5/FRT/TO (Thermo Fisher, catalog number: V652020)
Bacterial strains
E. coli TOP10 competent cells (homemade)
Equipment
Benchtop centrifuge (Eppendorf, catalog number: 5415D)
Refrigerated benchtop centrifuge (Eppendorf, catalog number: 5417R)
Refrigerated centrifuge suitable for 50 mL volume (Heraeus Megafuge 1.0 R)
Magnetic stirrer (IKA REO Basic C IKAMAG)
Thermocycler (Bio-Rad T100)
Shaker incubator (Infors HT Multitron 2)
Spectrophotometer (DeNovix DS-11)
Gel scanner (FLA-5100)
Humidified 37 °C, 5% CO2 incubator (Heracell 150)
Diagenode bioruptor
Digital rotary mixer (Labinco, LD-76)
Standard equipment for sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE)
Odyssey imaging system (LiCOR Biosciences)
Crosslinker, Stratalinker 1800 (Stratagene) with UV bulbs, λ = 254 nm (Ushio, catalog number: 3000016)
Geiger counter (Thermo Scientific, Mini monitor 900)
Phosphor imaging screens (Bio-Rad)
Typhoon imager (GE Healthcare)
Optional: Luminometer (Thermo Scientific, VL0000D0)
Procedure
S1m tagging
Optimal site for S1m insertion/fusion
The S1m aptamer sequence can be inserted in any region of the RNA of interest, but one has to make sure the tag will be available for purification. The streptavidin-binding aptamer must be properly folded, and the tertiary structure of the RNA and/or associated proteins must not interfere with the binding of streptavidin to the S1m aptamer. If the RNA structure and/or binding sites for RNA-binding proteins are known, place the S1m sequence in a non-conserved, exposed RNA region (Walker et al., 2008). In addition, the junction sites for the 5′- and 3′-ends of the S1m aptamer should be close in space to allow proper folding of the aptamer.
We advise trying at least two distinct aptamer insertion sites and assessing which one leads to the most efficient purification. In case of RNase MRP, fusion to the 5′ or 3′ termini of the RNase MRP RNA appeared to be more efficient than insertion at an internal position. It should be noted that placing the affinity tag at the 5′ or 3′ position might decrease the RNA stability due to exonuclease degradation.
Moreover, it is necessary to adjust the promoter to the gene of interest (pol I, II, or III) and to consider the required expression level. An inducible system such as a doxycycline-inducible RNA polymerase promoter might be desirable to avoid overexpression and unwanted effects.
Tag gene of interest with S1m tag
Insert the S1m aptamer sequence at the selected location(s) in the gene of interest, using a standard PCR-based strategy. The sequence of the S1m aptamer is AUGCGGCCGCCGACCAGAAUCAUGCAAGUGCGUAAGAUAGUCGCGGGUCGGCGGCCGCAU. This sequence can be commercially synthesized as an oligonucleotide and cloned by the use of restriction enzymes, as it was done for RMRP (for details, see Derksen et al., 2022). Another method is to add the aptamer sequence to the primers used for the amplification of the gene of interest and use PCR amplification and fusion-based cloning (such as In-Fusion®) to add the tag directly in the sequence of the gene of interest. This second method is faster, easier, and allows the introduction of the aptamer at any desired location.
Confirm insertion of the aptamer by restriction digestion and gel electrophoresis.
Note: We decided to use the S1m aptamer, which is a modified S1 aptamer containing an extended perfect strand complementarity in comparison with the original S1 basal stem, leading to a 2-fold higher affinity for streptavidin (Leppek and Stoecklin, 2014).
Bacterial transformation and purification
Once you obtain plasmids containing the insert, transform competent bacteria (e.g., E. coli TOP10) with the tagged gene following standard bacterial transformation protocols (Green and Rogers, 2013). Grow bacteria overnight on LB agar plates containing the appropriate antibiotics.
The next day, pick single colonies and perform colony PCR to verify that they contain the correct plasmid.
Inoculate bacteria from a single colony in 100 mL of selective medium and grow overnight at 37 °C in a shaking incubator at 200 rpm.
Purify the plasmid from the bacterial culture using the Qiagen Plasmid Midi kit, following the manufacturer’s instructions.
Confirm the sequence of the construct obtained by sequencing (e.g., Sanger sequencing).
Check the concentration and purity of the plasmid by spectrometry and agarose gel electrophoresis.Note: Expected 260/280 and 260/230 ratios are >1.8 and >2.0, respectively. A lower 260/280 ratio may indicate protein or phenol contamination, and a lower 260/230 ratio may indicate contamination by organic compounds.
Store purified plasmids at -20 °C or use directly for transfection (step B2).
Cell culture and transfection
Seed adherent HEK293T cells at 10% confluency in 150 cm2 dishes with 40 mL of complete DMEM medium.
Around 24 h after seeding, when cells are approximately 20% confluent, transfect them with the S1m-containing construct generated in step A:
Per 150 cm dish, mix in this order: 4 mL of Opti-MEM, 40 μg of DNA, and 120 μL of 1 mg/mL of PEI.
Note: A PEI:DNA ratio of 3:1 (w/w) was optimal for transfection of RNase MRP constructs, but this has to be optimized for each construct to reach the optimal expression levels. Adapt the quantities for transfection in accordance with the surface area, considering 1 μg of DNA per milliliter of culture medium.
As a mock control, transfect a dish with the same quantities of Opti-MEM and PEI without DNA.
Incubate at room temperature for 30 min.
Add the mixture dropwise to the cells and mix by swirling gently.
Optional: If the transfected plasmid expresses Gaussia or Renilla luciferase, assess transfection efficiency by measuring luciferase expression level. Twenty-four to forty-eight hours after transfection, harvest 200 μL of culture medium and assess luciferase activity using the BioLux® Gaussia Luciferase assay kit following the manufacturer’s instructions.
Forty-eight hours after transfection, harvest the cells by trypsinization.
Remove the culture medium.
Add 150 µL of 1× trypsin-EDTA per 10 cm2 dropwise to the cells at room temperature and incubate for 2–3 min at room temperature until more than 90% of the cells are detached.
Add at least four volumes of pre-warmed complete DMEM medium to deactivate the trypsin and centrifuge at 500–1000 × g for 5 min.
Wash the pellet once with at least three volumes of PBS.
Note: The cell pellet can be stored at -20 °C.
Purification of S1m-tagged complexes
Streptavidin purification
Lyse the harvested cells by resuspending the pellet in 1 mL of S1m lysis buffer and incubating on ice for 5 min.
Sonicate 10 times in the bioruptor on high power for 30 s to shear genomic DNA.
Centrifuge at 11,000 × g for 10 min at 4 °C and transfer the supernatant to a new tube to separate from insoluble material. Insoluble material can be discarded.
Add 1 mL of S1m incubation buffer.
Transfer 10% of the lysate to a new tube labeled as Input material for western and northern blotting analyses.
Note: The amount of material to save at this stage depends on the expression level of the complex of interest, purification efficiency, and number of analyses. For RNase MRP, we used 0.6% of input material for western blotting analysis, and 1% for northern blotting analysis.
Add the lysate and 3 μL of RNasin to 40 μL of packed pre-equilibrated streptavidin Sepharose beads. To pre-equilibrate the beads, wash them three times with 0.5 mL of S1m wash buffer and centrifuge at 800 × g for 2 min.
Incubate overnight at 4 °C under agitation using a rotary mixer.
Centrifuge at 800 × g for 2 min at 4 °C and transfer 10% of the supernatant to a new tube labeled as Non-bound material for western and northern blotting analyses.
Note: Save the same amount as for the Input.
Wash the beads three times for 5 min with 1 mL of S1m wash buffer by incubation at 4 °C under agitation and centrifugation at 800 × g for 2 min at 4 °C. The wash fractions can be discarded.
During the last wash, split the resuspended beads into two tubes, to use one for protein extraction and the other for RNA extraction.
Protein extraction from purified material
Centrifuge one of the two tubes to pellet the beads and discard the supernatant.
Add 20 µL of 4× protein sample buffer directly to the beads.
Note: The amount of material to load on each gel depends on the purification efficiency. For RNase MRP, we used 10%–25% of bound material for western blotting analysis.
Store the purified material at -20 °C or use directly for western blotting (step D2).
RNA extraction from purified material
Centrifuge the second tube to pellet the beads and discard the supernatant.
Add 1 mL of TRIzol reagent directly to the beads and isolate the RNA following the manufacturer’s instructions.
Note: We recommend performing an additional extraction step of the aqueous phase with one volume of chloroform and adding 10 µg of glycogen or GlycoBlue as a carrier during precipitation.
Dissolve the RNA in 10–30 µL of RNase-free water.
Store the purified RNA at -80 °C.
Identification of the composition of purified material
Northern blotting
Verify the presence of your tagged RNA of interest by northern blot hybridization. The probe used to detect the RNA of interest should lead to the detection of both the endogenous RNA and the tagged RNA, in order to compare their expression levels. The analysis of a non-related RNA is recommended. This can serve as a loading control for the input fractions and to have a reference for the specificity of RNA purification.
Here, we describe the procedure for northern blotting using an α-32P-labeled RNA probe (riboprobe), the most sensitive and specific type of radiolabeled probe. For RNAs expressed at a high level, 32P 5′-end-labeled DNA oligonucleotides or non-radioactive (e.g., fluorescently labeled) RNA or DNA probes can also be used. For detailed protocols describing these methods, see Tabor and Struhl (2001) and Franke et al. (2015), respectively.
Probe cloning
Insert (part of) the sequence of the RNA of interest in a plasmid, using a standard PCR-based strategy. The probe should be at least 100 nucleotides long. The vector should contain an in vitro transcription promoter (such as the promoter for T7 or SP6 RNA polymerase) upstream of the sense sequence of the RNA of interest, to generate a complementary probe after transcription. For RMRP, the pGEM-3Zf plasmid containing the complete sense sequence of RMRP downstream of the T7 promotor was used.
Once you obtain the plasmid containing the probe sequence, follow steps A3a–g to amplify and purify the plasmid.
In vitro transcription
Linearize the plasmid by digestion with the appropriate restriction enzyme cleaving downstream of the probe sequence.
Purify the linearized plasmid by 25:24:1 phenol:chloroform:isoamyl alcohol extraction and ethanol precipitation [for details on the protocol, see Suchan (2020)].
Confirm linearization by agarose gel electrophoresis. Load both the circular and linear plasmid on the same gel and compare the size of the bands. The linear plasmid should migrate at a different position compared to the circular plasmid.
Mix in the following order: RNase-free water (volume resulting in a total reaction volume of 20 µL), 2 µL of 100 mM DTT, 4 µL of 5× transcription optimized buffer, 2 µL of IVT NTP mix, 40 U of RNA polymerase, 2 µL of RNasin (80 units), 2 µL 32P-α-UTP (3000 Ci/mmol), and 1 µg of linearized plasmid.
Incubate for 1 h at 37 °C.
Purify the in vitro–transcribed probe using a Sephadex G50 column following the manufacturer’s instructions.
Measure the amount of radioactivity in the purified probe and the column using a Geiger counter.
Note: The radioactivity level should be higher in the purified RNA probe than in the column; the opposite indicates sub-optimal production of the labeled probe.
Gel electrophoresis
This protocol describes the procedure for the use of a polyacrylamide gel, but an agarose gel can also be used. Additional information can be found in more detailed protocols (Petrov et al., 2013).
Prepare a polyacrylamide RNA denaturing gel.
Pre-run the gel in 1× TBE for 30 min at 30 mA.
Add one volume of RNA sample buffer to the Input, Non-bound, and purified RNA samples from transfected and non-transfected cells and incubate for 5 min at 95 °C to denature the RNA. Place samples back on ice immediately.
After pre-running, clean the slots using a syringe and load the samples.
Run the gel at 30 mA.
Note: The running time depends on the size of the RNA molecules of interest. Use the colored dyes to determine when to stop running the gel (Petrov et al., 2013). For RNase MRP RNA, the gel should be run until the dark blue dye (bromophenol blue) reaches the bottom of the gel.
RNA blotting and detection
Transfer the RNA to a Hybond N+ membrane in northern blotting buffer for 1 h at 500 mA at room temperature.
Allow the blot to dry and crosslink the RNA to the membrane by UV irradiation at 700 mJ/cm2.
Note: Store the blot dry between filter paper at 4 °C if needed.
Block the blot for 1 h at 65 °C with pre-hybridization buffer.
Add the RNA probe to the pre-hybridization buffer and incubate at 65 °C overnight.
Wash the blot twice with 1× SSC and 0.2% SDS, using enough buffer to cover the whole blot.
Wash the blot once or twice with 0.1× SSC until no radioactivity is detected anymore at regions of the blot where no signal is expected.
Wrap the blot in plastic foil and put it in a cassette with a Phosphor Screen.
Develop with a phosphor imager to visualize the result (for an example see Figure 2).
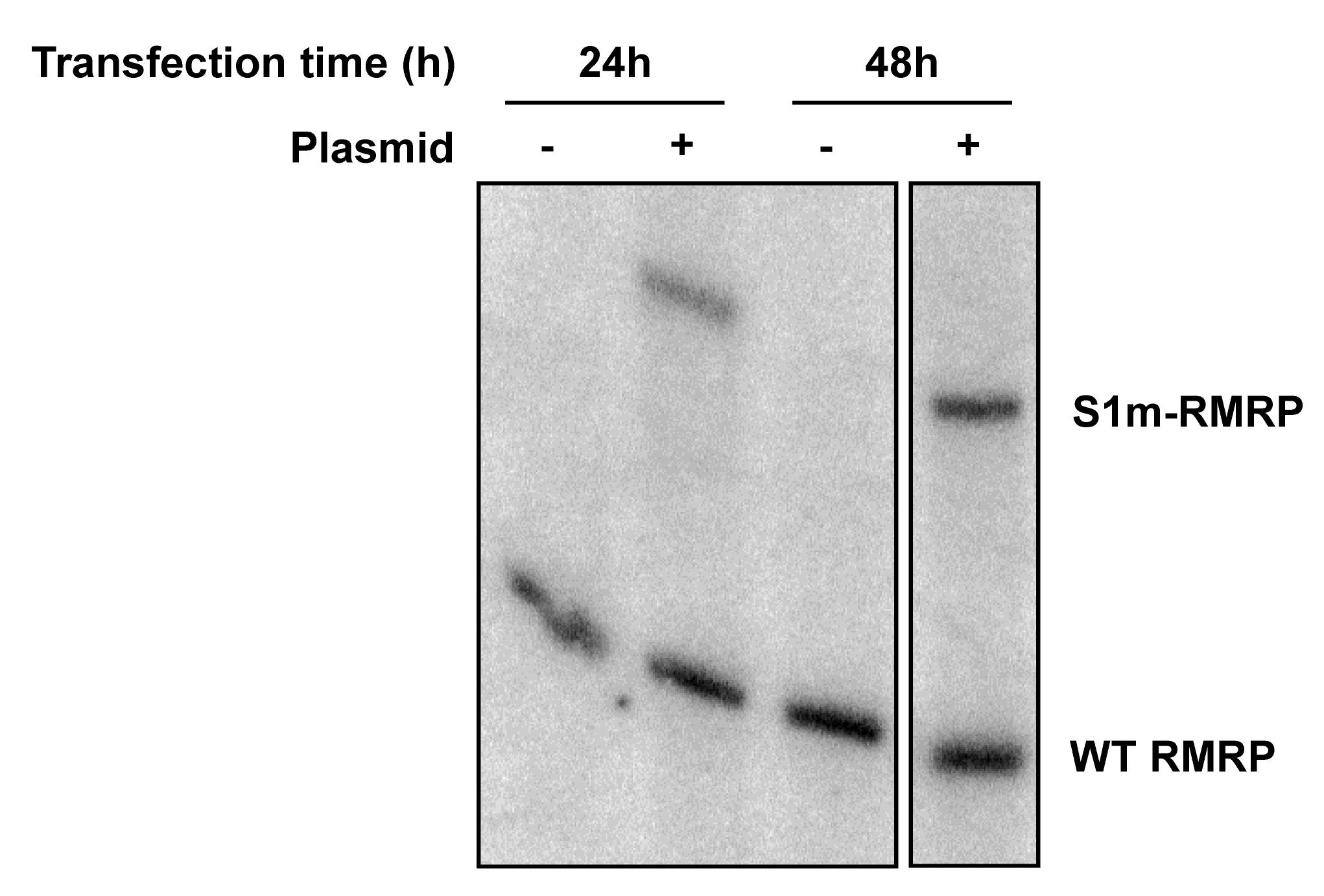
Figure 2. Northern blot analysis of S1m-tagged RNA. HEK293 cells were transfected with a S1m-RMRP construct to transiently express the tagged RNA and cultured for 24 or 48 h before extracting total RNA with TRIzol. As a control (- plasmid), cells subjected to the transfection procedure without the plasmid construct were used. Equal amounts of RNA were separated by denaturing polyacrylamide gel electrophoresis, and both wild-type and S1m-tagged RMRP were detected using a radiolabeled probe complementary to RMRP.
Western blotting
Protein gel electrophoresis
Prepare an SDS-PAGE gel [see Recipes and detailed protocol in Sambrook and Russell (2006)].
After polymerization, remove the comb carefully and clean the slots using a syringe.
Mix one volume of Input and Non-bound material with three volumes of 4× protein sample buffer.
Load the Input, Non-bound, and purified protein samples after 2–5 min incubation at 95 °C and centrifugation to remove insoluble material.
Note: Load up to 15 μL for a 0.75 mm thick gel and up to 30 μL for a 1.5 mm thick gel.
Run the gel in SDS-PAGE running buffer at 90 V constant for approximately 15 min, until the dye reaches the bottom of the stacking gel. Then, run at 150 V constant for approximately 50–60 min, until the dye reaches the bottom of the running gel.
Remove the gel from the running apparatus, carefully remove one of the glass plates and cut off the stacking gel.
Protein blotting
Assemble the transfer cassette: anode (-); foam mat; two sheets of Whatman paper; gel; nitrocellulose membrane; two sheets of Whatman paper, foam mat; cathode (+).
Note: Soak all Whatman papers and the membrane in western blotting buffer before assembling the blotting sandwich. Avoid any air bubbles between the anode and cathode and gently remove any using a roller before closing the cassette to ensure efficient transfer on the whole surface of the blot.
Insert the cassette into a blotting apparatus. Add an ice pack in the apparatus or keep the apparatus cold during the transfer.
Transfer the proteins to a nitrocellulose membrane in western blotting buffer with magnetic stirring for at least 1 h at constant 100 V or overnight at constant 250 mA.
Note: For thick gels (1.5 mm), transfer for at least 1.5 h.
After transfer, remove the blot from the cassette and briefly incubate in Ponceau-S solution until clear bands appear for the Input and Non-bound fractions. Rinse the blot a few times with demineralized water and make an image of the stained blot.
Immunodetection
Block non-specific antibody binding by incubating the membrane in 10 mL of blot blocking solution at room temperature for at least 30 min with gentle shaking.
Discard the blocking solution and add the primary antibody diluted to working concentration in 5–10 mL of blocking solution. Incubate for at least 1 h at room temperature or ideally overnight at 4 °C, with shaking or rotation.
Wash the membrane three times for 5 min with 10–20 mL of PBST.
Add the IRDye® 800CW- or 680RD-conjugated secondary antibody diluted to working concentration in 5–10 mL of blocking solution. Incubate for at least 1 h at room temperature, protected from the light.
Note: Ensure that the secondary antibody used is targeted against immunoglobulins of the organism of origin of the primary antibody.
Wash the membrane two times for 5 min with 10–20 mL of PBST.
Briefly wash the membrane with 10 mL of PBS and keep it in PBS protected from the light until imaging.
Note: We observe that after rinsing the membrane with PBS, less background fluorescence is observed when imaging.
Detect the fluorescence using the Odyssey imaging system or Typhoon imager with the 700 nm or 800 nm channel, depending on the secondary antibody used.
Data analysis
Confirm by northern blotting that the band corresponding to the endogenous non-tagged RNA is visible in material from both transfected and non-transfected cells, in Input and Non-bound fractions. The RNA containing the aptamer should be visible in Input and purified material from transfected cells only. Normalize the expression level of the RNA of interest in different conditions to the control RNA.
After western blotting, no or very weak bands are expected in the bound material, while the Ponceau-S staining is expected to be highly similar in Input and Non-bound fractions.
Confirm the efficiency of the purification by immunodetection of known protein partners in the purified samples of transfected cells (for an example see Figure 3).
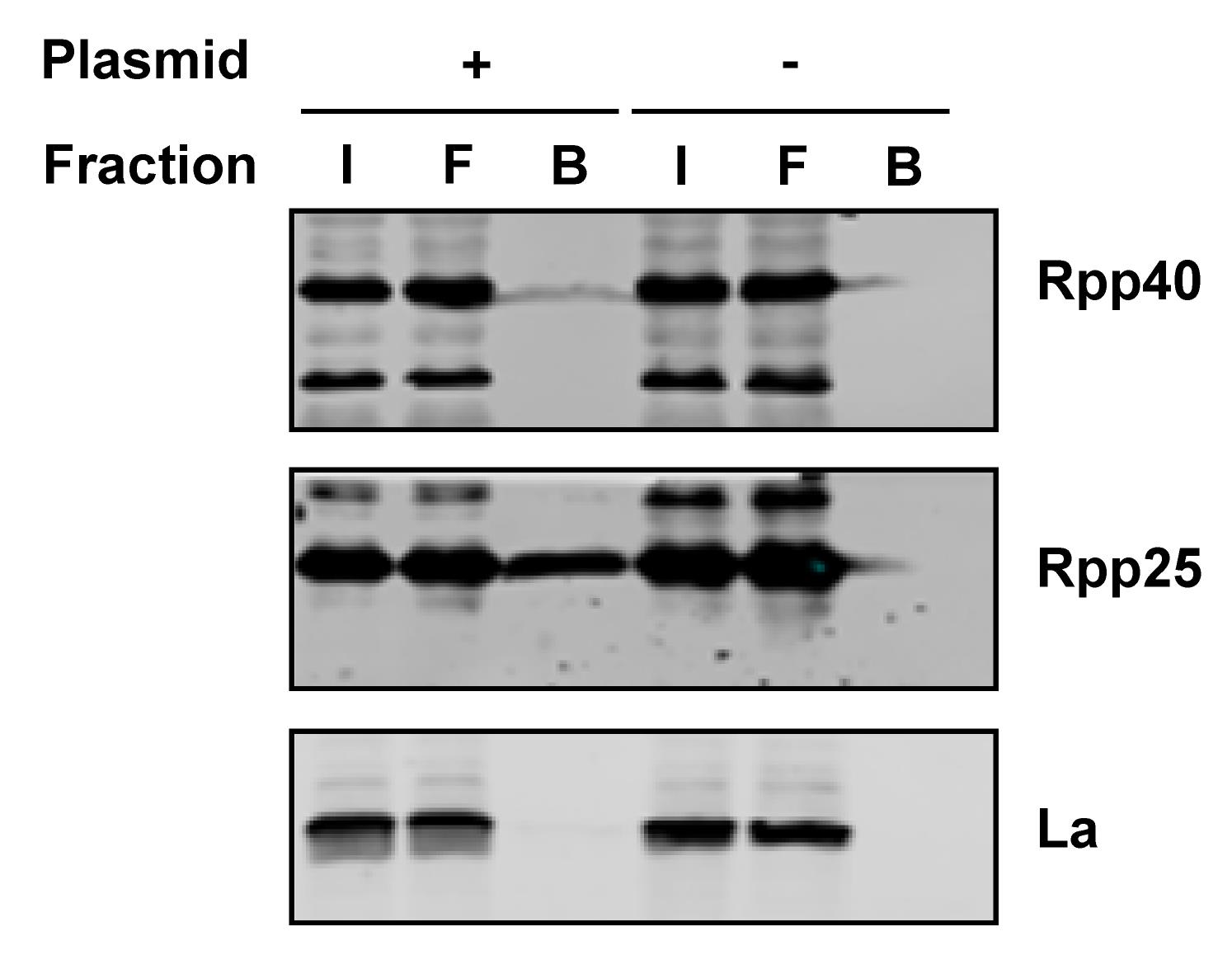
Figure 3. Identification of proteins in the isolated complexes by western blotting. HEK293 cells were transfected with a S1m-RMRP construct to transiently express the tagged RNA and cultured for 48 hours. As a control (- plasmid), cells subjected to the transfection procedure without the plasmid construct were used. The cells were lysed, tagged ribonucleoprotein complexes were purified with streptavidin Sepharose beads, and proteins were eluted with protein sample buffer. Equal amounts of Input (I) and Flow-through (F) and three times more Bound (B) samples were separated by SDS-PAGE and analyzed by western blotting using specific antibodies. Rpp40 and Rpp25 are known components of the RNase MRP; the La protein, which does not interact with the complex, was used as a negative control.Recipes
Luria-Bertani (LB) broth and agar
1% (w/v) tryptone
1% (w/v) NaCl
0.5% (w/v) yeast extract
To prepare LB agar, add 1.5% (w/v) agar
Adjust pH to 7.0 with NaOH
Autoclave and store at room temperature
To prepare LB agar plates, warm up to melt the agar and let it cool to <60 °C before adding antibiotics. Pour 10–20 mL per Petri dish in a sterile environment, let it cool, and store at 4 °C
Complete DMEM medium
DMEM
10% FCS
1% penicillin-streptomycin mix
Store at 4 °C until use
10× tyrode solution
1.37 M NaCl
27 mM KCl
3.6 mM NaH2PO4·H2O
120 mM NaHCO3
111 mM glucose
Mix all components except the glucose, adjust pH to 7.2 if necessary and autoclave. Then, add the glucose and keep sterile.
Store at room temperature
10× Trypsin-EDTA solution
3.5% (w/v) trypsin
1% (w/v) EDTA
Dissolve in 10× tyrode solution by shortly incubating at 37 °C
Adjust pH to 7.4 with NaOH
Filtrate through a sterile 0.22 µm filter
Store at -20 °C or use directly to prepare 1× aliquots
To prepare 1× solution for use in cell culture, dilute 10 times with sterile ultrapure water and store aliquots at -20 °C
Polyethyleneimine (PEI) stock
Add ultrapure water to PEI so that the final concentration is 1 mg/mL
Incubate for 1 h at room temperature under agitation
Incubate for 30 min at 50 °C
Incubate for 30 min at room temperature under agitation
Filtrate through a sterile 0.22 µm filter
Store aliquots at -70 °C or -80 °C (up to three months)
S1m lysis buffer
50 mM Tris-HCl, pH 7.6
10 mM MgCl2
100 mM KCl
10% glycerol
0.1% NP-40
Make fresh and add cOmplete protease inhibitor and 1 mM of DTT right before use
S1m incubation buffer
50 mM Tris-HCl, pH 7.6
10 mM MgCl2
100 mM KCl
10% glycerol
Make fresh and add cOmplete protease inhibitor and 1 mM of DTT right before use
S1m wash buffer
50 mM Tris-HCl, pH 7.6
10 mM MgCl2
100 mM KCl
10% glycerol
0.05% NP-40
Make fresh and add cOmplete protease inhibitor and 1 mM of DTT right before use
4× protein sample buffer
200 mM Tris-HCl, pH 6.8
8% SDS
40% glycerol
5% β-mercaptoethanol
50 mM EDTA
0.05% bromophenol blue
Store at 4 °C
IVT NTP mix
1 mM ATP
1 mM GTP
1 mM CTP
0.1 mM UTP
Store at -20 °C
1× TBE
100 mM Tris
100 mM boric acid
2 mM EDTA
Store at room temperature
RNA denaturing gel
1× TBE
8 M urea
X% 19:1 acrylamide:bisacrylamide
0.1% TEMED
0.1% fresh APS (prepare 10% APS and use immediately or store single-use aliquots at -20 °C)
Choose the percentage of acrylamide based on the size of the RNA of interest.
Note: Prepare 500 mL of RNA denaturing gel mix without TEMED and APS and store at 4 °C if you plan to make multiple RNA gels.
Before use, warm up at 37 °C to dissolve the urea if needed and add TEMED and APS. Beware: the gel will polymerize faster when the solution is warm.
2× RNA sample buffer
8 M urea
1× TBE
0.05% XCFF
0.05% BFB
Store at room temperature
Northern blotting buffer
18.4 mM NaH2PO4
6.5 mM Na2HPO4
Store at room temperature
20× SSC
3.0 M NaCl
0.3 M sodium citrate
Adjust to pH 7.0 with HCl
Store at room temperature
100× Denhardt’s
2% BSA
2% Ficoll® 400
2% polyvinylpyrrolidone
Store at -20 °C
10 mg/mL sheared herring sperm DNA
Dissolve herring sperm DNA at 10 mg/mL in ultrapure water
Sonication is required for the proper preparation of the solution
Store at -20 °C
Pre-hybridization buffer
6× SSC
0.1 mg/mL sheared herring sperm DNA
0.2% SDS
10× Denhardt’s
Store at room temperature
SDS-PAGE running gel
0.4 M Tris-HCl, pH 8.8
X% 37:1 acrylamide:bisacrylamide
0.1% SDS
0.1% TEMED
0.1% fresh APS (prepare 10% APS and use immediately or store single-use aliquots at -20 °C)
Choose the percentage of acrylamide based on the size of the proteins of interest.
SDS-PAGE stacking gel
70 mM Tris-HCl, pH 6.8
4% 37:1 acrylamide:bisacrylamide
0.1% SDS
0.1% TEMED
0.1% fresh APS (Prepare 10% APS and use immediately or store single-use aliquots at -20 °C)
SDS-PAGE running buffer
Tris-glycine (25 mM Tris, 192 mM glycine)
0.1% SDS
Store at room temperature
Western blotting buffer
Tris-glycine (25 mM Tris, 192 mM glycine)
20% methanol
0.1% SDS
Store at room temperature
Blot blocking solution
PBS
0.1% Tween-20
5% non-fat dry milk
Store single-use aliquots at -20 °C
PBST
PBS
0.1% Tween-20
Store at room temperature
Acknowledgments
This procedure was adapted from the original research article (Derksen et al., 2022), where we demonstrate the efficient isolation of RNase MRP and the possibility to use this method to study protein binding and substrate cleavage activity of wild-type and mutant RNase MRP RNA.
Competing interests
The authors declare no conflict of interest.
References
- Bachler, M., Schroeder, R. and von Ahsen, U. (1999). StreptoTag: a novel method for the isolation of RNA-binding proteins. RNA 5(11): 1509-1516.
- Chang, D. D. and Clayton, D. A. (1987). A novel endoribonuclease cleaves at a priming site of mouse mitochondrial DNA replication. EMBO J 6(2): 409-417.
- Coughlin, D. J., Pleiss, J. A., Walker, S. C., Whitworth, G. B. and Engelke, D. R. (2008). Genome-wide search for yeast RNase P substrates reveals role in maturation of intron-encoded box C/D small nucleolar RNAs. Proc Natl Acad Sci U S A 105(34): 12218-12223.
- Derksen, M., Mertens, V., Visser, E. A., Arts, J., Vree Egberts, W. and Pruijn, G. J. M. (2022). A novel experimental approach for the selective isolation and characterization of human RNase MRP. RNA Biol 19(1): 305-312.
- Franke, C., Grafe, D., Bartsch, H. and Bachmann, M. P. (2015). Use of Nonradioactive Detection Method for North- and South-Western Blot. Methods Mol Biol 1314: 63-71.
- Hermanns, P., Tran, A., Munivez, E., Carter, S., Zabel, B., Lee, B. and Leroy, J. G. (2006). RMRP mutations in cartilage-hair hypoplasia. Am J Med Genet A 140(19): 2121-2130.
- Gill, T., Cai, T., Aulds, J., Wierzbicki, S. and Schmitt, M. E. (2004). RNase MRP cleaves the CLB2 mRNA to promote cell cycle progression: novel method of mRNA degradation. Mol Cell Biol 24(3): 945-953.
- Green, R. and Rogers, E. J. (2013). Transformation of chemically competent E. coli. Methods Enzymol 529: 329-336.
- Guerrier-Takada, C., Gardiner, K., Marsh, T., Pace, N. and Altman, S. (1983). The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell 35(3 Pt 2): 849-857.
- Hou, S., Shi, L. and Lei, H. (2016). Biotin-Streptavidin Affinity Purification of RNA-Protein Complexes Assembled In Vitro. Methods Mol Biol 1421: 23-34.
- Lan, P., Zhou, B., Tan, M., Li, S., Cao, M., Wu, J. and Lei, M. (2020). Structural insight into precursor ribosomal RNA processing by ribonuclease MRP. Science 369(6504): 656-663.
- Leppek, K. and Stoecklin, G. (2014). An optimized streptavidin-binding RNA aptamer for purification of ribonucleoprotein complexes identifies novel ARE-binding proteins. Nucleic Acids Res 42(2): e13.
- Li, Y. and Altman, S. (2002). Partial reconstitution of human RNase P in HeLa cells between its RNA subunit with an affinity tag and the intact protein components. Nucleic Acids Res 30(17): 3706-3711.
- Lygerou, Z., Allmang, C., Tollervey, D. and Seraphin, B. (1996). Accurate processing of a eukaryotic precursor ribosomal RNA by ribonuclease MRP in vitro. Science 272(5259): 268-270.
- Mattijssen, S., Welting, T. J. and Pruijn, G. J. (2010). RNase MRP and disease. Wiley Interdiscip Rev RNA 1(1): 102-116.
- Mattijssen, S., Hinson, E. R., Onnekink, C., Hermanns, P., Zabel, B., Cresswell, P. and Pruijn, G. J. (2011). Viperin mRNA is a novel target for the human RNase MRP/RNase P endoribonuclease. Cell Mol Life Sci 68(14): 2469-2480.
- Park, O. H., Ha, H., Lee, Y., Boo, S. H., Kwon, D. H., Song, H. K. and Kim, Y. K. (2019). Endoribonucleolytic Cleavage of m(6)A-Containing RNAs by RNase P/MRP Complex. Mol Cell 74(3): 494-507 e498.
- Petrov, A., Tsa, A. and Puglisi, J. D. (2013). Analysis of RNA by analytical polyacrylamide gel electrophoresis. Methods Enzymol 530: 301-313.
- Pluk, H., van Eenennaam, H., Rutjes, S. A., Pruijn, G. J. and van Venrooij, W. J. (1999). RNA-protein interactions in the human RNase MRP ribonucleoprotein complex. RNA 5(4): 512-524.
- Ramanathan, M., Porter, D. F. and Khavari, P. A. (2019). Methods to study RNA-protein interactions. Nat Methods 16(3): 225-234.
- Randau, L., Schroder, I. and Soll, D. (2008). Life without RNase P. Nature 453(7191): 120-123.
- Reiner, R., Ben-Asouli, Y., Krilovetzky, I. and Jarrous, N. (2006). A role for the catalytic ribonucleoprotein RNase P in RNA polymerase III transcription. Genes Dev 20(12): 1621-1635.
- Reiner, R., Krasnov-Yoeli, N., Dehtiar, Y. and Jarrous, N. (2008). Function and assembly of a chromatin-associated RNase P that is required for efficient transcription by RNA polymerase I. PLoS One 3(12): e4072.
- Robertson, H. D., Altman, S. and Smith, J. D. (1972). Purification and properties of a specific Escherichia coli ribonuclease which cleaves a tyrosine transfer ribonucleic acid precursor. J Biol Chem 247(16): 5243-5251.
- Sambrook, J. and Russell, D. W. (2006). SDS-Polyacrylamide Gel Electrophoresis of Proteins. CSH Protoc 2006(4).
- Srisawat, C. and Engelke, D. R. (2001). Streptavidin aptamers: affinity tags for the study of RNAs and ribonucleoproteins. RNA 7(4): 632-641.
- Srisawat, C. and Engelke, D. R. (2002). RNA affinity tags for purification of RNAs and ribonucleoprotein complexes. Methods 26(2): 156-161.
- Suchan, T. (2020). Phenol-chloroform DNA purification. protocols.io: DOI: dx.doi.org/10.17504/protocols.io.re6d3he.
- Tabor, S. and Struhl, K. (2001). Enzymatic labeling of DNA. Curr Protoc Hum Genet Appendix 3: Appendix 3E.
- Walker, S. C., Scott, F. H., Srisawat, C. and Engelke, D. R. (2008). RNA affinity tags for the rapid purification and investigation of RNAs and RNA-protein complexes. Methods Mol Biol 488: 23-40.
- Welting, T. J., Kikkert, B. J., van Venrooij, W. J. and Pruijn, G. J. (2006). Differential association of protein subunits with the human RNase MRP and RNase P complexes. RNA 12(7): 1373-1382.
- Welting, T. J., Mattijssen, S., Peters, F. M., van Doorn, N. L., Dekkers, L., van Venrooij, W. J., Heus, H. A., Bonafe, L. and Pruijn, G. J. (2008). Cartilage-hair hypoplasia-associated mutations in the RNase MRP P3 domain affect RNA folding and ribonucleoprotein assembly. Biochim Biophys Acta 1783(3): 455-466.
- Yoon, J. H. and Gorospe, M. (2016). Identification of mRNA-Interacting Factors by MS2-TRAP (MS2-Tagged RNA Affinity Purification). Methods Mol Biol 1421: 15-22.
- Yoon, J. H., Srikantan, S. and Gorospe, M. (2012). MS2-TRAP (MS2-tagged RNA affinity purification): tagging RNA to identify associated miRNAs. Methods 58(2): 81-87.
Note: The amount of material to load on each gel depends on the purification efficiency. For RNase MRP, we used 15% of bound material for northern blotting analysis.
Article Information
Copyright
© 2023 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Charteau, V., Derksen, M. and Pruijn, G. J. (2023). A Novel Method to Isolate RNase MRP Using RNA Streptavidin Aptamer Tags. Bio-protocol 13(4): e4615. DOI: 10.21769/BioProtoc.4615.
Category
Biochemistry > Protein > Isolation and purification
Molecular Biology > RNA > RNA detection
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.