- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Analysis of N6-methyladenosine RNA Modification Levels by Dot Blotting
Published: Vol 12, Iss 23, Dec 5, 2022 DOI: 10.21769/BioProtoc.4565 Views: 4872
Reviewed by: Chiara AmbrogioWilly R Carrasquel-UrsulaezTanxi CaiYoshihiro Adachi
Original research article
The authors used this protocol in:

May 2022
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
N6-methyladenosine (m6A) is the most prevalent internal modification of eukaryotic messenger RNAs (mRNAs), affecting their fold, stability, degradation, and cellular interaction(s) and implicating them in processes such as splicing, translation, export, and decay. The m6A modification is also extensively present in non-coding RNAs, including microRNAs (miRNAs), ribosomal RNAs (rRNAs), and transfer RNAs (tRNAs). Common m6A methylation detection techniques play an important role in understanding the biological function and potential mechanism of m6A, mainly including the quantification and specific localization of m6A modification sites. Here, we describe in detail the dot blotting method for detecting m6A levels in RNA (mRNA as an example), including total RNA extraction, mRNA purification, dot blotting, and data analysis. This protocol can also be used to enrich specific RNAs (such as tRNA, rRNA, or miRNA) by isolation technology to detect the m6A level of single RNA species, so as to facilitate further studies of the role of m6A in biological processes.
Keywords: Dot blotBackground
N6-methyladenosine (m6A) is the most prevalent internal RNA modification in eukaryotic mRNAs and long non-coding RNAs (Jia et al., 2013). The m6A modification refers to the methylation of the nitrogen atom at position 6 of the RNA adenosine, mainly located on the common motif of RRm6ACH (R denotes A or G, H denotes A, C, or U) (Csepany et al., 1990). The reversible activity of m6A modification is regulated by the combined action of methylases and demethylases (Niu et al., 2013). The m6A writers with methyltransferase activity consist of three individual proteins: METTL3, METTL14, and WTAP (Wiedmer et al., 2019); FTO and ALKBH5 are m6A demethylases (Nachtergaele and He, 2018). Another protein family is the m6A readers, which can recognize the m6A modification to modulate the fate of mRNA (Harcourt et al., 2017). Recent studies suggest that m6A plays a pivotal role during various biological processes including virus infection (Winkler et al., 2019), stress (Engel et al., 2018), heat shock (Zhou et al., 2015), and DNA damage (Xiang et al., 2017; E. Li et al., 2022). In mammals, m6A methylation plays a variety of key roles including embryonic development, neurogenesis, circadian rhythm, stress responses, sex determination, and tumorigenesis (Sun et al., 2019). The m6A modification is vital during stem cell proliferation, with METTL3 depletion reducing the differentiation of embryonic stem cells (Batista et al., 2014). The correlation between the level of m6A modification and clinicopathological features has been shown in diverse tumors, which may provide prognostic value in these diseases.
Detection of m6A modification in vitro can help identify the precise regulatory forms and synergistic roles of m6A modifications in cancer and other diseases. Also, detection of m6A is important for studying its biological functions and mechanisms. Currently, a variety of methods have been developed to identify m6A modifications in cells, which can be divided into three categories: semi-quantitative, quantitative, and precise location detection. Semi-quantitative methods include dot blot technology (Z. Li et al., 2017), methyl-sensitivity of MazF RNA endonucleases (Imanishi et al., 2017), and immuno-northern blotting (Mishima et al., 2015). Quantification methods include RNA photo-crosslinkers and quantitative proteomics (Arguello et al., 2017), electrochemical immunosensors (Yin et al., 2017), and support vector machine–based methods (Chen et al., 2016). Precise location detection includes methylated RNA immunoprecipitation sequencing (MeRIP-seq) (Liu et al., 2018), m6A individual-nucleotide-resolution cross-linking and immunoprecipitation (miCLIP) (Linder et al., 2015), and high-performance liquid chromatography (HPLC) (Rana and Tuck, 1990). Although many methods have been developed to detect m6A methylation, in many cases dot blot hybridization remains the method of choice for analyzing the global changes of m6A levels in total RNA or single RNA species. The dot blotting technique significantly saves time because it does not require chromatography, gel electrophoresis, or complex gel closure procedures, and is relatively low in cost (Wang et al., 2018). In this protocol, we describe in detail how to detect m6A content in mRNA by dot blot (Figure 1).
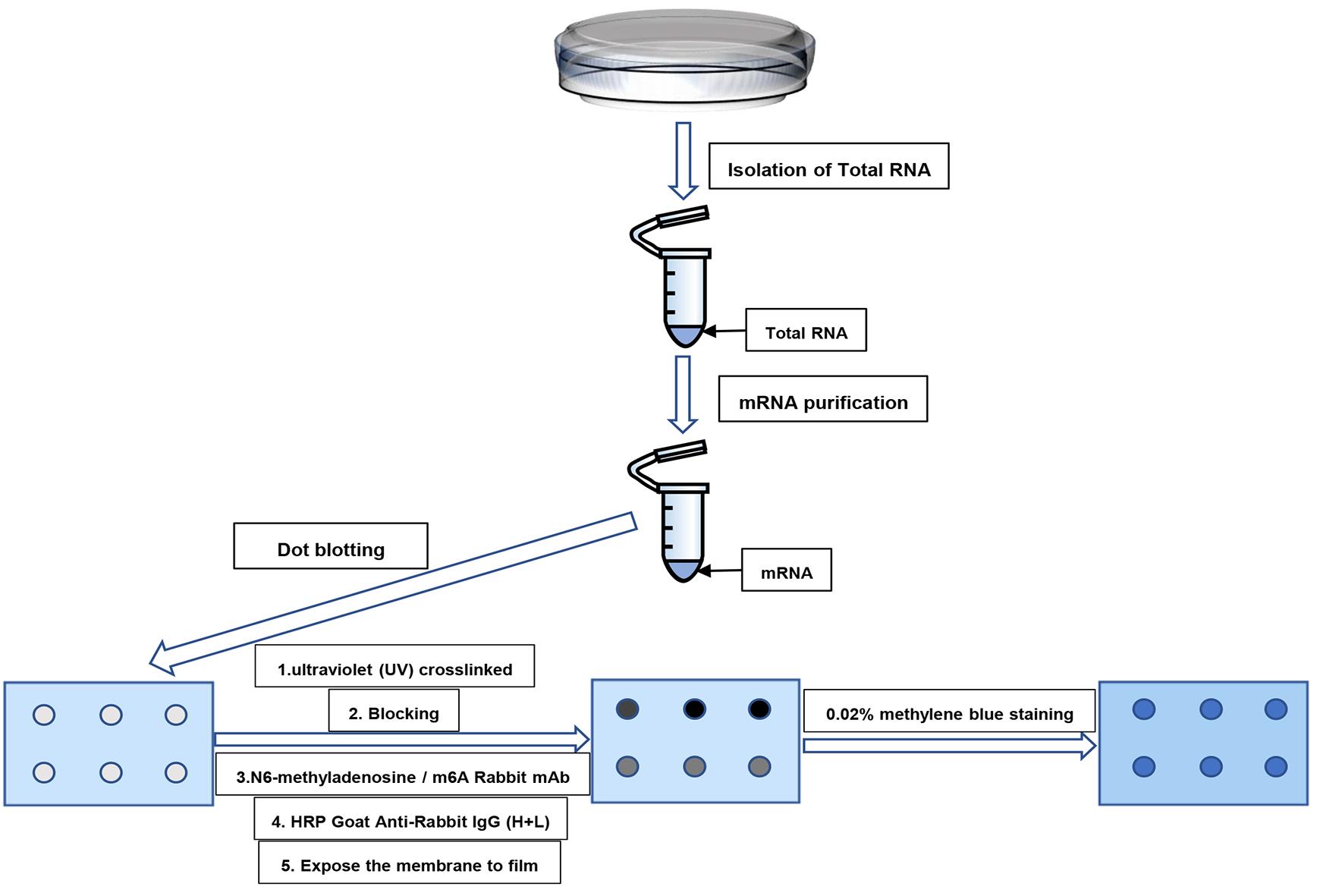
Figure 1. Schematic diagram of the major steps of dot blot analysis for the separation and purification of m6A in mRNA
Materials and Reagents
100 mm culture dish (Corning, Falcon, catalog number: 353003)
Pipette tips (Axygen, catalog numbers: T-300, T-200Y, T-1000B)
1.5 mL RNase/DNase-free microcentrifuge tube (Axygen, catalog numbers: MCT-50-C)
Cell scraper (any brand)
TRIzol reagent (Invitrogen, catalog number:15596026)
Chloroform (General-Reagent, catalog number: G75915B)
Isopropanol (Sangon Biotech, catalog number: A507048)
Ethanol absolute (Sangon Biotech, catalog number: A500737)
RNase-free water
GenElute mRNA miniprep kit (Sigma, catalog number: DMN10)
AmershamTM HybondTM-N+ membrane (GE Healthcare, catalog number: RPN203B )
N6-methyladenosine/m6A rabbit mAb (ABclonal, catalog number: A19841)
HRP goat anti-rabbit IgG (H+L) (ABclonal, catalog number: AS014)
ECL western blotting substrate (Thermo Fisher Scientific, catalog number: 32106)
Saran wrap (any brand)
Tween-20 (Sigma-Aldrich, catalog number: P1379)
Non-fat milk (any brand)
Na2HPO4 (Sangon Biotech, catalog number: A501727)
KH2PO4 (Sangon Biotech, catalog number: A100781)
KCl (Sangon Biotech, catalog number: A100395)
NaCl (Sangon Biotech, catalog number: A501218)
10× phosphate-buffered saline (PBS) (see Recipes)
1× phosphate-buffered saline (PBS) (see Recipes)
1× phosphate-buffered saline/0.1% Tween-20 (PBST) (see Recipes)
Blocking buffer (see Recipes)
Antibody dilution buffer (see Recipes)
Methylene blue dye buffer (Solarbio, catalog number: G1301)
Equipment
Refrigerated centrifuge (Eppendorf, model: 5424 R)
NanoDrop (Thermo Fisher Scientific, model: ND-1000 spectrophotometer)
UV cross-linker or UV torch with 254 nm wavelength UV (UVP, model: CL1000)
Chemiluminescent imaging system (Tanon, model: Tanon 5200)
Camera (any brand)
Software
GraphPad Prism (GraphPad Software)
ImageJ (https://imagej.nih.gov/ij)
Procedure
Isolation of total RNA
For mammalian cells we recommend using approximately 1 × 107 cells for mRNA purification.
Remove the medium and rinse the cells twice with 1–2 mL of ice-cold PBS.
Remove PBS and lyse the cells directly in the culture dish by adding 1 mL of TRIzol reagent per 100 mm culture dish and scraping with a cell scraper.
Pass the cell lysate through a pipette several times. Then, transfer to 1.5 mL RNase-free microcentrifuge tubes. Vortex thoroughly.
Incubate the homogenized samples for 5 min at room temperature.
Add 0.2 mL of chloroform, vortex for 30 s, and incubate for 2–3 min at room temperature.
Centrifuge at 12,000 × g and 4 °C for 15 min.
Carefully transfer the upper, aqueous phase to a fresh tube without disturbing the interface.
Add 0.5 mL of isopropanol to the aqueous phase and mix well. Incubate the samples for 10 min at room temperature.
Centrifuge at 12,000 × g and 4 °C for 10 min.
Decant the supernatant and completely remove any traces of liquid by aspiration.
Wash the pellet with 1 mL of ice-cold 75% ethanol.
Centrifuge at 12,000 × g and 4 °C for 15 min.
Remove all traces of ethanol. Air-dry or vacuum-dry the RNA pellet for 5–10 min.
Resuspend the pellet in 10–30 μL of RNase-free water.
mRNA purification
Isolate mRNA from total RNA using the GenElute mRNA miniprep kit following the manufacturer’s instructions (other brands can also be used for the purification of mRNA).
Mix the oligo (dT) beads thoroughly by vortexing and inverting until resuspended and homogenous.
Add the resuspended oligo (dT) beads to the total RNA, cap the tube, and mix thoroughly by vortexing.
Incubate the mixture at room temperature for 10 min. No mixing or rocking is necessary.
Resuspend the pellet in 350 μL of wash solution by vortexing.
Transfer the suspension into a GenElute spin filter–collection tube assembly by pipetting. Spin for 1–2 min at maximum speed.
Remove the spin filter, discard the flow through liquid, then place the spin filter back into the same collection tube.
Pipette 350 μL of low salt wash solution into the column. Spin for 1–2 min at maximum speed.
Remove the spin filter, discard the flow through liquid, then place the spin filter back into the same collection tube.
Pipette another 350 μL of low salt wash solution into the column. Spin for 1–2 min at maximum speed.
Transfer the spin filter into a fresh collection tube. Pipette 10–20 μL of preheated (65 °C) elution solution onto the spin filter, ensuring that it comes into contact with the bead–mRNA complex. Incubate for 2–5 minutes at 65 °C. Spin for 1–2 min at maximum speed. Save the flow through liquid; it contains most of the purified mRNA.
Determine the concentration of purified mRNA with NanoDrop.
Dilute different concentrations of mRNA to 50 ng/μL.
Dot blotting
The isolated mRNA is first denatured by heating at 95 °C for 5 min to disrupt the secondary structure.
Chill on ice for 5 min immediately after denaturation to prevent re-formation of the mRNA secondary structure.
Have the nitrocellulose membrane ready.
Using a narrow-mouth pipette tip, drop 2 μL of mRNA sample directly onto the Hybond-N+ membrane at the center of the grid (Figure 2).

Figure 2. Dot sample exampleAir dry at room temperature for 5 min.
Crosslink spotted RNA to the membrane using the UV cross linker (irradiate under ultraviolet lamp with 254 nm wavelength for 5 min).
Incubate the membrane with blocking buffer for 1 h at room temperature.
After blocking, incubate the membrane with anti-m6A antibody (1:1,000 dilution) in 5 mL of antibody dilution buffer overnight at 4 °C with gentle shaking.
Wash three times with PBST (3 × 5 min).
Incubate the membrane with HRP goat anti-rabbit IgG (1:10,000 dilution) in 5 mL of antibody dilution buffer for 1 h at room temperature with gentle shaking.
Wash three times with PBST (3 × 5 min).
Incubate with ECL substrate for 1 min (according to the manufacturer’s instructions, 0.125 mL ECL solution per cm2 of the membrane is recommended), cover with Saran wrap aiming to remove the excess solution from the surface, and expose the film in the darkroom (Figure 3B). Try different exposure times to get clear results.
After exposure, transfer the membrane to a solution containing 10 mL of 0.02% methylene blue dye buffer, gently shake the membrane at room temperature, and incubate for 30 min.
Lastly, clean the membrane with dH2O until the background is clean (Figure 3C).
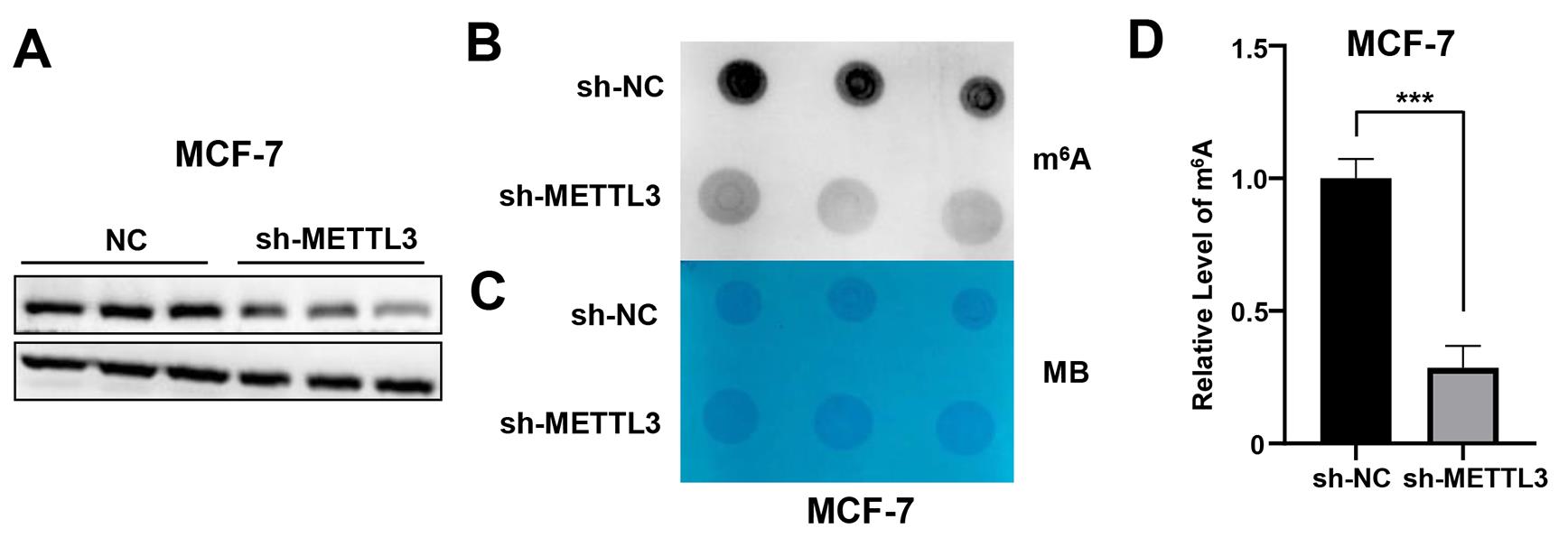
Figure 3. Representative data of m6A level detected by dot blot. (A) Western blotting assay of METTL3 in the METTL3-KD stable cell lines in MCF-7. (B) Dot blot to detect the m6A level of mRNA isolated from total RNA of METTL3-KD MCF-7 cells. (C) Methylene blue (MB) staining served as a loading control. (D) Measurement and normalization of m6A levels in ImageJ.
Data analysis
Perform densitometric analysis using ImageJ (result calculation method: m6A grayscale value/MB grayscale value). Use a minimum of three biological replicates to perform statistical analysis of the m6A levels between samples (Figure 3D).
For statistical analysis, use GraphPad Prism software.
Notes
This protocol can be modified for individual species of RNA, such as messenger RNA, tRNA, rRNA, or microRNA, by employing isolation techniques for the enrichment of the specific RNAs to be analyzed.
For the preparation of wash buffer, blocking buffer, and antibody dilution buffer, the use of nuclease-free water is not necessary.
Recipes
10× phosphate-buffered saline (PBS) (1,000 mL)
Weigh 14.4 g of Na2HPO4, 2.4 g of KH2PO4, 2.0 g of KCl, and 80.0 g of NaCl
Add 800 mL of sterile water
Stir until dissolved
Bring the final volume to 1,000 mL using sterile water
1× phosphate-buffered saline (PBS) (500 mL)
50 mL of 10× PBS
Bring final volume to 500 mL using sterile water
1× phosphate-buffered saline/0.1% Tween-20 (PBST) (500 mL)
50 mL of 1× PBS
1 mL of 100% Tween-20
Bring final volume to 500 mL using sterile water
Blocking buffer and antibody dilution buffer
50 mL of PBST
5 g non-fat milk
Acknowledgments
This work was supported by a grant from the National Natural Science Foundation of China (32171407) and Priority Academic Program Development of Jiangsu Higher Education Institutions. The protocol presented here was adapted from those in Terenghi (1998) and Jia et al. (2011).
Competing interests
The author declares there are no competing interests.
References
- Arguello, A. E., DeLiberto, A. N. and Kleiner, R. E. (2017). RNA Chemical Proteomics Reveals the N6-Methyladenosine (m6A)-Regulated Protein-RNA Interactome. J Am Chem Soc 139(48): 17249-17252.
- Batista, P. J., Molinie, B., Wang, J., Qu, K., Zhang, J., Li, L., Bouley, D. M., Lujan, E., Haddad, B., Daneshvar, K., et al. (2014). m6A RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell 15(6): 707-719.
- Chen, W., Feng, P., Ding, H. and Lin, H. (2016). Identifying N6-methyladenosine sites in the Arabidopsis thaliana transcriptome. Mol Genet Genomics 291(6): 2225-2229.
- Csepany, T., Lin, A., Baldick, C. J., Jr. and Beemon, K. (1990). Sequence specificity of mRNA N6-adenosine methyltransferase. J Biol Chem 265(33): 20117-20122.
- Engel, M., Eggert, C., Kaplick, P. M., Eder, M., Roh, S., Tietze, L., Namendorf, C., Arloth, J., Weber, P., Rex-Haffner, M., et al. (2018). The Role of m6A/m-RNA Methylation in Stress Response Regulation. Neuron 99(2): 389-403 e389.
- Harcourt, E. M., Kietrys, A. M. and Kool, E. T. (2017). Chemical and structural effects of base modifications in messenger RNA. Nature 541(7637): 339-346.
- Imanishi, M., Tsuji, S., Suda, A. and Futaki, S. (2017). Detection of N6-methyladenosine based on the methyl-sensitivity of MazF RNA endonuclease. Chem Commun (Camb) 53(96): 12930-12933.
- Jia, G., Fu, Y. and He, C. (2013). Reversible RNA adenosine methylation in biological regulation. Trends Genet 29(2): 108-115.
- Jia, G., Fu, Y., Zhao, X., Dai, Q., Zheng, G., Yang, Y., Yi, C., Lindahl, T., Pan, T., Yang, Y. G., et al. (2011). N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol 7(12): 885-887.
- Li, E., Xia, M., Du, Y., Long, K., Ji, F., Pan, F., He, L., Hu, Z. and Guo, Z. (2022). METTL3 promotes homologous recombination repair and modulates chemotherapeutic response in breast cancer by regulating the EGF/RAD51 axis. Elife 11: e75231.
- Li, Z., Weng, H., Su, R., Weng, X., Zuo, Z., Li, C., Huang, H., Nachtergaele, S., Dong, L., Hu, C., et al. (2017). FTO Plays an Oncogenic Role in Acute Myeloid Leukemia as a N6-Methyladenosine RNA Demethylase. Cancer Cell 31(1): 127-141.
- Linder, B., Grozhik, A. V., Olarerin-George, A. O., Meydan, C., Mason, C. E. and Jaffrey, S. R. (2015). Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat Methods 12(8): 767-772.
- Liu, H., Wang, H., Wei, Z., Zhang, S., Hua, G., Zhang, S. W., Zhang, L., Gao, S. J., Meng, J., Chen, X., et al. (2018). MeT-DB V2.0: elucidating context-specific functions of N6-methyl-adenosine methyltranscriptome. Nucleic Acids Res 46(D1): D281-D287.
- Mishima, E., Jinno, D., Akiyama, Y., Itoh, K., Nankumo, S., Shima, H., Kikuchi, K., Takeuchi, Y., Elkordy, A., Suzuki, T., et al. (2015). Immuno-Northern Blotting: Detection of RNA Modifications by Using Antibodies against Modified Nucleosides. PLoS One 10(11): e0143756.
- Nachtergaele, S. and He, C. (2018). Chemical Modifications in the Life of an mRNA Transcript. Annu Rev Genet 52: 349-372.
- Niu, Y., Zhao, X., Wu, Y. S., Li, M. M., Wang, X. J. and Yang, Y. G. (2013). N6-methyl-adenosine (m6A) in RNA: an old modification with a novel epigenetic function. Genomics Proteomics Bioinformatics 11(1): 8-17.
- Rana, A. P. and Tuck, M. T. (1990). Analysis and in vitro localization of internal methylated adenine residues in dihydrofolate reductase mRNA. Nucleic Acids Res 18(16): 4803-4808.
- Sun, T., Wu, R. and Ming, L. (2019). The role of m6A RNA methylation in cancer. Biomed Pharmacother 112: 108613.
- Terenghi, G. (1998). Detecting mRNA in tissue sections with digoxigenin-labeled probes. Methods Mol Biol 86: 137-142.
- Wang, Y., Li, Y., Yue, M., Wang, J., Kumar, S., Wechsler-Reya, R. J., Zhang, Z., Ogawa, Y., Kellis, M., Duester, G., et al. (2018). N6-methyladenosine RNA modification regulates embryonic neural stem cell self-renewal through histone modifications. Nat Neurosci 21(2): 195-206.
- Wiedmer, L., Eberle, S. A., Bedi, R. K., Sledz, P. and Caflisch, A. (2019). A Reader-Based Assay for m6A Writers and Erasers. Anal Chem 91(4): 3078-3084.
- Winkler, R., Gillis, E., Lasman, L., Safra, M., Geula, S., Soyris, C., Nachshon, A., Tai-Schmiedel, J., Friedman, N., Le-Trilling, V. T. K., et al. (2019). m6A modification controls the innate immune response to infection by targeting type I interferons. Nat Immunol 20(2): 173-182.
- Xiang, Y., Laurent, B., Hsu, C. H., Nachtergaele, S., Lu, Z., Sheng, W., Xu, C., Chen, H., Ouyang, J., Wang, S., et al. (2017). RNA m6A methylation regulates the ultraviolet-induced DNA damage response. Nature 543(7646): 573-576.
- Yin, H., Wang, H., Jiang, W., Zhou, Y. and Ai, S. (2017). Electrochemical immunosensor for N6-methyladenosine detection in human cell lines based on biotin-streptavidin system and silver-SiO2 signal amplification. Biosens Bioelectron 90: 494-500.
- Zhou, J., Wan, J., Gao, X., Zhang, X., Jaffrey, S. R. and Qian, S. B. (2015). Dynamic m6A mRNA methylation directs translational control of heat shock response. Nature 526(7574): 591-594.
Article Information
Copyright
![]() Du et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Du et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Du, Y., Xia, M. and Hu, Z. (2022). Analysis of N6-methyladenosine RNA Modification Levels by Dot Blotting. Bio-protocol 12(23): e4565. DOI: 10.21769/BioProtoc.4565.
- Li, E., Xia, M., Du, Y., Long, K., Ji, F., Pan, F., He, L., Hu, Z. and Guo, Z. (2022). METTL3 promotes homologous recombination repair and modulates chemotherapeutic response in breast cancer by regulating the EGF/RAD51 axis.Elife 11: e75231.
Category
Molecular Biology > RNA > RNA detection
Molecular Biology > RNA > RNA extraction
Cancer Biology > General technique > Biochemical assays > RNA
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.