- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Infection of the Developing Central Nervous System of Drosophila by Mammalian Eukaryotic and Prokaryotic Pathogens
Published: Vol 12, Iss 23, Dec 5, 2022 DOI: 10.21769/BioProtoc.4563 Views: 2307
Reviewed by: Nafisa M. JadavjiKai YuanAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Nov 2020
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Pathogen invasion of the central nervous system (CNS) is an important cause of infection-related mortality worldwide and can lead to severe neurological sequelae. To gain access to the CNS cells, pathogens have to overcome the blood–brain barrier (BBB), a protective fence from blood-borne factors. To study host–pathogen interactions, a number of cell culture and animal models were developed. However, in vitro models do not recapitulate the 3D architecture of the BBB and CNS tissue, and in vivo mammalian models present cellular and technical complexities as well as ethical issues, rendering systematic and genetic approaches difficult. Here, we present a two-pronged methodology allowing and validating the use of Drosophila larvae as a model system to decipher the mechanisms of infection in a developing CNS. First, an ex vivo protocol based on whole CNS explants serves as a fast and versatile screening platform, permitting the investigation of molecular and cellular mechanisms contributing to the crossing of the BBB and consequences of infection on the CNS. Then, an in vivo CNS infection protocol through direct pathogen microinjection into the fly circulatory system evaluates the impact of systemic parameters, including the contribution of circulating immune cells to CNS infection, and assesses infection pathogenicity at the whole host level. These combined complementary approaches identify mechanisms of BBB crossing and responses of a diversity of CNS cells contributing to infection, as well as novel virulence factors of the pathogen.
Graphical abstract

Procedures flowchart. Mammalian neurotropic pathogens could be tested in two Drosophila central nervous system (CNS) infection setups (ex vivo and in vivo) for their ability to: (1) invade the CNS (pathogen quantifications), (2) disturb blood–brain barrier permeability, (3) affect CNS host cell behaviour (gene expression), and (4) alter host viability.
Background
Infections of the central nervous system (CNS) are devastating yet remain relatively rare due to the presence of specific protective layers. In particular, the blood–brain barrier (BBB) is a selective physical and chemical filter, and performs its neuroprotective role by controlling molecular import into the CNS and blocking the entry of circulating cells (Daneman and Prat, 2015). In higher vertebrates, brain microvascular endothelial cells, harbouring intercellular tight junctions, form the core structure of the BBB. In addition, pericytes, astrocytes, and an extracellular matrix basal membrane regulate BBB integrity and functions (Obermeier et al., 2013; Daneman and Prat, 2015; Saunders et al., 2016). However, several pathogens have developed strategies to overcome the BBB and invade the CNS (Coureuil et al., 2017). To study interactions between pathogens and the BBB, various cell culture systems have been developed, from simple endothelial monolayers to more complex multicellular cultures, combining endothelial cells and pericytes (Sivandzade and Cucullo, 2018). While these in vitro models have proposed a number of BBB crossing mechanisms, they remain limited in recapitulating the 3D architecture of the BBB and its interactions with the complexity of CNS cells (Sivandzade and Cucullo, 2018; Jackson et al., 2019). Moreover, organoids also present architectural limitations (Bergmann et al., 2018). To bypass this restriction, a number of animal models, in particular rodents and zebrafish, have been used (Dando et al., 2014; Jackson et al., 2019), allowing the investigation of infection mechanisms in vivo. However, in addition to cost and ethical issues, their genetic and cellular complexity prevents systematic, screen-based approaches. Separating the systemic from the local impact of these immune challenges is also difficult in such context, and infections can have lethal consequences for the host. In addition, culturing CNS tissue of these animal models is still difficult.
Drosophila melanogaster represents an original and powerful model to study CNS infection, from identifying novel mechanisms of BBB-pathogen interactions to understanding the extent of consequences on the diversity of CNS cell populations. The fly BBB is composed of two glial layers exhibiting conserved neuroprotective mechanisms with the mammalian BBB. The subperineurial glia (SPG), forming an epithelium-like structure with septate junctions, represent the core structure and physical filter of the BBB (Stork et al., 2008; Hindle and Bainton, 2014; Weiler et al., 2017; Babatz et al., 2018). The perineurial glia, a hemolymph sensor, and the extracellular matrix both cover the SPG (DeSalvo et al., 2014; Parkhurst et al., 2018). Many transporter proteins expressed by the BBB to allow the selective shuttling of diverse solutes and xenobiotics are actually conserved, including solute carriers, ATP-binding cassette transporters, and lipoprotein receptors (DeSalvo et al., 2014; Hindle and Bainton, 2014; Weiler et al., 2017). Moreover, many aspects of mammalian neurogenesis are conserved in flies (Mira and Morante, 2020), enabling us to probe the impact of infection on such process. Drosophila neural stem cells self-renew via asymmetric cell division, switch from quiescence to proliferation, and are present within a specific microenvironment sharing neurogenic features and common constituents with the mammalian niche, including glial populations and the BBB (Homem and Knoblich, 2012; Otsuki and Brand, 2017). Finally, due to Drosophila unrivalled genetics, each of the BBB cell layers and the different CNS populations can be manipulated independently, in parallel with each other, and in a temporally controlled and spatially restricted manner. These features provide a simpler, genetically tractable model to study CNS infection, from pathogen entry to downstream consequences for the host.
We have devised a double, complementary approach to generate CNS infection in Drosophila:
An ex vivo protocol that mimics CNS infection. Here, an intact larval CNS explant is cultured in a specific medium inoculated with pathogens (Benmimoun et al., 2020). This explant setup is first designed to allow fast screening of pathogens, mutant strains, and culture conditions for the generation and outcome of CNS infection. In this frame, it has first allowed the identification of a number of prokaryotic and eukaryotic neurotropic pathogens, known to trigger meningitis and/or encephalitis in mammals, that are able to cross the fly BBB. On the prokaryotic side, this included Streptococcus agalactiae (Group B Streptococcus, GBS), Streptococcus pneumoniae, Listeria monocytogenes, and the strictly human Neisseria meningitidis. The eukaryotic Candida albicans and Candida glabrata were also found to reach the Drosophila CNS, but not Cryptococcus neoformans. This ex vivo platform was further used to screen a collection of GBS mutant strains, by which surface lipoproteins were identified—more precisely the lipoprotein Blr—as virulence factors crucial for BBB crossing (Benmimoun et al., 2020). Finally, its combination with Drosophila genetics revealed the Drosophila lipoprotein receptor (LpR2), which is expressed on the surface of the BBB layer on the SPGs, as a host receptor for Blr, and important for CNS invasion by GBS (Benmimoun et al., 2020).
Another asset of the explant protocol is to study the effect of pathogenic infection on the different cell populations outside of the systemic context (for example, outside of systemic inflammation, a common hallmark of infection), potentially revealing other layers of response that could be otherwise hidden. For example, the lactic acidosis generated in the milieu by GBS could be quenched by buffer solutions, revealing the contribution of this phenomenon to BBB weakening (Benmimoun et al., 2020).
An in vivo setup through direct microinjection of the pathogen into the fly circulatory system (Benmimoun et al., 2020; Winkler et al., 2021). This approach aims to evaluate the impact of infection i) on the CNS, this time including the contribution of systemic components, such as circulating immune cells, and ii) at the level of the whole organism, by assessing a diversity of host–pathogen interactions, in particular virulence. Comparison with the explant system allows to confirm or infirm the importance of molecular and cellular mechanisms in a whole-body context, offering the possibility to discriminate between local and systemic effects, and to identify the hierarchy between and dominance of specific mechanisms.
This protocol was first used to determine the conservation of GBS Blr and Drosophila LpR2 interactions in BBB crossing mechanism and CNS invasion, initially identified through the explant setup (Benmimoun et al., 2020). Moreover, assessing survival overtime of the injected larvae in this setup demonstrated that Blr is a virulence factor in Drosophila, a property further validated in a mouse model of GBS hematogenous brain infection (Benmimoun et al., 2020). This whole in vivo CNS infection protocol also participated in revealing a new inflammatory mechanism, in which glial cells direct CNS infiltration by macrophages in response to GBS infection (Winkler et al., 2021).
Here, we detail these two complementary platforms to elicit infection of the Drosophila developing CNS by mammalian neurotropic pathogens. By taking advantage of the Swiss knife of Drosophila genetics, the architectural preservation of the CNS tissue, and the similarity of many fundamental cellular and molecular mechanisms with mammals, these approaches allow more systematic screening of wild-type and mutant pathogen strains, as well as targeted genetic manipulations, to infer precise causal relationships between cellular layers or between molecular pathways during infection. Ultimately, these setups can be used to study all steps of the CNS infection, from crossing of the BBB to downstream consequences on the host, including neuroinflammation and impact on neurogenesis.
Although these methods are described for prokaryotic and eukaryotic pathogens, they could be adapted for other pathogens (virus, parasites) as well as for studying systemic extrinsic stresses, and for investigating the entry of therapeutic molecules into the CNS.
Materials and Reagents
Fly strain and Drosophila larval culture
mdr65-mtd: Tomato (Benmimoun et al., 2020 and Figure 1)
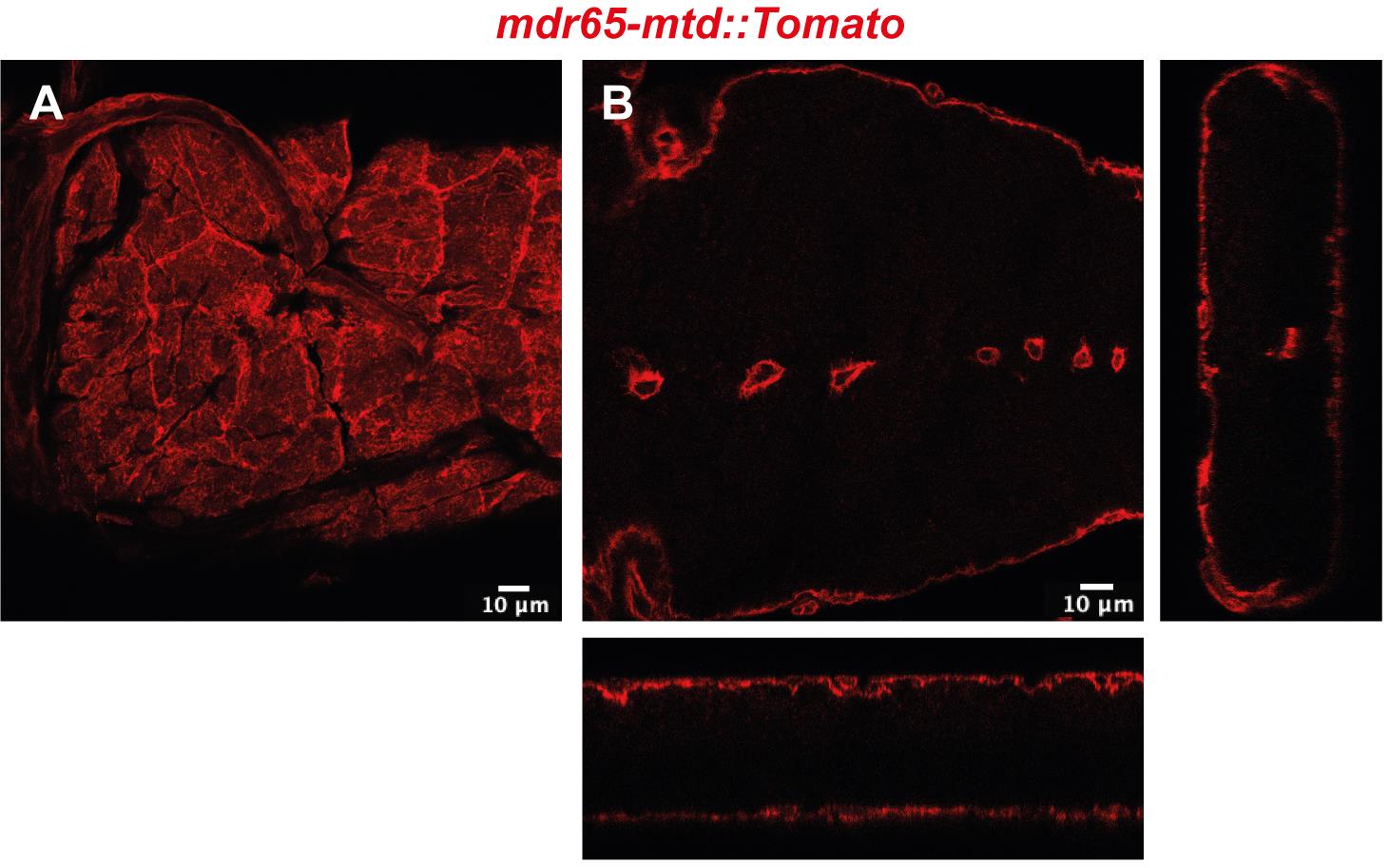
Figure 1. Confocal image of top view (A) and cross-section with orthogonal views (B) of the Drosophila BBB in a portion of the ventral nerve cordEgg laying plates (50 mm, Deep) (Thermo Scientific, catalog number: 124-17), see Figure 2A
Fly egg laying cages (homemade, see Figure 2B)
Larval collection plates (60 mm Petri dishes) (Corning-Falcon, catalog number: 353004), see Figure 2C
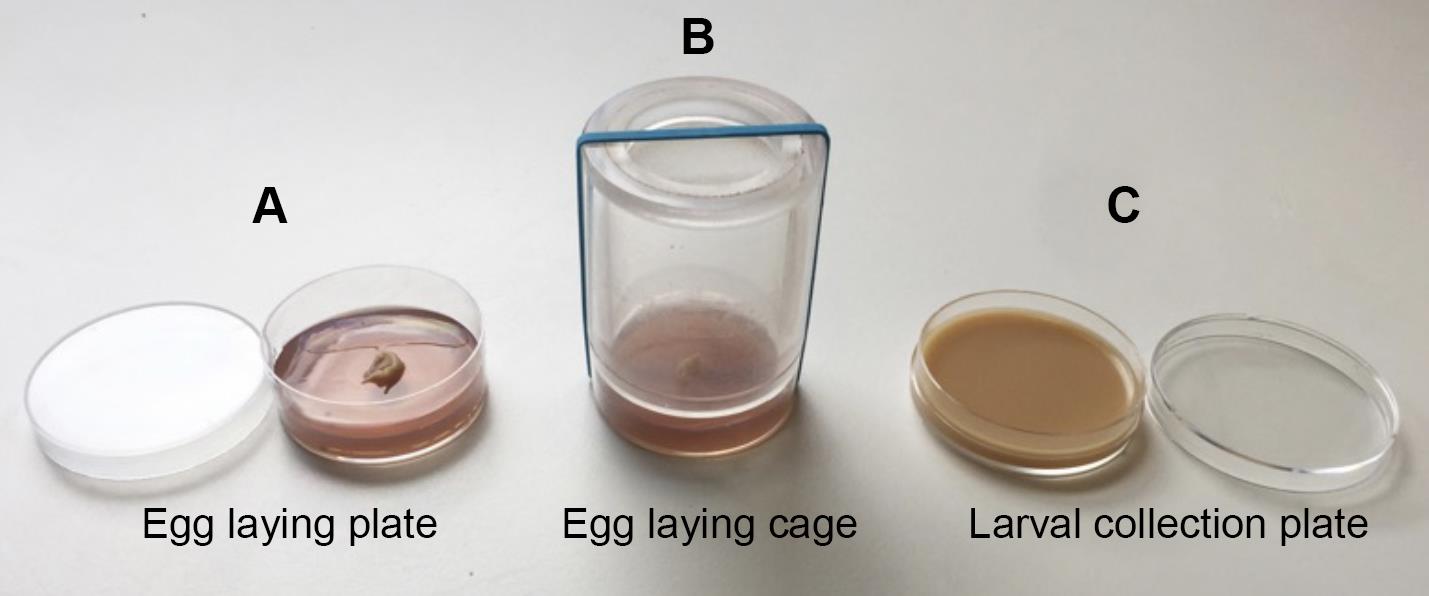
Figure 2. Fly larval collection and culture materialsActive dry yeast (Genesee Scientific, catalog number: 62-103)
Corn flour (Limagrain, catalog number: WFMZ01HS)
Agar (Sigma-Aldrich, catalog number: A7002-1KG)
Sugar (Dominique Dutscher, catalog number: 067508B)
Organic grape juice
Propionic acid (Sigma-Aldrich, catalog number: P5561-1L)
Methyl-4-hydroxybenzoate (Sigma-Aldrich, catalog number: H3647)
Ethanol (Sigma-Aldrich, catalog number: 24105)
Egg laying medium (see Recipes)
Fly food (see Recipes)
Pathogen preparation
Pathogen growth media [Brain Heart Infusion broth (BHI), Yeast extract Peptone Dextrose (YPD), or de Man Rogosa Sharpe (MRS)] (BD Difco, Millipore, catalog numbers: 237500, 242820 or 288130)
Round-bottom tubes with cap (Fisher Scientific, Falcon, catalog number: 352059)
Micro cuvette for spectrophotometer (Dutscher, Greiner Bio-One, catalog number: 613101)
Microcentrifuge tubes (Fisher Scientific, Eppendorf, catalog number: 10708704)
Dulbecco’s phosphate-buffered saline (PBS) (Sigma-Aldrich, catalog number: D1408)
Drosophila Schneider’s medium (Fisher Scientific, Gibco, catalog number: 21720-024)
Drosophila dissection
Fine forceps (Fine Science Tools, Dumont #5, catalog number: 11252-20)
Tissue culture dishes (Fisher Scientific, Corning, Falcon, catalog number: 3530001)
Glass cavity dish (Atom Scientific, catalog number: SDCE4040-1)
Dulbecco’s phosphate-buffered saline (PBS) (Sigma-Aldrich, catalog number: D1408)
Ethanol (Sigma-Aldrich, catalog number: 493511)
Drosophila Schneider’s medium (Fisher Scientific, Gibco, catalog number: 21720-024)
CNS explant culture
24-well cell culture plate (Fisher Scientific, Corning Falcon, catalog number: 353047)
Drosophila Schneider’s medium (Fisher Scientific, Gibco, catalog number: 21720-024)
l-Glutamine (Fisher Scientific, Gibco, catalog number: 25030-032)
Sodium l-ascorbate (Sigma-Aldrich, catalog number: A4034)
Fetal bovine serum (Sigma-Aldrich, catalog number: F4135)
CNS culture medium I (see Recipes)
CNS culture medium II (see Recipes)
In vivo CNS infection
Fine forceps (Fine Science Tools, Dumont #5, catalog number: 11252-20)
Pipette Drummond Scientific 3.5’’ (Dutsher, Drummond Scientific, catalog number: 3-000-203-G/X)
Nano-injector Nanoject III (Dutsher, Drummond Scientific, catalog number: 075250)
Mineral oil (Sigma-Aldrich, catalog number: M5904)
Microscope slides (Fisher Scientific, Fisherbrand, catalog number: 10219280)
Permeability assay
Drosophila Schneider’s medium (Fisher Scientific, Gibco, catalog number: 21720-024)
Dextran, Texas Red, lysine fixable 10 KDa (ThermoFisher, catalog number: 11530236, Invitrogen D1863)
Dulbecco’s phosphate-buffered saline (PBS) (Sigma-Aldrich, catalog number: D1408)
4% methanol-free formaldehyde (ThermoFisher Scientific, catalog number: 28908)
Immunohistochemistry
4% methanol-free formaldehyde (ThermoFisher Scientific, catalog number: 28908)
Bouin’s solution (Sigma-Aldrich, catalog number: HT10132)
Dulbecco’s phosphate-buffered saline (PBS) (Sigma-Aldrich, catalog number: D1408)
Triton X-100 (Sigma-Aldrich, catalog number: T9284)
Bovine serum albumin (Sigma-Aldrich, catalog number: A3608)
Normal goat serum (Invitrogen, catalog number: 10000C)
Mowiol mounting medium (homemade), or equivalent
Microscope slides (Fisher Scientific, Fisherbrand, catalog number: 10219280)
Coverslips (Fisher Scientific, Fisherbrand, catalog number: 15707592)
Anti-GBS antibody (Benmimoun et al., 2020)
Gene expression
Fine forceps (Fine Science Tools, Dumont #5, catalog number: 11252-20)
Dulbecco’s phosphate-buffered saline (PBS) (Sigma-Aldrich, catalog number: D1408)
Glass cavity dish
RNeasy Mini kit (QIAGEN, catalog number: 74104)
QuantiTect Rev. Transcription Kit (QIAGEN, catalog number: 205311)
TaqMan gene expression assay (ThermoFisher, catalog number: 4331182)
TaqMan Universal PCR Master Mix II (ThermoFisher, catalog number: 4440042)
96-well PCR plates (e.g., Bio-Rad, catalog number: MLL9601)
Seals for PCR plate (e.g., Bio-Rad, catalog number: MSB1001)
Equipment
Incubator with regulated temperature and humidity (Memmert, HPP110, or equivalent)
Agitator Titramax 100 (Heidolph Instruments, catalog number: 544-11200-00)
Spectrophotometer (Pharmacia, Novaspec II)
Centrifuge (Sigma Bioblock scientific, 1-15K)
Microbiological Safety Post (HeraSafe KS9)
Flaming/Brown micropipette puller (Sutter Instrument Model P-97)
Nano-injector Nanoject III (Dutsher, Drummond Scientific, catalog number: 075250)
Laser scanning confocal microscope [Zeiss LSM 880 with Zen software (2012 S4)]
Real-Time PCR machine (e.g., ThermoFisher, StepOne Real-Time PCR System, catalog number: 4376357)
Software
Zen software (2012 S4)
Fiji (ImageJ version 2020 2.1.0/1,53c and version 1.52p; available at: https://imagej.net/software/fiji/)
GraphPad Prism software [version 7 and version 2020 8.4.2 (464)]
R, SPSS, or comparable software
Procedure
Drosophila larval culture
Set fly crosses for 2–3 h in cages with grape juice egg-laying plates (supplemented with a bit of yeast paste, see Figure 2A and 2B) at appropriate temperature in order to collect fly embryos (Figure 3A).
After 21 h at 25 °C (or 48 h at 18 °C), remove the already hatched larvae from the plate. Then, at each hour, collect and transfer equivalent numbers (approximately 100) of hatching first instar larvae to standard food plates at appropriate temperature (29 °C for RNAi knockdown, for example) (Figures 2C and 3B-C).
Let the larvae grow and then collect them from the appropriate developmental stage to perform experiments. Of note, pathogenic infection has so far been done with third instar larvae (+72 h at 25 °C) (Figure 3D).
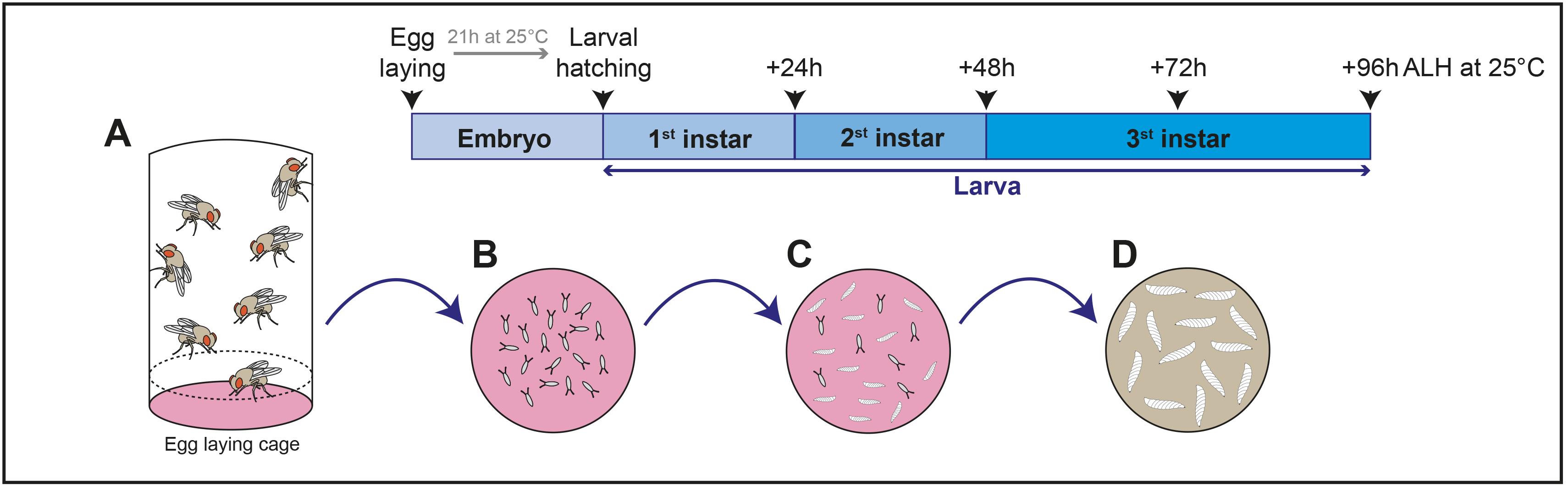
Figure 3. Schematic representation of Drosophila larval culture procedure
Pathogen preparation
Set an overnight bacteria or a 36 h yeast preculture from -80 °C glycerol stocks on appropriate growth medium (BHI, YPD, MRS, …) and at adequate temperature (30 °C or 37 °C) (Figure 4A).
a) For bacteria, dilute the overnight preculture at 1/20 in corresponding growth medium, and allow them to grow till the mid-exponential phase (~2 h 30 min at 37 °C for GBS) (to adapt depending on the bacterial strains) (Figure 4B).
b) For fungi, dilute the overnight preculture at OD600 = 0.2, and allow them to grow at 30 °C for 5–6 h until OD600 of 1 (to adapt depending on the strains) (Figure 4B).
Pellet the pathogens, after OD600 measurement (Figure 4C), by centrifugation at 3,500 × g and 4 °C for 5 min.
Wash pathogenic culture in PBS, then pellet the pathogens by centrifugation at 3,500 × g and 4 °C for 5 min. Do this step twice (Figure 4D).
Wash pathogenic culture in Drosophila Schneider’s medium, then pellet the pathogens by centrifugation at 3,500 × g and 4 °C for 5 min (Figure 4E).
a) For the ex vivo CNS infection protocol: Resuspend the pathogens in 750 µL of Drosophila Schneider’s medium (Figure 4F), to prepare a 10× infectious dose and keep them at 4 °C until use (for example: 10 × 108 CFU/mL for Streptococcus agalactiae, Streptococcus pneumoniae, Listeria innocua, and Listeria monocytogenes; 10 × 107 CFU/mL for Neisseria meningitidis, Candida albicans, and Candida glabrata, and 10 × 105 CFU/mL for Cryptococcus neoformans).
b) For the in vivo CNS infection protocol: Resuspend the pathogens in 100 µL of Drosophila Schneider’s medium (Figure 4F), to reach the infectious dose in the hemolymph (estimated at 2 μL) by injecting 20 nL of concentrated pathogen (for example: 4.4 × 1010 GBS/mL Streptococcus agalactiae).
Note: Pathogen concentration was calculated by OD600 correlation (Streptococcus agalactiae, Streptococcus pneumoniae, Listeria innocua, and Listeria monocytogenes: 1 OD600 = 8.8 × 108 CFU/mL; Neisseria meningitidis: 1 OD600 = 109 CFU/mL; Candida albicans and Candida glabrata: 1 OD600 = 3 × 107 CFU/mL; Cryptococcus neoformans: 1 OD600 = 6 × 107 CFU/mL).
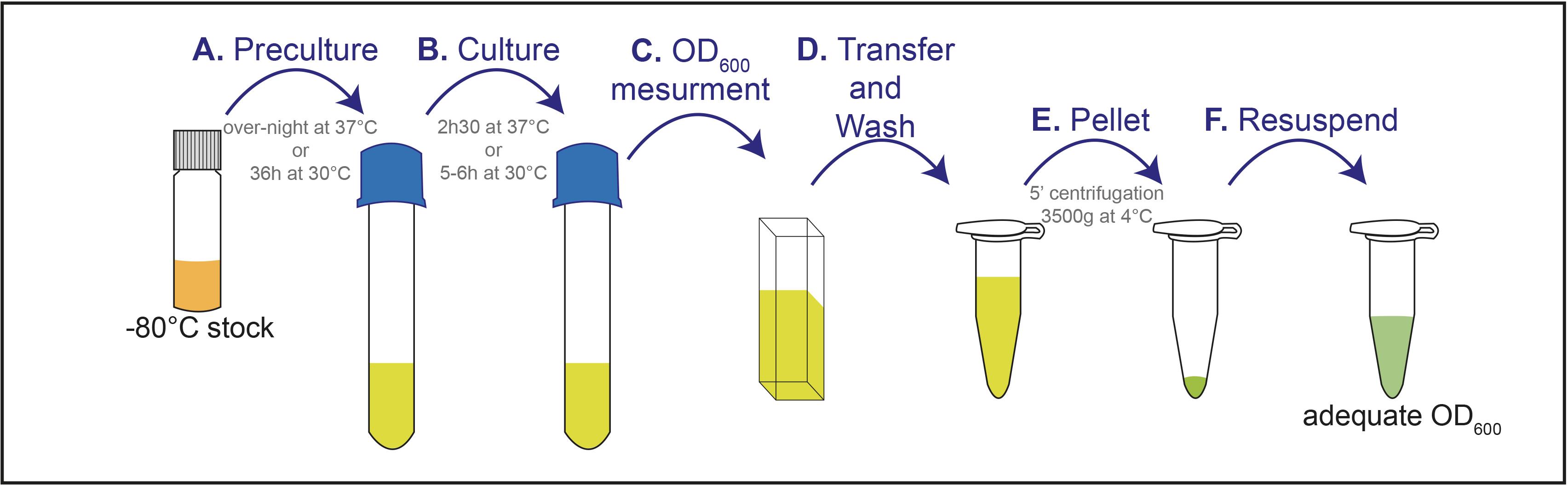
Figure 4. Schematic representation of pathogen preparation procedureCNS explant culture and ex vivo CNS infection
Wash staged larvae successively in PBS and ethanol 70% v/v in water and transfer them into cold Drosophila Schneider’s medium inside dissection wells.
Dissect larvae by (a) cutting at approximately a quarter from the posterior spiracle, to minimise damage to motor nerves, (b) turning the anterior part inside-out to expose the CNS [Figure 5A and see video (Hafer and Schedl, 2006)]. Keep all larval tissues except for the gut (to avoid contamination with intestinal symbiotic pathogens).
Transfer eight larvae to one well (24-well cell culture plate) and culture them under gentle rotary agitation (275 rpm) in 750 μL of CNS medium I at 30 °C and 60% humidity (Figure 5B).
Note: A temperature of 30 °C was chosen as a compromise temperature, to allow Drosophila development while culturing mammalian pathogens closer to their usual environment.
Dilute 1/10 of the prepared 10× infectious dose of each pathogen in the CNS culture medium I (Figure 5C), to reach the appropriate infectious dose (see step B6a).
Replace the infected culture medium with Drosophila Schneider’s medium supplemented with 2 mM l-Glutamine, 0.5 mM sodium l-ascorbate, and 1% fetal bovine serum. Place the culture at 30 °C and 60% humidity under gentle rotary agitation (275 rpm) for a defined time (3 h for GBS; the same time must be kept for the control condition without pathogenic inoculum).
Replace the medium after 3 h and every 10 h, with a fresh culture medium (Drosophila Schneider’s medium supplemented with 2 mM l-Glutamine, 0.5 mM sodium l-ascorbate, and 1% fetal bovine serum)
Note: In these conditions, CNS explants can be kept for up to 48 h, at 30 °C.
Proceed with the treatment required for each different analysis (D, E, and F).
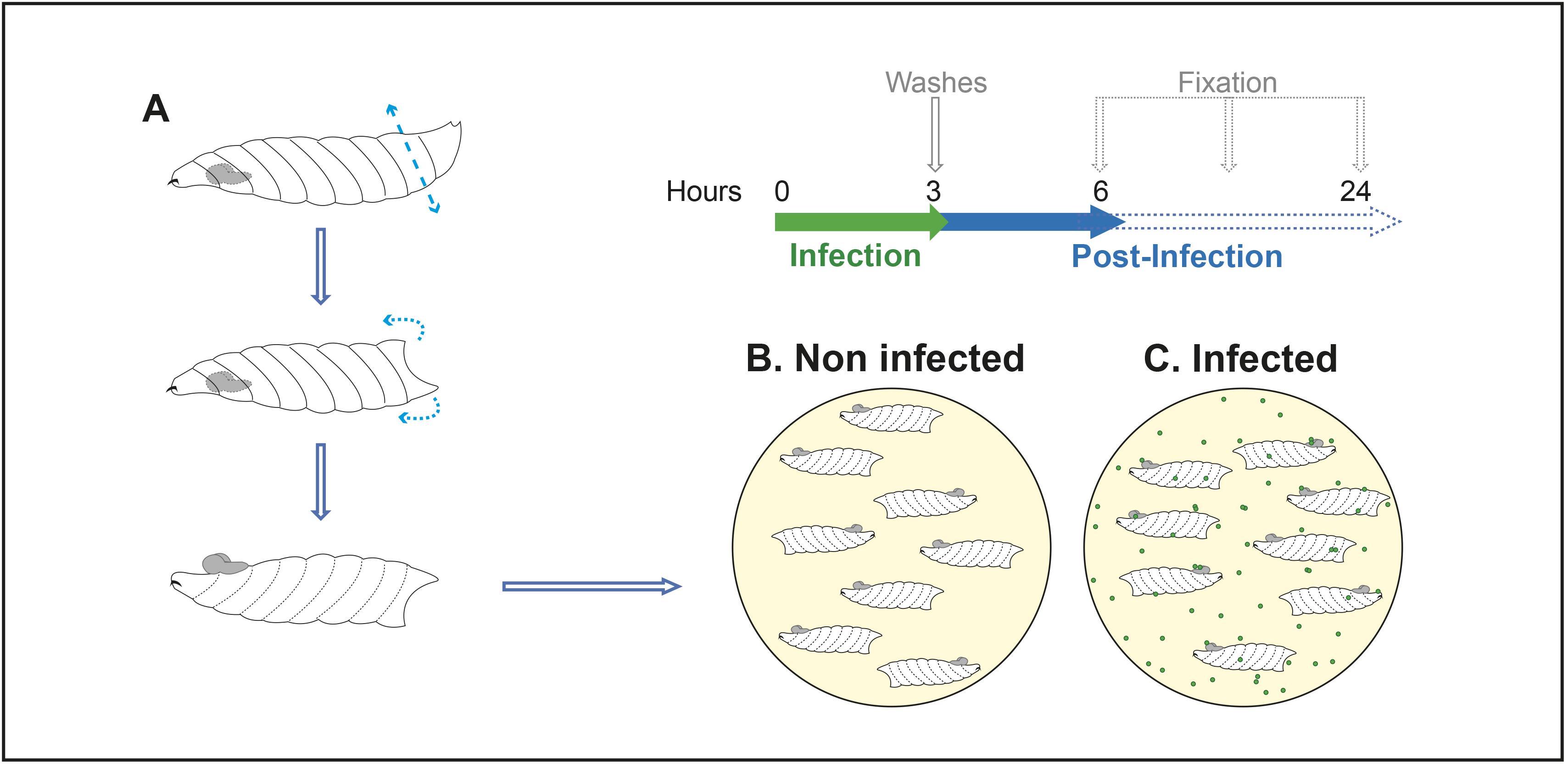
Figure 5. chematic representation of the ex vivo CNS infection procedurePathogenic microinjection and in vivo CNS infection
Pull micropipettes for injection (Heat: 350–360, Pull: 60–80, Velocity: 100, Time: 150). This step can be done in advance.
Fill the completely prepared micropipette with mineral oil, using a needle and syringe (Figure 6A).
Attach the micropipette filled with mineral oil to the injector head (Figure 6A).
Eject part of the mineral oil and replace it by filling the micropipette with prepared concentrated pathogens (Figure 6A) (this will avoid contamination of the injector head with pathogens, by forming two distinct phases in the micropipette).
Wash staged larvae successively in PBS and ethanol 70% v/v in water and transfer them into cold Drosophila Schneider’s medium inside dissection wells.
One by one, dry the larvae, place them on one slide, and gently inject 20 nL of Drosophila Schneider’s medium (mock control) or prepared pathogen in its posterior part, using the nanoinjector (Figure 6B). This injection should be done very gently to only puncture the cuticle without affecting other tissues. We have chosen to inject the posterior part as it is far from the CNS.
Keep injected larvae on standard fly food plates in an incubator at 30 °C and 60% humidity until analysis.
Notes:
All injected larvae will present a melanization spot, which seals and heals the cuticle punctured by the injection.
Mock injection results in lethality per se, due to a combination of unsuccessful healing of the punctured cuticle, potential damage to tissues neighbouring the injection point, and potential temperature-induced stress (30 °C). In our hands, approximately 10% of GBS-injected larvae survive 4 h post-injection (all animals died between 4 and 6 h post-injection), while approximately 40% of mock-injected larvae survive 4 h post-injection (Benmimoun et al., 2020).
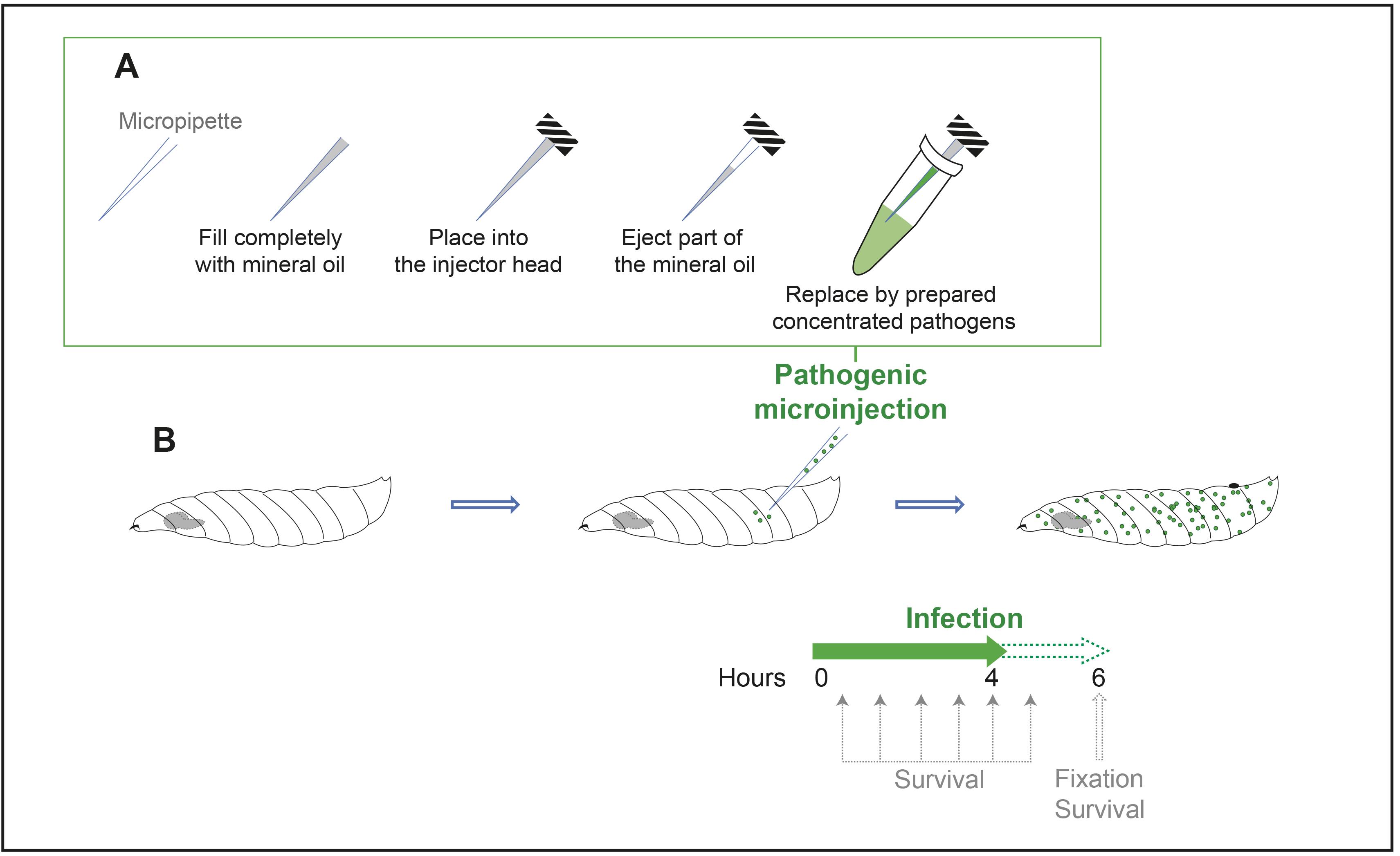
Figure 6. Schematic representation of the in vivo CNS infection procedureBlood–brain barrier permeability assay
Prepare a solution of 10 KDa dextran Texas Red (50 mM) in CNS culture medium II.
Change CNS culture medium and incubate CNS explants with this solution under agitation (275 rpm) for 30 min.
CNS explants are immediately fixed 4 × 5 min (to wash out excess dextran) in 4% methanol-free formaldehyde.
Wash 3 × 10 min with PBS.
Dissect the CNS and mount samples in mounting medium.
Visualise with a laser scanning confocal microscope, with an optimal distance between each slice of 0.38 μm.
Immunohistochemistry
This protocol is a classic, general protocol to perform immunochemistry on the Drosophila larval CNS. For infections, we have used it to assess pathogen localization inside the CNS, by using specific antibodies against the strains of interest (Benmimoun et al., 2020), as well as cell fate (anti-Deadpan, anti-Repo, and anti-ElaV antibodies), morphology, and proliferation (anti-phosphohistone 3). It can also detect fusion proteins (anti-GFP antibody). A variation (using Bouin’s solution, see below) exists for antigens and antibodies, which do not work well with the classical formaldehyde fixation (for example, anti-Neurexin-IV antibody, Babatz et al., 2018).
Fix (inside-out larvae ex vivo, or dissect and turn inside-out in vivo injected larvae).
In 4% methanol-free formaldehyde at room temperature for 30 min, or overnight at 4 °C.
In Bouin’s solution at room temperature for 3 min.
Wash 3 × 10 min in PBS.
Permeabilize 3 × 10 min in PBS-Triton 0.3%.
Incubate with primary antibodies at 4 °C in blocking solution (PBS-Triton 0.3%, bovine serum albumin 5%, normal goat serum 2%) for 18–36 h.
Of note: medium acidification could occur upon infection for some pathogenic strains (e.g., lactic pathogens, such as GBS or S. pneumoniae). If so, all GFP fusions should be detected with an anti-GFP antibody.
Wash 3 × 10 min with PBS-Triton 0.3%.
Incubate with secondary antibodies in blocking solution at room temperature for 3 h, or at 4 °C for 18–24 h.
Wash 3 × 10 min with PBS-Triton 0.3%.
Dissect the CNS and mount samples in Mowiol mounting medium.
Visualise with a laser scanning confocal microscope, with an optimal distance between each slice of 0.38 μm (Figure 7).
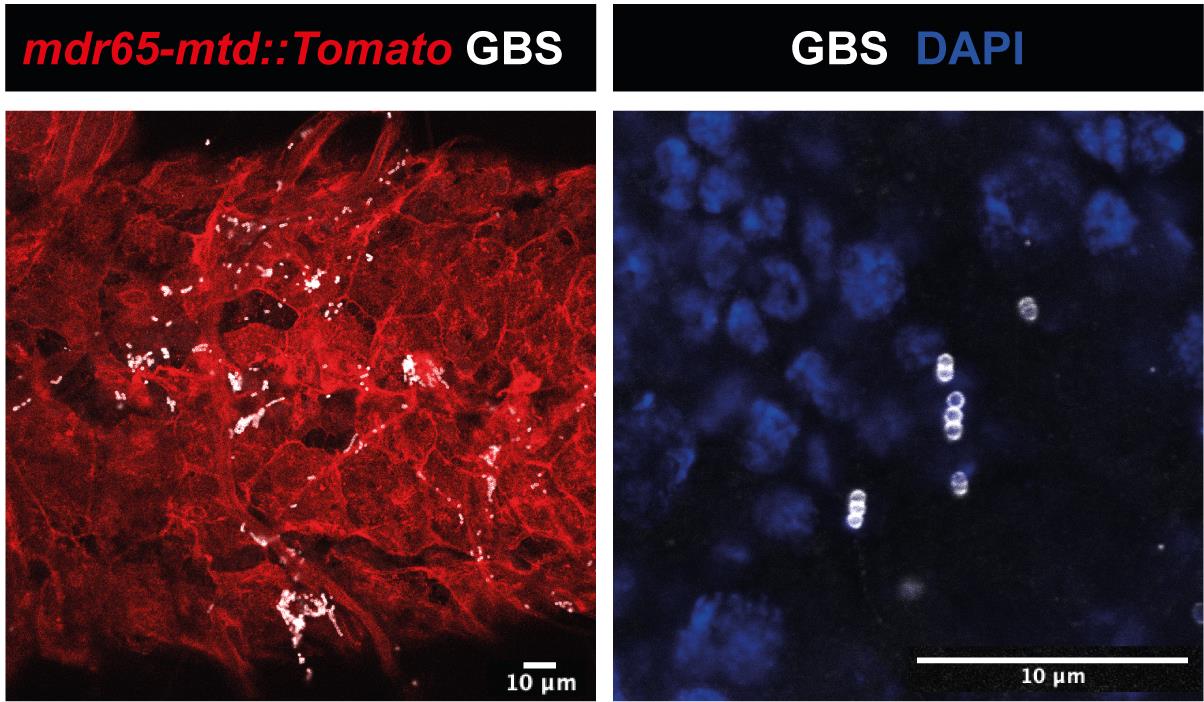
Figure 7. Confocal images showing GBS (attached to the BBB or inside the brain) using a specific GBS antibodyPathogenic load (CFU quantifications)
Without tissue fixation, wash five injected flies (by condition) on paper with ethanol 70%.
Open and bleed the larvae in 10 μL of PBS, and pool their hemolymph (keep on ice).
Dissect, transfer, and homogenise the CNS in 10 μL of PBS (keep on ice).
Transfer and homogenise the rest of the larval carcass (other tissues except for the gut) in 10 μL of PBS (keep on ice).
Prepare seven serial tenfold dilutions with 2 μL in 20 μL of PBS for each tissue tested (hemolymph, CNS, carcasses).
Plate 2 x 4 μL for each dilution on pathogen culture medium agarose plate(s).
Incubate the plates at 37 °C for 16 h prior to the quantifications.
Note: While these steps are described for the in vivo infection, only CNS bacterial load is determined for ex vivo infection. For this, follow steps 3 and 5–6 only.
qPCR for gene expression
Dissect 9–12 CNS with fine forceps at the desired time (3 h or 6 h post infection for GBS) and transfer each dissected CNS directly into the lysis buffer of the RNeasy Mini kit on ice.
Isolate RNA according to the manufacturer’s instructions.
Directly proceed with the cDNA synthesis. Use 450–600 ng RNA per reaction. Store the remaining isolated RNA at -80 °C.
Set up qPCR reactions with a final volume of 10 µL.
Seal plates carefully and spin them down in a centrifuge at 3,000 rpm for approximately 2 min.
Run qPCR.
Analyse qPCR data (see Data analysis).
Notes:
50 ng of cDNA for each reaction is generally sufficient to get reasonable cycle threshold (Ct) values.
Rpl32 can be used as a housekeeping gene.
Data analysis
Pathogen quantifications in infected Drosophila CNS
After imaging (immunohistochemistry)
Manually determine the exact number of pathogens for each analysed CNS by counting each individual bacterium contained within the boundary of the BBB (mdr65-mtd:: Tomato) through confocal acquisition.
Compare between pathogen entry into the CNS using Student’s t-test (two conditions) or one-way ANOVA test followed by Tukey’s post-hoc analysis (more than two conditions), when values follow a normal distribution (assessed by Shapiro–Wilk normality test). Otherwise, using non-parametric Mann–Whitney tests (two conditions) or Kruskal–Wallis tests (more than two conditions).
After pathogenic load (CFU quantifications)
Count the bacterial colonies (CFUs) for each dilution (when possible, i.e., when isolated single colonies are detectable).
Calculate an average of CFU/µL (bacterial load) by condition.
Represent the bacterial loads per larva in a log10 scale.
For in vivo infection, calculate the ratios from raw counting and represent them on a log10 scale:
Ratio CNS/hemolymph = log10 (cfu per CNS/cfu per hemolymph)
Ratio CNS/other tissues = log10 (cfu per CNS/cfu per other tissues)
Ratio CNS/(hemolymph + other tissues) = log10 [cfu per CNS/(cfu per hemolymph + cfu per other tissues)].
Blood–brain barrier permeability assay
Calculate Texas Red pixel intensity of three selected equal-sized areas (300 × 300 µm) from each CNS.
Calculate the average of the mean pixel intensity.
Calculate background intensity (empty equal-sized area 300 × 300 µm).
Calculate permeability index by subtracting background intensity from the average of the mean pixel intensity.
Compare between permeability index using Student’s t-test (two conditions) or one-way ANOVA test followed by Tukey’s post-hoc analysis (more than two conditions) when values follow a normal distribution (assessed by Shapiro–Wilk normality test). Otherwise, using non-parametric Mann–Whitney tests (two conditions) or Kruskal–Wallis tests (more than two conditions).
Quantification of gene expression levels
Perform qPCR with at least three biological and three technical replicates.
Calculate the average for the technical replicates for each sample.
Calculate the ∆Ct, by subtracting the average Ct of the housekeeping gene from the gene of interest.
Calculate the ∆∆Ct, by calculating the average of the ∆Ct of the control group (uninfected) as a calibrator. Then, subtract the calibrator from each calculated ∆Ct of each biological replicate.
For easier representation of the expression level, the data can be normalised to the uninfected control, by calculating the fold gene expression levels. For this, calculate two to the power of negative ∆∆Ct (2-∆∆Ct) for each biological replicate.
For statistical analysis, first test the calculated fold gene expression levels for normal distribution (e.g., by using the Shapiro–Wilk test). When normally distributed, perform a Student’s t-test; if not normally distributed, perform a Mann–Whitney U Test. Represent data in a box plot.
Survival of in vivo infected Drosophila
Score survival overtime (each 30 min) and compare survival curves using the log-rank test. When more than two conditions are considered, p-values should be adjusted by determining their statistical significance (alpha = 0.05) through stacked p-values analysis, through the Holm–Sidak method.
Represent data as Kaplan–Meier curves with error bars corresponding to standard errors (SE).
Recipes
Egg laying medium
Reagent Final concentration Amount Agar 22.2 g/L 22.2 g Sugar 25 g/L 25 g Organic grape juice 25% v/v 250 mL Methyl-4-hydroxybenzoate (20% in ethanol) 0.25% v/v 12.5 mL Purified water (reverse osmosis) n/a 900 mL Total n/a ~1 L Fly food
Reagent Final concentration Amount Active dry yeast n/a 300 g Corn flour n/a 211 g Agar n/a 46 g Sugar n/a 300 g Propionic acid 0.40% v/v 25 mL Methyl-4-hydroxybenzoate (20% in ethanol) 0.25% v/v 75 mL Purified water (reverse osmosis) n/a 6.3 L Total n/a 6 L (after cooking) CNS culture medium I
Reagent Final concentration Amount l-Glutamine (200 mM) 2 mM 100 μL Sodium l-ascorbate (500 mM) 0.5 mM 10 μL Drosophila Schneider’s medium n/a 9.89 ml Total n/a 10 mL CNS culture medium II
Reagent Final concentration Amount l-Glutamine (200 mM) 2 mM 100 μL Sodium l-ascorbate (500 mM) 0.5 mM 10 μL Fetal bovine serum 1% 100 μL Drosophila Schneider’s medium n/a 9.79 mL Total n/a 10 mL
Acknowledgments
We thank S. Dramsi, C. d’Enfert, G. Janbon, F. Leulier, M.-K. Taha, O. Disson, M. Lecuit, and F. Schweisguth for reagents and strains. We thank S. Liégeois for his help on hemolymph injection of Drosophila larvae and CFU counting. We thank D. Briand for his technical help on qPCR. This work has been funded by a starting package from Institut Pasteur/ LabEx Revive and a JCJC grant from Agence Nationale de la Recherche (NeuraSteNic, ANR- 17-CE13-0010-01) to P.S. B.B. has been supported by a Roux-Cantarini (Institut Pasteur) and a LabEx Revive post-doctoral fellowships.
This protocol was adapted from previous work (Benmimoun et al., 2020; Winkler et al., 2021).
Competing interests
The authors declare no conflicts of interest.
References
- Babatz, F., Naffin, E. and Klambt, C. (2018). The Drosophila Blood-Brain Barrier Adapts to Cell Growth by Unfolding of Pre-existing Septate Junctions. Dev Cell 47(6): 697-710 e693.
- Benmimoun, B., Papastefanaki, F., Perichon, B., Segklia, K., Roby, N., Miriagou, V., Schmitt, C., Dramsi, S., Matsas, R. and Speder, P. (2020). An original infection model identifies host lipoprotein import as a route for blood-brain barrier crossing. Nat Commun 11(1): 6106.
- Bergmann, S., Lawler, S. E., Qu, Y., Fadzen, C. M., Wolfe, J. M., Regan, M. S., Pentelute, B. L., Agar, N. Y. R. and Cho, C. F. (2018). Blood-brain-barrier organoids for investigating the permeability of CNS therapeutics. Nat Protoc 13(12): 2827-2843.
- Coureuil, M., Lecuyer, H., Bourdoulous, S. and Nassif, X. (2017). A journey into the brain: insight into how bacterial pathogens cross blood-brain barriers. Nat Rev Microbiol 15(3): 149-159.
- Dando, S. J., Mackay-Sim, A., Norton, R., Currie, B. J., St John, J. A., Ekberg, J. A., Batzloff, M., Ulett, G. C. and Beacham, I. R. (2014). Pathogens penetrating the central nervous system: infection pathways and the cellular and molecular mechanisms of invasion. Clin Microbiol Rev 27(4): 691-726.
- Daneman, R. and Prat, A. (2015). The blood-brain barrier. Cold Spring Harb Perspect Biol 7(1): a020412.
- DeSalvo, M. K., Hindle, S. J., Rusan, Z. M., Orng, S., Eddison, M., Halliwill, K. and Bainton, R. J. (2014). The Drosophila surface glia transcriptome: evolutionary conserved blood-brain barrier processes. Front Neurosci 8: 346.
- Hafer, N., and Schedl, P. (2006). Dissection of larval CNS in Drosophila melanogaster. J Vis Exp 9: (1):85.
- Hindle, S. J. and Bainton, R. J. (2014). Barrier mechanisms in the Drosophila blood-brain barrier. Front Neurosci 8: 414.
- Homem, C. C. and Knoblich, J. A. (2012). Drosophila neuroblasts: a model for stem cell biology. Development 139(23): 4297-4310.
- Jackson, S., Meeks, C., Vezina, A., Robey, R. W., Tanner, K. and Gottesman, M. M. (2019). Model systems for studying the blood-brain barrier: Applications and challenges. Biomaterials 214: 119217.
- Mira, H. and Morante, J. (2020). Neurogenesis From Embryo to Adult - Lessons From Flies and Mice. Front Cell Dev Biol 8: 533.
- Obermeier, B., Daneman, R. and Ransohoff, R. M. (2013). Development, maintenance and disruption of the blood-brain barrier. Nat Med 19(12): 1584-1596.
- Otsuki, L. and Brand, A. H. (2017). The vasculature as a neural stem cell niche. Neurobiol Dis 107: 4-14.
- Parkhurst, S. J., Adhikari, P., Navarrete, J. S., Legendre, A., Manansala, M. and Wolf, F. W. (2018). Perineurial Barrier Glia Physically Respond to Alcohol in an Akap200-Dependent Manner to Promote Tolerance. Cell Rep 22(7): 1647-1656.
- Saunders, N. R., Habgood, M. D., Mollgard, K. and Dziegielewska, K. M. (2016). The biological significance of brain barrier mechanisms: help or hindrance in drug delivery to the central nervous system? F1000Res 5.
- Sivandzade, F. and Cucullo, L. (2018). In-vitro blood-brain barrier modeling: A review of modern and fast-advancing technologies. J Cereb Blood Flow Metab 38(10): 1667-1681.
- Stork, T., Engelen, D., Krudewig, A., Silies, M., Bainton, R. J. and Klambt, C. (2008). Organization and function of the blood-brain barrier in Drosophila. J Neurosci 28(3): 587-597.
- Weiler, A., Volkenhoff, A., Hertenstein, H. and Schirmeier, S. (2017). Metabolite transport across the mammalian and insect brain diffusion barriers. Neurobiol Dis 107: 15-31.
- Winkler, B., Funke, D., Benmimoun, B., Speder, P., Rey, S., Logan, M. A. and Klambt, C. (2021). Brain inflammation triggers macrophage invasion across the blood-brain barrier in Drosophila during pupal stages. Sci Adv 7(44): eabh0050.
Article Information
Copyright
© 2022 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Benmimoun, B., Winkler, B. and Spéder, P. (2022). Infection of the Developing Central Nervous System of Drosophila by Mammalian Eukaryotic and Prokaryotic Pathogens. Bio-protocol 12(23): e4563. DOI: 10.21769/BioProtoc.4563.
Category
Neuroscience > Development
Microbiology > Microbe-host interactions > Ex vivo model
Biological Sciences > Biological techniques
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.