- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Site-specific Incorporation of Phosphoserine into Recombinant Proteins in Escherichia coli
Published: Vol 12, Iss 21, Nov 5, 2022 DOI: 10.21769/BioProtoc.4541 Views: 3959
Reviewed by: Petru-Iulian TrasneaSrujana Samhita YadavalliNoelia ForesiAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jul 2019

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
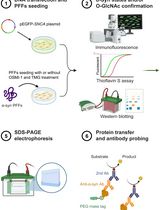
Abstract
This protocol describes the recombinant expression of proteins in E. coli containing phosphoserine (pSer) installed at positions guided by TAG codons. The E. coli strains that can be used here are engineered with a ∆serB genomic knockout to produce pSer internally at high levels, so no exogenously added pSer is required, and the addition of pSer to the media will not affect expression yields. For “truncation-free” expression and improved yields with high flexibility of construct design, it is preferred to use the Release Factor-1 (RF1) deficient strain B95(DE3) ∆A ∆fabR ∆serB, though use of the standard RF1-containing BL21(DE3) ∆serB is also described. Both of these strains are serine auxotrophs and will not grow in standard minimal media. This protocol uses rich auto-induction media for streamlined and maximal production of homogeneously modified protein, yielding ~100–200 mg of single pSer-containing sfGFP per liter of culture. Using this genetic code expansion (GCE) approach, in which pSer is installed into proteins during translation, allows researchers to produce milligram quantities of specific phospho-proteins without requiring kinases, which can be purified for downstream in vitro studies relating to phosphorylation-dependent signaling systems, protein regulation by phosphorylation, and protein–protein interactions.
Graphical abstract:

Background
Cellular signaling pathways are tightly regulated by serine phosphorylation, yet, historically, it has been very challenging to produce site-specifically and homogenously phosphorylated proteins in sufficient quantity for biochemical, biophysical, and structural characterization. Aspartate or glutamate have often been used to mimic the negative charge of phosphoserine (pSer) and phosphothreonine; however, due to the differences in charge and structure of Asp and Glu compared to pSer, both Asp and Glu commonly fail to recapitulate the effects of authentic phosphorylation. Kinases can be used to install phosphates on proteins, but the complexities of kinase specificity, as well as their need to be phosphorylated to be active, limit their utility to the few that can be recombinantly expressed in an active form and have well-defined substrates. Further discussion of these challenges is provided in our earlier publication, on which we base the protocol described here (Zhu et al., 2019).
Genetic code expansion (GCE) technologies have been developed for the translational installation of pSer in response to amber (TAG) stop codons. These technologies require a phosphoserine amino-acyl tRNA synthetase (SepRS), its cognate tRNA with an anti-codon that can suppress TAG codons (Sep-tRNACUA), and an EFTu variant (EFSep) engineered to deliver the pSer-amino-acylated tRNA to the ribosome (Park et al., 2011; Rogerson et al., 2015). Additionally, all pSer expression hosts have their serB gene knocked out, which causes free pSer amino acid to build up inside the cell to levels sufficient to feed the GCE machinery (Park et al., 2011). A few such ∆serB strains have been developed, and the choice of which to use is an important factor in the success of pSer protein expression. We recently created a healthy Release Factor-1 (RF1—the protein responsible for terminating translation at TAG codons) deficient strain of E. coli for pSer protein expression, called B95(DE3) ∆A ∆fabR ∆serB (Zhu et al., 2019). As reported by Zhu et al. (2019), by coupling this expression host with the efficient pSer GCE machinery created by Chin and colleagues (Rogerson et al., 2015), this expression system produces pSer proteins at higher yields than when using the parent RF1-containing BL21(DE3) ∆serB strain, does so without buildup of truncated protein, and has been shown to express proteins with up to five pSer residues incorporated. Because little or no truncation is observed, target proteins can be expressed with N-terminal affinity and solubility tags (e.g., SUMO, GST, MBP) without co-purification of truncated protein. This pSer protein expression chassis grows healthily (doubling time of ~40–45 min at 37 °C), and avoids misincorporation of natural amino acids caused by near-cognate suppression seen in other RF1 deficient pSer protein expression systems. Target proteins are expressed from the well-established T7 promoter expression system for high levels of protein production, and expression is compatible with auto-induction media. Hydrolysis of the pSer moiety by endogenous E. coli phosphatases during protein expression can be an issue, depending on target protein and site of incorporation, and so expression conditions and times may require additional optimization. Described below is the general workflow for expressing pSer proteins with B95(DE3) ∆A ∆fabR ∆serB, as well as with BL21(DE3) ∆serB, should users decide to use this strain.
Materials and Reagents
Expression strain options
B95(DE3) ∆A ∆fabR ∆serB (Zhu et al., 2019). Available upon request.
BL21(DE3) ∆serB (Addgene # 34929) (Park et al., 2011; Rogerson et al., 2015)
Neither strain has any plasmids in them, and they are sensitive to all antibiotics.
The B95 ∆A ∆fabR ∆serB strain is RF1-deficient, and so little or no truncated protein is produced. Thus, N-terminal purification tags can be used with minimal concern of co-purifying truncated protein. This strain is highly preferred when (i) expressing homomultimeric proteins, (ii) C-terminal affinity tags hinder protein function, (iii) N-terminal solubility enhancing domains are needed (e.g., SUMO, GST, MBP, etc.), or (iv) seeking to achieve improved yields of multi-site pSer incorporation. These cells grow modestly, slower than the BL21 strain (described next), but yields from this strain are at least equivalent and in many cases higher than those of the BL21 strain (Zhu et al., 2019).
The BL21 (DE3) ∆serB strain contains Release Factor 1 (RF1), so truncated protein is produced along with full-length phospho-protein. To avoid co-purification of truncated protein, C-terminal rather than N-terminal purification tags are strongly recommended (Rogerson et al., 2015). Furthermore, for proteins that self-assemble to homomultimers (dimers, trimers, etc.), purification can be problematic due to the possible co-purification of truncated forms that are incorporated as subunits in the assembly. This is especially a concern when the TAG codon is near the C-terminus.
These strains can be made chemically competent using the Inoue method (Green and Sambrook, 2020). Aliquots of chemically competent cells can be stored at -80 °C for at least 3 years. Do not re-freeze aliquots.
The C321 ∆A strain, a different RF1-deficient strain of E. coli that has been used for recombinant pSer protein expression (Pirman et al., 2015), grows more slowly, is not compatible with T7-based protein expression systems or auto-induction media, and therefore should not be used with this protocol.
Plasmids
pKW2-EFSep (Addgene #173897)
Machinery plasmid for pSer incorporation, chloramphenicol resistance/pBR322 origin of replication (Rogerson et al., 2015). The pKW2-EFSep machinery plasmid has the same origin of replication as standard pET/pBad/pGEX vectors. Therefore, these vectors cannot be used to express target proteins with pSer, due to origin incompatibility with pKW2-EFSep. Target proteins must be expressed from vectors with a different origin of replication, such as p15a, CloDF, or RSF. The pRBC plasmid described below is recommended for target protein expression.
pRBC-sfGFP (Addgene #174075)
Expresses sfGFP wild-type (wt) control protein with C-terminal His6 tag, under a T7 transcriptional promoter, and ampicillin resistance/p15a origin of replication (Zhu et al., 2019).
pRBC-sfGFP 150TAG (Addgene #174076)
Same as above (including the C-terminal 6x-His tag), except for the sfGFP gene containing a TAG amber stop codon at site N150.
pRBC-[x] wild type (you must create)
Expresses your protein of interest [x] or wild type. It is important, at least for initial tests, to clone your wild-type protein into the pRBC plasmid, to ensure it will express as expected.
pRBC-[x] TAG (you must create)
Your protein of interest [x] with a TAG codon at the intended site (or sites) of pSer incorporation.
Media reagents and materials
LB/agar media (see Recipes)
ZY media (see Recipes)
Non-inducing ZY Media (see Recipes)
Auto-inducing ZY media (see Recipes)
SOC media (see Recipes)
25× M-salts (see Recipes)
1 M MgSO4 (see Recipes)
40% (w/v) glucose (see Recipes)
50× 5052 solution (see Recipes)
5,000× Trace metals solution (optional, see Recipes)
Tryptone (e.g., VWR, catalog number: 97063-386)
Yeast Extract (e.g., VWR, catalog number: 97064-368)
NaCl (e.g., VWR, catalog number: 97061-274)
Agar (e.g., VWR, catalog number: 97064-336)
MgSO4·7H2O (e.g., VWR, catalog number: 97062-134)
Na2HPO4 (sodium dibasic) (e.g., VWR, catalog number: AA13437-30)
KH2HPO4 (potassium monobasic) (e.g., VWR, catalog number: BDH9268)
NH4Cl (e.g., VWR, catalog number: 97062-050)
Na2SO4 or (NH4)2SO4 (e.g., VWR, catalog number: BDH9302 or BDH9216)
α-D-glucose (e.g., VWR, catalog number: 97061-168)
α-D-lactose (e.g., VWR, catalog number: 36218.A3)
Glycerol (e.g., VWR, catalog number: BDH24388.320)
Ampicillin (e.g., VWR, catalog number: 97061-442)
Chloramphenicol (e.g., VWR, catalog number: 97061-244)
Ethanol (e.g., VWR, catalog number: 89125-188)
Antifoam B (e.g., J.T. Baker, catalog number: B531-05)
Sodium Fluoride (e.g., VWR, catalog number: 470302-540)
Sodium pyrophosphate (e.g., VWR, catalog number: JT3850-1)
Sodium orthovanadate (e.g., VWR, catalog number: BT219470-25G)
Phos-tag acrylamide (e.g., VWR, catalog number: 101974-086)
Phosphatase inhibitor cocktail mix (e.g., Sigma, catalog number: P2850)
Phos-tag agarose resin (e.g., Wako Chemicals, catalog number: AG-501)
1.7 mL Eppendorf tubes (e.g., VWR, catalog number: 87003-294)
100 mm plates (e.g., VWR, catalog number: 470210-568)
50 mL conical tubes (e.g., VWR, catalog number: 89039-656)
250 mL baffled flasks (e.g., VWR, catalog number: 89095-266)
2.8 L baffled Fernbach flasks (e.g., Sigma, catalog number: CLS44232XL)
Equipment
Autoclave capable of sterilizing liquid media and culturing materials at 121 °C, and of a saturated steam pressure of 15 PSI.
Expression
Static incubator for growing LB/agar plates (set to 37 °C) (e.g., VWR, catalog number: 97025-630)
Shaker incubator for growing liquid cultures (e.g., New Brunswick I26R, Eppendorf, catalog number: M1324-0004)
Shaker should be able to rotate at 200–250 rpm
Refrigeration is necessary for expressions below room temperature (< 25 °C)
Shaker deck should have clamps to hold 250 mL and 2.8 L Fernbach flasks
Optical density 600 nm spectrophotometer (e.g., Ultrospec 10, Biochrome, catalog number: 80-2116-30)
Cell Harvesting
Centrifuge capable of speeds of at least 5,000 rcf and able to hold volumes commensurate with culture sizes (e.g., Beckman Coulter, model: Allegra 25R)
Centrifuge bottles that can withstand centrifugal forces and hold the volume of cultures grown for harvesting cells. The type of bottles depends on the centrifuge and rotor used.
Freezer (-80 °C), if cells are to be stored before protein is purified (e.g., VIP ECO ULT freezer, VWR, catalog number: 76305-596).
42 °C water bath (e.g., VWR, catalog number: 76308-830)
Fluorometer capable of reading sfGFP fluorescence (excitation 488 nm/emission 512 nm). Handheld fluorometers work well for routine fluorescence reads (e.g., PicoFluor from Turner Biosystems).
Procedure
4-Day expression process
Day 1: Transformation
Notes:
Fresh double plasmid transformations must be performed for each expression. Never freeze expression cells containing plasmids; after freezing for storage, the cells will grow with the antibiotics, but they will not express protein. Do not sequentially transform plasmids.
To standardize this protocol in your lab, run control expressions with sfGFP wt and sfGFP-150TAG first, to verify you can achieve the reference expression benchmarks, and ensure that pSer suppression of the TAG site is functioning as expected with a model protein. The protocol below contains information for a standard 50-mL test expression, as well as a 1-L expression, as is often used for target proteins once the system is known to be working. Expression volumes can be scaled as needed.
Ensure a water bath is available at 42 °C. A water bath is important to ensure rapid heat transfer for the heat shock step.
For each expression, add 2 μL each of pKW2-EFSep and the desired pRBC plasmid into a 1.7-mL Eppendorf tube.
This should be ~50–200 ng of each plasmid.
Place tube on ice for 5 min to pre-chill.
Thaw aliquot(s) of B95(DE3) ∆A ∆fabR ∆serB or BL21(DE3) ∆serB competent cells.
Add 50 μL of competent cells to each tube with plasmids, gently mix cells with plasmids, and place back on ice for 30 min.
After 30 min on ice, heat shock cells by placing the tube in the water bath at 42 °C for exactly 45 s.
Immediately place tube back on ice, and incubate for 2 min.
Add 1 mL of SOC media.
Note: make sure SOC is not contaminated from prior use.
Allow cells to recover with shaking at >200 rpm at 37 °C. It is convenient to simply tape Eppendorf tubes horizontally to shaker deck.
For B95(DE3) ∆A ∆fabR ∆serB, recover for 120 min.
For BL21(DE3) ∆serB cells, recover for 90 min.
Note: Longer recovery times for the B95(DE3) ∆A ∆fabR ∆serB are suggested, due to the slightly slower growth rates and metabolism of this strain.
Plate the recovered culture onto LB/agar/ampicillin/chloramphenicol plates appropriate for the chosen expression strain: 25 μg/mL ampicillin + 7 μg/mL chloramphenicol for B95(DE3) ∆A ∆fabR ∆serB, and 100 μg/mL ampicillin + 25 μg/mL chloramphenicol for BL21(DE3) ∆serB cells.
To ensure a sufficient number of colonies, plate all cells. To do this, centrifuge cells at 3000 rcf for 3 min, remove the top 900 μL of supernatant, resuspend the cell pellet by gentle pipetting in the remaining 100 μL of supernatant, then plate and spread the fully resuspended cells.
Let plate(s) dry with lid partially open for ~20 min near a flame (maintaining sterility), and then incubate plate(s) upside down at 37 °C overnight.
Day 2: Starter culture
Notes:
Volumes of media can be changed depending on the scale of expression desired. Described below are volumes for a 50-mL sfGFP test expression and a 1-L target protein expression.
This protocol requires overnight non-inducing starter cultures to reach stationary phase. Starter cultures that do not reach stationary phase may not express target protein appropriately in auto-inducing media.
Remove LB/agar plates from the incubator at 37 °C, and place them at room temperature or 4 °C for the day.
~20–100 colonies should have grown for B95(DE3) ∆A ∆fabR ∆serB cells (Figure 1). If no colonies grew, check that the correct antibiotic concentrations were used (25 μg/mL ampicillin, 7 μg/mL chloramphenicol). If correct concentrations were used, the cells may not have been made sufficiently competent.
Note: Concentrations of antibiotics for this strain are adjusted to facilitate faster-growth on LB/agar plates. These concentrations were chosen to be approximately one quarter (25%) that used with standard strains.
~100–500 colonies should have grown for BL21(DE3) ∆serB cells (Figure 1).
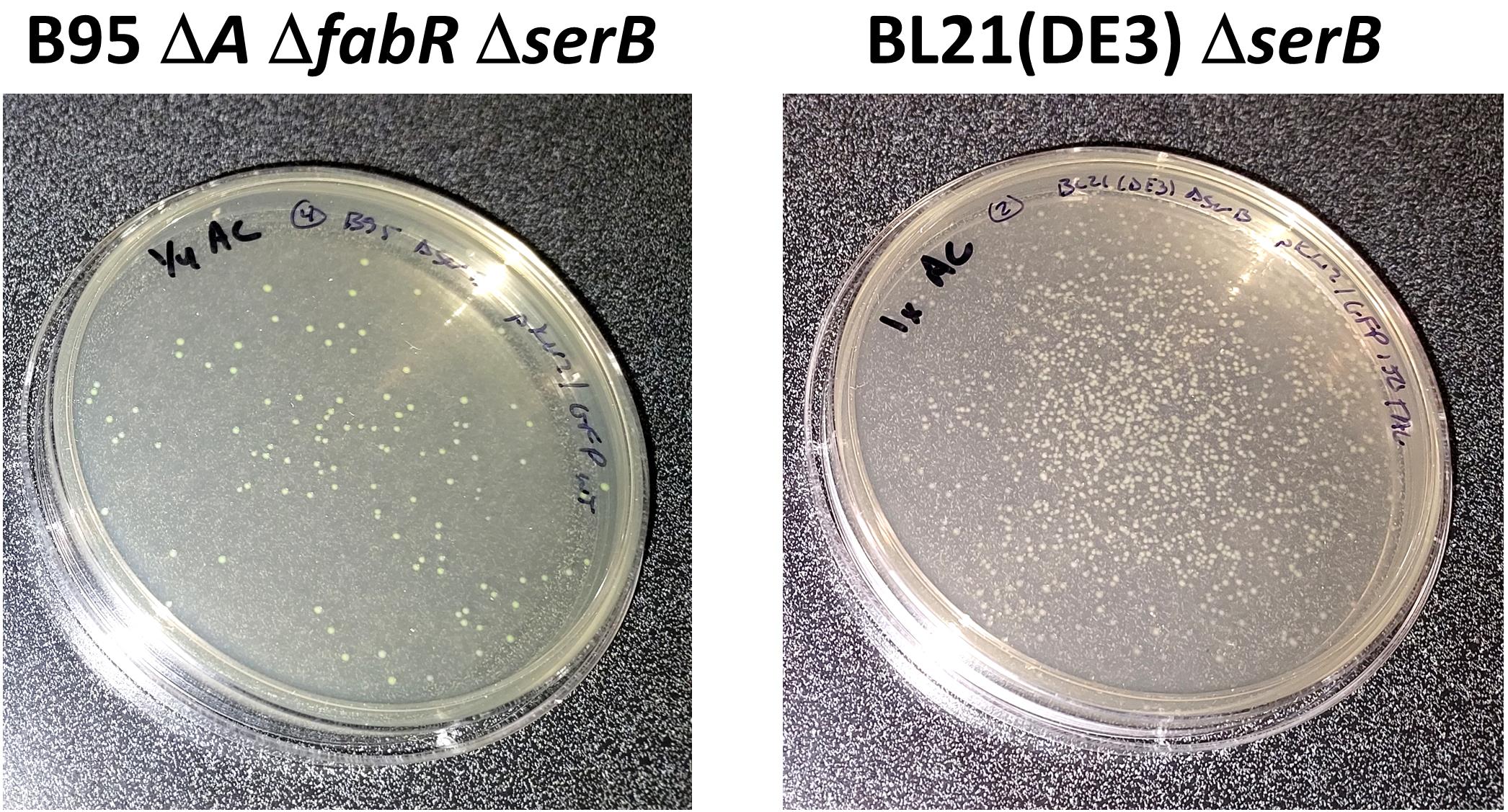
Figure 1. Representative LB/agar plates after co-transformation of pRBC-sfGFP and pKW2-EFSep into B95 ∆A ∆fabR ∆serB (left) and BL21(DE3) ∆serB strains and growth at 37 °C overnight (~18 h). Note that the B95 ∆A ∆fabR ∆serB strain contains ~10-fold fewer colonies, due to the lower transformation efficiency of this strain compared to BL21(DE3) ∆serB.At the end of the day (~3–5 p.m.), prepare ZY-NIM starter culture:
Prepare 50 mL of ZY-NIM with appropriate antibiotics.
For B95(DE3) ∆A ∆fabR ∆serB, use 50 μg/mL ampicillin and 13 μg/mL chloramphenicol.
Note: Concentrations of antibiotics for this strain in liquid media are adjusted to be approximately one half (50%) of that used with standard strains.
For BL21(DE3) ∆serB, use 100 μg/mL ampicillin and 25 μg/mL chloramphenicol.
Scrape several dozen colonies from overnight LB/agar plate, and use them to inoculate:
For a ≥ 1 L expression: 50 mL of ZY-NIM starter culture in a 250-mL baffled flask. Add 1–2 drops of antifoam B.
For a 50 mL expression: 5 mL of ZY-NIM in a 15-mL sterile culture tube.
Grow starter culture(s) with shaking at ~250 rpm at 37 °C overnight.
Day 3: Expression
Prepare expression culture:
Note: Baffled flasks are highly recommended, to ensure sufficient aeration for auto-induction cultures.
Overnight ZY-NIM should have grown to saturation overnight (OD600 ~3–8). If it did not reach OD > 3, either culture did not grow for long enough or media/antibiotic concentrations may not have been correct.
Prepare ZY-AIM according to the recipe below.
Note: It is recommended that you make all media required in a single batch and then divide into appropriate culture flasks, to ensure all expressions contain the same media.
For a 1-L expression: prepare ZY-AIM in a baffled 2.8-L Fernbach flask.
For a 50-mL test expression: prepare ZY-AIM in a 250-mL baffled flask.
Add antibiotics
For B95(DE3) ∆A ∆fabR ∆serB, use 50 μg/mL ampicillin and 13 μg/mL chloramphenicol.
For BL21(DE3) ∆serB, use 100 μg/mL ampicillin and 25 μg/mL chloramphenicol.
Add Antifoam B: ~5–7 drops for a 1-L culture, or 1–2 drops for a 50-mL culture.
Add ~1% inoculum of ZY-NIM culture to the ZY-AIM culture (e.g., 10 mL into 1 L, or 0.5 mL into 50 mL).
Grow at 200–250 rpm and 37 °C until OD600 is ~1.5.
This should take about 3–4 h for BL21 cells, or 4–5 h for B95 cells.
Note: The linear range of spectrophotometers is 0.1–1.0 when reading OD600, so a 5- or 10-fold dilution of the culture will be necessary for accurate measurements of cell density, once they grow above 1.0.
Once OD600 reaches ~1.5, add more antifoam: ~5 drops for a 1-L culture; or ~1–2 drops for a 50-mL culture.
Reduce temperature as needed for effective target protein expression. For sfGFP and sfGFP-150TAG expressions, cultures can be kept at 37 °C.
For B95(DE3) ∆A ∆fabR ∆serB, expressions can go as low as 18 °C, but 22–25 °C may be preferable for “cold temperature” expressions. Temperatures as high as 37 °C are acceptable.
For BL21(DE3) ∆serB, expressions as low as 18 °C and as high as 37 °C are acceptable.
Continue shaking at 200–250 rpm for ~16–20 h.
Day 4: Expression analysis and harvesting of cells
sfGFP control protein expression analysis. Approximately 16–20 h after cells reached OD ~1.5; the control culture cells should be visibly green. Measure OD600 and fluorescence of both sfGFP and sfGFP-150TAG cultures using a fluorometer. Since sfGFP chromophore formation requires synthesis of full-length protein, fluorescence of whole cells provides a convenient strategy to evaluate the efficiency of sfGFP TAG codon suppression, and therefore pSer incorporation. Fluorescence can be measured with any fluorimeter capable of detecting sfGFP fluorescence (ex/em: 488/510nm). Diluting the culture 1:10 to 1:100 in a buffer (e.g., 100 μL of culture + 1,900 μL of PBS for a 1:20 dilution) prior to fluorescence measurements may be necessary to obtain a signal within the detection limits of the fluorometer. The OD600 values will vary depending on target protein. Normal values will range from ~2.5 to 15. Final OD600 values below 2 are indicative of poor cell growth or toxicity due to target protein expression.
Test Expression Benchmarks: Carrying out 50-mL test expressions for the wild-type sfGFP and the sfGFP-150TAG constructs ensures that the protocol is properly implemented. The benchmark yields listed in Table 1 are based on 50-mL culture volumes, expressed exactly as described in this protocol (37 °C expression temperature, ZY-AIM).
Table 1. Benchmarks for sfGFP-WT and 150TAG protein expression yields, based on culture fluorescence
Strain Fluorescence of culture# mg of sfGFP per liter culture* OD600 wt sfGFP B95 9,550 (6,000–10,000) 550 (340–600) 8.0 sfGFP 150TAG B95 2,900 (2,000–3,000) 180 (120–200) 8.5 wt sfGFP BL21 8,500 (6,000–10,000) 480 (340–600) 11.0 sfGFP 150TAG BL21 1,300 (1,000–2,500) 80 (60–150) 12.1 #The range indicated in parenthesis is considered normal depending on the day and reagent preparation. Fluorescence values reported here were obtained on a hand-held PicoFluor fluorometer (Turner Biosystems) by diluting cells directly from the culture into PBS (1:20). Fluorescence values are arbitrary and will depend on the fluorometer used. Important is that the relative ratio of sfGFP-150TAG and sfGFP-WT culture fluorescence is consistent with the above values (i.e., the fluorescence of the sfGFP-150TAG protein expression cultures should be ~1/4 to 1/2 that of WT sfGFP).
*Yield of sfGFP in milligrams per liter was determined using a fluorescence standard curve of purified sfGFP.
Harvest cells by centrifugation if OD600 > ~2 at 5,000 rcf for 15 min. Pour off culture supernatant and resuspend in appropriate buffer.
The choice of buffer depends on the protein of interest, the downstream purification strategy, and the application, and should be determined by the user.
Adding a cryoprotectant [e.g., 10% (v/v) glycerol] to this buffer can help to minimize adverse effects associated with freezing for sensitive or unstable proteins. The addition of phosphatase inhibitors (see Recipes) to the buffer is recommended for all steps of purification. Cells can be flash frozen in liquid nitrogen and stored at -80 °C, or you can proceed with purification.
For His6 tagged proteins to be purified via TALON resin, a recommended resuspension/lysis buffer would be 50 mM Tris pH 7.5, 500 mM NaCl, 10% (v/v) glycerol, 5 mM imidazole, 50 mM NaF, 5 mM sodium pyrophosphate, and 1 mM sodium orthovanadate.
Data analysis
Analysis of Protein Phosphorylation
For each expression, it is important to confirm faithful incorporation of pSer into the target protein. While E. coli does not harbor the extensive network of protein phosphatases found in eukaryotic cells, they do have phosphatases that can hydrolyze the target protein during protein expression, resulting in a mixture of phosphorylated and non-phosphorylated protein. The susceptibility of pSer hydrolysis depends greatly on the target protein, site of pSer incorporation, and culturing conditions. When expressing sfGFP-150TAG using the above-described methods at 37 °C, the purified protein should be 90–95% phosphorylated (see Figure 2 and Zhu et al., 2019).
Several methods can be used to evaluate the phosphorylation status of the purified target protein.
Phos-tag electrophoresis: Phos-tag gel electrophoresis is a modified form of SDS-PAGE, in which phosphorylated proteins migrate with attenuated electrophoretic mobility compared to their non-phosphorylated counterparts; the more sites of phosphorylation on a protein, the slower it will migrate, allowing one to distinguish between no, single-, and multi-pSer containing proteins (Kinoshita et al., 2006). Further, by measuring the density of the phosphorylated vs. non-phosphorylated protein bands, one can qualitatively evaluate the percentage of protein that is phosphorylated. Other advantages of Phos-tag include the ability to evaluate multiple samples at once, requiring only a standard SDS-PAGE electrophoresis setup, and being economical. Important to note is that wild-type (non-phosphorylated) protein must be run side-by-side with the phosphorylated protein, to observe relative shifts in electrophoretic mobility. Do not run molecular weight markers on Phos-tag gels, as they contain EDTA, which is not compatible with Phos-tag. See Figure 2, as well as Zhu et al. (2019), for examples of sfGFP control protein SDS-PAGE and Phos-tag gels.
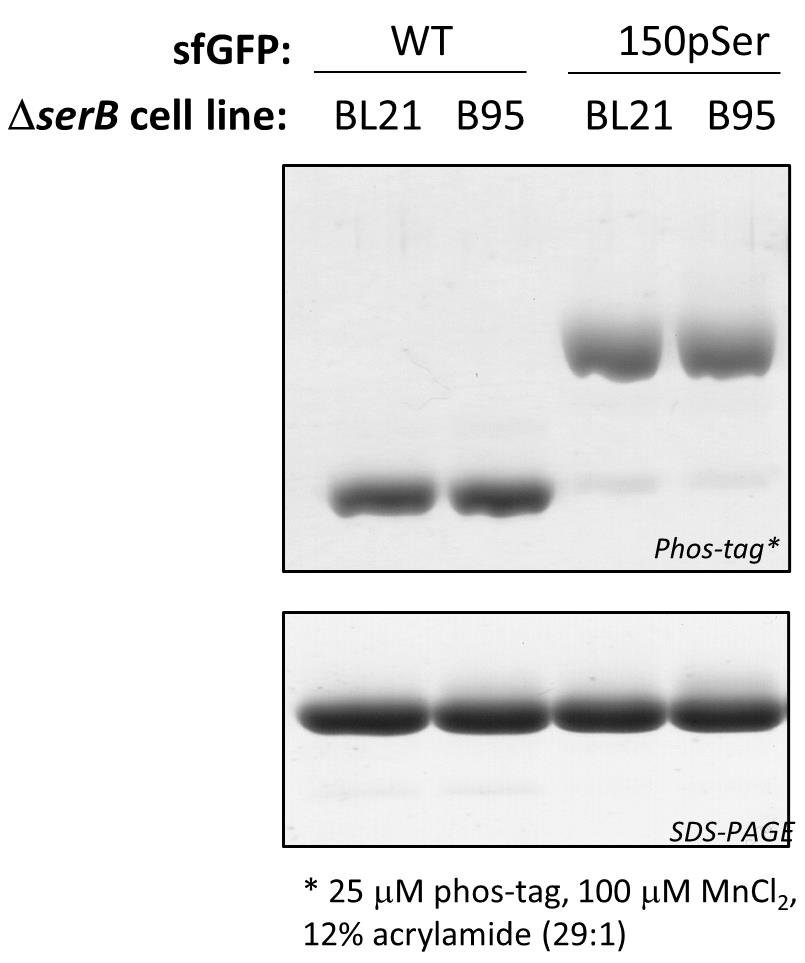
Figure 2. Assessing sfGFP phosphorylation status by Phos-tag gel electrophoresis. Phos-tag (top) and SDS-PAGE (bottom) gels of purified WT sfGFP and sfGFP-150pSer proteins using the above described expression methods at 37 °C. Because they migrate differently on Phos-tag gels, phosphorylated and non-phosphorylated proteins can be resolved from one another and the fraction of phosphorylated protein can be easily evaluated. In this example, the 150pSer-sfGFP protein samples are >90% phosphorylated, having only trace amounts of non-phosphorylated protein that migrate identically as the wild-type sfGFP protein. All proteins migrate identically on SDS-PAGE, indicating the electrophoretic shifts observed in Phos-tag are due to phosphorylation status and not differences in protein size. These samples were incubated at 95 °C for 5 min in standard Laemmli buffer prior to their loading on gel.Mass spectrometry: Whole-protein mass spectrometry (MS) can be the most convincing methodology for confirming protein phosphorylation, if facilities are available and the target protein is amenable to MS analysis.
Important note: tryptic-digest followed by MS fragmentation analysis (i.e., “bottom up” MS/MS analysis) of phospho-proteins is useful for confirming pSer incorporation and the site of incorporation, but it should not be used to evaluate or make conclusions regarding the degree of phosphorylation.
Troubleshooting tips if the purified target protein is partially hydrolyzed.
Increase concentrations of phosphatase inhibitors in purification buffers, or consider a more extensive phosphatase inhibitor cocktail mix (e.g., Sigma #P2850).
Lower temperature of expression to 18–22 °C.
Shorten expression time from 16–20 h to 10–12 h, though care should be taken to ensure culture reached OD600 > 2.5 for sufficient time to allow auto-induction and expression to occur. This will result in less, but potentially better-quality protein.
Filter all purification buffers through a 0.2 μm filter, to ensure they are clean and free of contaminating bacterial growth. Perform purifications at 4 °C.
Develop purification strategies to separate phosphorylated protein from non-phosphorylated protein, e.g., via anion-exchange chromatography or Phos-tag agarose resin (e.g., Wako Chemicals #AG-501).
Consider incorporating the non-hydrolyzable mimic of phospho-serine containing a carbon-phosphorus bond instead of the labile oxygen-phosphorus bond. This is a distinct GCE expression system, and its methodology is outside the scope of this protocol (Zhu et al., 2021).
Recipes
Note: Ampicillin antibiotic stocks can be made at 100 mg/mL in water. Chloramphenicol stocks can be made at 25 mg/mL in ethanol. Store in 1-mL aliquots at -20 °C.
LB/agar (1 L)
10 g Tryptone, 5 g yeast extract, 5 g NaCl, 15 g agar
After mixing thoroughly, autoclave on the standard liquid setting.
After autoclaving, gently swirl the bottle to ensure melted agar is evenly mixed.
Notes:
Store LB/agar bottle in a 55 °C oven and pour plates on an as-needed basis. LB/agar can be stored in molten form for ~2 weeks, if sterility is maintained.
If an oven is not available, plates can be poured with antibiotics once LB/agar is sufficiently cooled to touch. Plates can be stored at 4 °C for 3–5 days.
For B95(DE3) ∆A ∆fabR ∆serB plates, use 25 μg/mL ampicillin (for pRBC plasmid) and 7 μg/mL chloramphenicol (for pKW2-EFSep plasmid).
For BL21(DE3) ∆serB plates, use 100 μg/mL ampicillin (for pRBC plasmid) and 25 μg/mL chloramphenicol (for pKW2-EFSep plasmid).
Starter culture media
ZY-non inducing media (ZY-NIM), adapted from Studier (2005).
Each component in Table 2 below should be prepared separately and sterilized separately. Once sterilized, each component can be stored separately at room temperature indefinitely, provided sterility is maintained. Components should only be mixed when at room temperature and immediately before use.
Table 2. Y-NIM formulation
For 100 mL ZY media 94.5 mL 1 M MgSO4 0.2 mL 25× M-Salts 4.0 mL 40% (w/v) α-D-glucose 1.25 mL Trace Metals solution (5,000×)* 20 µL Ampicillin (100 mg/mL stock) 50 μL for B95 cells, 100 μL for BL21 cells Chloramphenicol (25 mg/mL stock) 50 μL for B95 cells, 100 μL for BL21 cells *Trace metals are not essential, but can help when expressing proteins that require metal co-factors.
ZY media (1 L)
10 g Tryptone, 5 g yeast extract
Autoclave
1 M MgSO4(50 mL)
12.3 g of heptahydrate salt, add MilliQ H2O to 50 mL total volume
Autoclave or sterile filter
25× M-salts (1 L)
0.625 M Na2HPO4 (sodium dibasic), 88.73 g anhydrous salt
0.625 M KH2HPO4 (potassium monobasic), 85.05 g anhydrous salt
1.25 M NH4Cl, 66.86 g
0.125 M Na2SO4, 17.75 g anhydrous salt
Autoclave
NOTE: If you do not have sodium sulfate but you do have ammonium sulfate, use the following alternate recipe:
0.625 M Na2HPO4 (sodium dibasic), 88.73 g
0.625 M KH2HPO4 (potassium monobasic), 85.05 g
1.0 M NH4Cl, 53.49 g
0.125 M (NH4)2 SO4, 16.52 g
Autoclave
40% (w/v) glucose (50 mL)
20 g α-D-glucose, add MilliQ H2O to 50 mL total volume
Autoclave or sterile filter.
5,000× Trace metals (see Table 3)
Table 3. Trace metal mix formulation
Amount for 30 mL stock solution Amount of 30 mL stock solution to make 50 mL of a 5,000× solution 1× media concentration CaCl2·2H2O (8.82 g) 500 μL 4 μM MnCl2·4H2O (5.93 g) 500 μL 2 μM ZnSO4·7H2O (8.62 g) 500 μL 2 μM CoCl2·6H2O (1.32 g) 500 μL 0.4 μM CuCl2 (807 mg) 500 μL 0.4 μM NiCl2 (777 g) 500 μL 0.4 μM Na2MoO4·2H2O (1.45 g) 500 μL 0.4 μM Na2SeO3 (1.03 g) 500 μL 0.4 μM H3BO3 (371 mg) 500 μL 0.4 μM FeCl3 (486 mg) 25 mL 10 μM Dilute to 50 mL with sterile water Expression media
ZY-auto inducing media (ZY-AIM), adapted from (Studier, 2005).
Each component in Table 4 below should be prepared separately and sterilized separately. Once sterilized, each component can be stored separately at room temperature indefinitely, provided sterility is maintained. Components should only be mixed when at room temperature and immediately before use. Note that recipes for all components, except the 50× 5052 solution, are in the section above describing “starter culture media.”
Table 4. ZY-AIM formulation
For 1 L ZY media 940 mL MgSO4 (1M) 2 mL 25× M-Salts 40 mL 50× 5052 solution 20 mL Trace Metals solution (5,000×)* 200 µL Ampicillin (100 mg/mL stock) 500 μL for B95 cells, 1 mL for BL21 cells Chloramphenicol (25 mg/mL stock) 500 μL for B95 cells, 1 mL for BL21 cells *Trace metals are not essential but can help when expressing proteins that require metal co-factors.
50× 5052 solution (500 mL)
2.5% (w/v) α-D-glucose, 12.5 g
10% (w/v) lactose, 50 g
25% (v/v) glycerol, 125 mL of glycerol or 250 mL of a 50% solution
Note: The lactose will not dissolve immediately. Heating solution in a microwave for ~2–3 min will warm solution sufficiently to dissolve lactose. One dissolved, lactose will stay in solution indefinitely. Lactose must be in solution before autoclaving.
Sterilize by autoclaving
Note: Due to the high viscosity of pure glycerol, it is easier to first make a 50% (v/v) glycerol solution as follows:
Add an appropriately sized, clean stir bar to a 500-mL graduated cylinder.
Add 250 mL of MilliQ H2O to the graduated cylinder.
Place on stir plate and begin stirring water in the graduated cylinder.
Slowly pour 100% glycerol into the graduated cylinder while stirring, until final volume is 500 mL.
Continue stirring for 5–10 min until glycerol is thoroughly mixed.
SOC media (1 mL needed for each transformation)
Prepare the following individual sterile components
2× YT media: 16 g tryptone, 10 g yeast extract, 5 g NaCl per liter (autoclave)
40% (w/v) glucose (see above)
1 M MgSO4 (see above)
Combine the following for 50 mL of SOC
49 mL of 2× YT
450 μL of 40% glucose (20 mM final concentration)
500 μL of 1 M MgSO4 (20 mM final concentration)
Note: It is very easy to contaminate SOC. We suggest breaking this into 5 × 10-mL aliquots before use, or make smaller batches. If sterility is maintained, SOC can be stored at room temperature indefinitely. It can also be stored at -20 °C, but avoid repeated freeze/thaw cycles.
Phosphatase inhibitors for protein purification
STOCK SOLUTIONS
1 L of 1 M NaF (sodium fluoride)
Dissolve 42 g of NaF with 900 mL of MilliQ H2O with stirring.
Once dissolved, adjust to 1 L final volume.
This solution is stable indefinitely at room temperature.
250 mM sodium pyrophosphate pH 7.5 (NaPPi)
Dissolve 27.7 g NaPPi (dibasic, MW = 221.9) in 450 mL.
pH adjust to 7.5 with 5 M NaOH or 5 M HCl.
Adjust total volume to 500 mL.
Aliquot 45 mL into 50 mL conicals, store frozen at -20 °C until use.
200 mM Sodium orthovanadate (Na3VO4)
Add 1.85 g Na3VO4 to 45 mL of MilliQ water and dissolve with stirring.
Slowly add 1 M HCl dropwise with stirring, to adjust pH to 10. Adding HCl will make the solution yellow, indicative of vanadate oligomers forming.
Bring solution to a gentle boil by heating in a microwave. After boiling for 5–15 sec, the solution will become clear and colorless.
Cool (on ice will speed up the process) until the solution reaches room temperature.
At this point, the pH might be greater than 10. Add a small amount (several drops, with stirring) of 1 M HCl to adjust solution back to pH 10.
If solution turns yellow, repeat steps (iii–v). After several cycles of boiling, cooling, and adjusting pH, the solution should reach a point where the pH stabilizes at ~10.
Make 1-mL aliquots in 1.5 mL Eppendorf tubes and store activated (colorless and monomeric) Na3VO4 at -20 °C.
WORKING SOLUTION
Dilute inhibitor stocks into your buffers at the following working concentrations:
20–50 mM NaF
5–10 mM NaPPi
0.5–1 mM orthovanadate
Acknowledgments
This protocol was adapted from previous work by Zhu et al. ( 2019). This research was funded in part by the GCE4All Biomedical Technology Development and Dissemination Center, supported by National Institute of General Medical Science grant RM1-GM144227 as well as National Institutes of Health grant 1R01GM131168-01.
Competing interests
The authors declare no conflicts of interest or competing interests.
References
- Green, M. R. and Sambrook, J. (2020). The Inoue Method for Preparation and Transformation of Competent Escherichia coli: "Ultracompetent" Cells. Cold Spring Harb Protoc 2020(6): 101196.
- Kinoshita, E., Kinoshita-Kikuta, E., Takiyama, K. and Koike, T. (2006). Phosphate-binding tag, a new tool to visualize phosphorylated proteins. Mol Cell Proteomics 5(4): 749-757.
- Park, H. S., Hohn, M. J., Umehara, T., Guo, L. T., Osborne, E. M., Benner, J., Noren, C. J., Rinehart, J. and Söll, D. (2011). Expanding the genetic code of Escherichia coli with phosphoserine. Science 333(6046): 1151-1154.
- Rogerson, D. T., Sachdeva, A., Wang, K., Haq, T., Kazlauskaite, A., Hancock, S. M., Huguenin-Dezot, N., Muqit, M. M., Fry, A. M., Bayliss, R., et al. (2015). Efficient genetic encoding of phosphoserine and its nonhydrolyzable analog. Nat Chem Biol 11(7): 496-503.
- Studier, F. W. (2005). Protein production by auto-induction in high density shaking cultures. Protein Expr Purif 41(1): 207-234.
- Zhu, P., Franklin, R., Vogel, A., Stanisheuski, S., Reardon, P., Sluchanko, N. N., Beckman, J. S., Karplus, P. A., Mehl, R. A. and Cooley, R. B. (2021). PermaPhos (Ser) : autonomous synthesis of functional, permanently phosphorylated proteins. bioRxiv. doi: 10.1101/2021.10.22.465468.
- Zhu, P., Gafken, P. R., Mehl, R. A. and Cooley, R. B. (2019). A Highly Versatile Expression System for the Production of Multiply Phosphorylated Proteins. ACS Chem Biol 14(7): 1564-1572.
Article Information
Copyright
© 2022 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Zhu, P., Mehl, R. A. and Cooley, R. B. (2022). Site-specific Incorporation of Phosphoserine into Recombinant Proteins in Escherichia coli. Bio-protocol 12(21): e4541. DOI: 10.21769/BioProtoc.4541.
Category
Biological Engineering > Synthetic biology
Biological Engineering > Biomedical engineering
Biochemistry > Protein > Posttranslational modification
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.