- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

OxiDIP-Seq for Genome-wide Mapping of Damaged DNA Containing 8-Oxo-2'-Deoxyguanosine
Published: Vol 12, Iss 21, Nov 5, 2022 DOI: 10.21769/BioProtoc.4540 Views: 3284
Reviewed by: Istvan Boldogh Avinash Chandra PandeyAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Apr 2022
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
8-oxo-7,8-dihydro-2′-deoxyguanosine (8-oxodG) is considered to be a premutagenic DNA lesion generated by 2'-deoxyguanosine (dG) oxidation due to reactive oxygen species (ROS). In recent years, the 8-oxodG distribution in human, mouse, and yeast genomes has been underlined using various next-generation sequencing (NGS)–based strategies. The present study reports the OxiDIP-Seq protocol, which combines specific 8-oxodG immuno-precipitation of single-stranded DNA with NGS, and the pipeline analysis that allows the genome-wide 8-oxodG distribution in mammalian cells. The development of this OxiDIP-Seq method increases knowledge on the oxidative DNA damage/repair field, providing a high-resolution map of 8-oxodG in human cells.
Keywords: 8-oxo-7,8-dihydro-2′-deoxyguanosineBackground
DNA, as a highly dynamic molecule, is constantly exposed to mutation events (Lindahl, 1993). Among DNA mutant factors, reactive oxygen species (ROS) are the most prominent source of DNA modifications that may trigger processes such as neurodegeneration (Kim et al., 2015), ageing (Beckman and Ames, 1998), and cancer (Klaunig and Kamendulis, 2004). The 2'-deoxyguanosine (dG) is the most ROS-targeted nucleobase due to its lower oxidation potential (Steenken and Jovanovic, 1997; Baik et al., 2001). The best characterized product of dG oxidation induced by ROS is 8-oxo-7,8-dihydro-2′-deoxyguanosine (8-oxodG; Figure 1A) (Baik et al., 2001; Cooke et al., 2003; Evans and Cooke, 2004; van Loon et al., 2010). This altered base is chemically generated by C8 oxidation and subsequent addition of a hydrogen atom on the N7 in the imidazole ring of deoxyguanosine.
The 8-oxodG is considered a premutagenic DNA lesion that may bond 2′-deoxyadenosine, thus causing a dC:dG to dA:dT premutagenic transversion (Shibutani et al., 1991; Maga et al., 2007; Batra et al., 2010; Koga et al., 2013; Boiteux et al., 2017). Human cells can survive this mutational phenomenon through the base excision repair (BER), a multi-layer defence machinery (Lindahl, 1990; Lindahl and Barnes, 2000).
The OGG1 glycosylase/AP(apurinic/apyrimidinic) lyase initiates the BER pathway, specifically recognizing and removing the 8-oxodGs from the sugar backbone. The OGG1 activity creates an abasic site, which is subsequently incised by either the OGG1 intrinsic AP lyase activity, or by the AP endonuclease 1, APE1. The lyases’ activity generates a single strand DNA (ssDNA) break. Then, the short patch BER carries on with the gap filling, mediated by the DNA polymerase beta, while the long patch BER generates a 5′ overhanging flap that is removed by FEN1 (flap structure–specific endonuclease 1). Finally, the DNA ligase I (short patch) or DNA ligase III (long patch) complete the repair process by fixing the nicked strand (Frosina et al., 1996; Fortini et al., 1999; van Loon et al., 2010).
Several next-generation sequencing (NGS)–based strategies have recently been developed by different laboratories to provide a high-resolution mapping of 8-oxodG in human and mouse genomes (Ding et al., 2017; Poetsch et al., 2018; Wu et al., 2018; Amente et al., 2019; Liu et al., 2019, 2019; Cao et al., 2020; Fang and Zou, 2020; Poetsch, 2020). The previous study developed a highly sensitive methodology named OxiDIP-Seq to isolate and map oxidized DNA fragments in mammalian cells (Amente et al., 2019; Gorini et al., 2020; Scala et al., 2022). OxiDIP-Seq combines the immuno-precipitation of single-stranded DNA through specific anti-8-oxodG antibodies with NGS, with a resolution of approximately 200–300 bp (Amente et al., 2019; Gorini et al., 2020; Scala et al., 2022).
Briefly, with this methodology, genomic DNA containing 8-oxodG residues were first extracted and then fragmented by sonication. The DNA fragments were then denatured at 95 °C to expose the 8-oxodG on the single-stranded DNA. Then, the single-stranded DNA containing 8-oxodG residues was immunoprecipitated using a specific antibody targeting 8-oxodG. The so formed immunocomplexes were then pulled down through specific magnetic beads. The eluted DNA was enriched in fragments containing 8-oxodGs that can be analyzed by qPCR and/or sequenced by high-throughput sequencing (Figure 1B). In this protocol, crucial steps were carried out in low light conditions and in the presence of free radical scavengers to preserve the oxidized state of DNA extracted from cells and to prevent the introduction of possible new nonspecific 8-oxodGs during DNA handling.

Figure 1. OxiDIP-Seq technique. (A) ROS oxidation of 2′-deoxyguanosine generates 8-oxo-7,8-dihydro-deoxyguanosine (8-oxodG). (B) Schematic protocol of the OxiDIP-Seq technique.
Sequencing was performed adopting the SBS (sequencing by synthesis) chemistry and using the Illumina HiSeq 2000 platform. Reads with an average length of 50 bp were obtained. Raw sequenced data were collected in a FASTQ file and subjected to further analyses.
With the OxiDIP-Seq approach, it has been demonstrated that in the genome of normal human cells 42% of the identified 8-oxodG peaks map within gene loci, specifically in the promoter and gene body regions (Amente et al., 2019). Moreover, it has been revealed that 8-oxodG regions show a specific G4-enrichment and that there is a complex relationship between 8-oxodG and guanine–cytosine (GC) content. In particular, the promoter regions with high (>47%) GC content display low levels of 8-oxodG (Gorini et al., 2020). This suggests that other mechanisms, such as the epigenetic regulation of transcription and replication, may be involved in the accumulation of 8-oxodG (Amente et al., 2019; Gorini et al., 2020). Moreover, a set of oxidized enhancers in human epithelial cells have been recently identified and characterized, which could be classified as super-enhancers. Specifically, it has been demonstrated that these oxidized enhancers are associated with bidirectional-transcribed enhancer RNAs and DNA damage response activation. Additionally, it has been revealed that the oxidized enhancer is physically associated with promoter regions in specific CTCF-mediated chromatin loops (Scala et al., 2022).
In conclusion, the OxiDIP-Seq technique allowed us to demonstrate that 8-oxodG accumulation in enhancers–promoters regions occur in a transcription-dependent manner, providing novel mechanistic insights on the intrinsic fragility of chromatin loops containing oxidized enhancers–promoters interactions (Scala et al., 2022).
Materials and Reagents
100 mm2 plate for cell culture (Corning, catalog number: CC430167)
96-well plate for StepOnePlusTM Real-Time PCR System
Primo® filter pipette tips, 0.5–10 μL (Euroclone, catalog number: ECTD00010)
Corning® filtered polypropylene IsoTipTM pipet tips, 1–200 μL (Corning, catalog number: 4810)
Corning® filtered polypropylene IsoTipTM pipet tips, 100–1,000 μL (Corning, catalog number: 4809)
1.5 mL Eppendorf safe-lock tubes (Eppendorf, catalog number: 0030 120.086)
InvitrogenTM QubitTM assay tubes (Invitrogen, catalog number: Q32856)
PBN, N-tert-butyl-α-phenylnitrone (Sigma-Aldrich, catalog number: B7263)
DNeasy Blood & Tissue kit (QIAGEN, catalog number: 69504)
Ethylenediaminetetraacetic acid (EDTA) solution, 0.5 M in H2O (Sigma-Aldrich, catalog number: E7889)
Agarose low EEO (agarose standard) (AppliChem, catalog number: A2114)
8-hydroxydeoxyguanosine antibody (Millipore, catalog number: AB5830)
Dynabeads® magnetic beads protein G (Thermo Fisher Scientific, catalog number: 10003D)
Bovine serum albumin (BSA) (Sigma-Aldrich, catalog number: B6917-100MG)
Mini Elute® PCR purification kit (QIAGEN, catalog number: 28004)
Random primers DNA labeling system (Thermo Fisher Scientific, catalog number: 18187-013)
QubitTM dsDNA HS assay kit (Invitrogen, catalog number: Q32851)
Nuclease-free water (not DEPC-treated) (Ambion, catalog number: AM9930)
Proteinase K, recombinant, PCR grade (Thermo Fisher Scientific, catalog number: EO0492)
TruSeq ChIP library preparation kit (Illumina)
N-acetyl cysteine (see Recipes)
NaPi buffer (1 M, pH 7.4) (see Recipes)
Adjust buffer (see Recipes)
Tris EDTA (TE) buffer (see Recipes)
IP buffer (see Recipes)
Washing buffer (see Recipes)
Elution buffer (see Recipes)
Notes:
Pipette tips used in this protocol should be low-retention, RNase-free, and DNase-free, with aerosol filter. All steps have to be performed using these tips.
PCR tubes and microtubes used in this protocol should be low-retention, RNase-free, and DNase-free. All steps have to be performed using these tubes.
Optional:
MCF10A cell line (ATCC, catalog number: CRL-10317)
Dulbecco's modified Eagle medium (DMEM) (Euroclone, catalog number: ECM0728L)
Ham's nutrient mixture F-12 without L-glutamine (Euroclone, catalog number: ECB7502L)
Horse serum (Euroclone, catalog number: ECS0090L)
Hydrocortisone (Millipore, catalog number: 3867)
Cholera toxin (Sigma-Aldrich, catalog number: C8052)
Insulin (Sigma-Aldrich, catalog number: I5500)
Recombinant human epidermal growth factor (Thermo Fisher Scientific, catalog number: PHG0311)
Penicillin/streptomycin 100× (Euroclone, catalog number: ECB3001D)
Trypsin-EDTA 1× in PBS w/o calcium w/o magnesium w/o phenol red (Euroclone, catalog number: ECB3052D)
Dulbecco's phosphate (PBS) buffer saline w/o calcium w/o magnesium (Euroclone, catalog number: ECB4004L)
N-acetyl cysteine (Sigma-Aldrich, catalog number: A7250)
DNA oligos (Integrated DNA Technologies)
Luna® Universal qPCR master mix (BIOLABS, catalog number: M3003)
Growth medium for the MCF10A cell line (see Recipes)
Equipment
Qubit® 2.0 fluorometer (Thermo Fisher)
Eppendorf micro centrifuge (Eppendorf, model: 5418 R)
Diagenode Bioruptor® Plus B01020001 (Diagenode, model: UCD-300TM)
Eppendorf® Thermomixer® R dry block heating and cooling shaker (Eppendorf, catalog number: T3442)
Wide mini-sub cell GT system (Bio-Rad, catalog number: 1704405EDU)
Wide mini-sub cell GT mini handcasting kit (Bio-Rad, catalog number: 1704497)
PowerPac HC power supply (Bio-Rad, catalog number: 1645052EDU)
Vortex mixer (IKA 3340000)
PureProteomeTM magnetic stand (Millipore, catalog number: LSKMAGS08)
SavantTM SpeedVacTM DNA 130 integrated vacuum concentrator system (Thermo Fisher Scientific, catalog number: 15819206)
StepOnePlusTM Real-Time PCR system upgrade (Applied BiosystemsTM, catalog number: 4379216)
Nanodrop microvolume spectrophotometers (Thermo Fisher Scientific)
(Optional) UV crosslinker (Stratalinker, model: 1800)
Software
Fastqc
Trimmomatic
Burrows-Wheeler Aligner (BWA, version: 0.7.12-r1039, http://bio-bwa.sourceforge.net/, free)
Samtools (v1.7, http://www.htslib.org/, free)
Bedtools (v2.17.0, https://bedtools.readthedocs.io/en/latest/, free)
Deeptools (https://deeptools.readthedocs.io/en/develop/, free)
Homer (http://homer.ucsd.edu/homer/ngs/peaks.html, free)
Procedure
This protocol can be easily applied to any cell line. The current study also includes the optimized procedure for preparation and analysis of untreated and UV- and N-acetyl cysteine (NAC)–treated MCF10A OxiDIP-Seq samples. UV- and NAC-treated MCF10A cells could be used as a positive and negative control, respectively, as described in Amente et al. (2019), Gorini et al. (2020), and Scala et al. (2022). Compared to untreated cells, a significantly higher level of 8-oxodG has been detected in UV-treated and a lower level of 8-oxodG in NAC-treated MCF10A cells, which demonstrates the sensibility and specificity of our method to detect 8-oxodG. Please refer to the above mentioned original papers for more information.
Day I
Optional:
Treat MCF10A cells with 1 mM NAC for 1 h, as negative control.
Irradiate MCF10A cells as positive control, as follows:
Remove the medium from the 100 mm2 plate and put it in a new 15 mL tube.
Insert plate into the Stratalinker® UV crosslinker equipped with a 254 nm UV light.
Irradiate cells by setting 40 J/m2 on the instrument.
After irradiation, add the medium recovered at step 2a on the irradiated cells and incubate cells at 37 °C for 30 min.
Extraction of genomic DNA
Trypsinize untreated and (optionally) treated cells (at least 5 × 106 cells; optimal cell number should be experimentally determined) by adding 1 mL of 0.05% trypsin to a 100 mm2 plate. Incubate in a 5% CO2 humidified incubator at 37 °C for 10 min.
Add 5 mL of growth medium to neutralize trypsin and transfer the cells to a 15 mL centrifuge tube.
Centrifuge the cells at 300 × g and room temperature (RT) for 3 min.
Carefully aspirate the supernatant, resuspend the cells in 200 μL PBS of 1× PBS, and add 20 μL of Proteinase K.
Add 200 μL of buffer AL (see DNeasy Blood & Tissue kit), mix thoroughly by vortexing, and incubate samples at 37 °C for 4 h.
Add 200 μL of ethanol 100%. Mix thoroughly by vortexing.
Pipet the mixture into a DNeasy mini spin column (see DNeasy Blood & Tissue kit) placed in a 2 mL collection tube.
Centrifuge at 6,000 × g (8,000 rpm) for 1 min. Discard the flow-through and collection tube.
Place the DNeasy mini spin column in a new 2 mL collection tube. Add 500 μL of buffer AW1 (see DNeasy Blood & Tissue kit).
Centrifuge at 6,000 × g (8,000 rpm) for 1 min. Discard the flow-through and collection tube.
Place the spin column in a new 2 mL collection tube. Add 500 μL of buffer AW2 (see DNeasy Blood & Tissue kit).
Centrifuge at 6,000 × g (8,000 rpm) for 1 min. Discard the flow-through and collection tube.
Repeat step A12.
Transfer the DNeasy mini spin column to a new 1.5 mL microcentrifuge tube.
Elute the DNA by adding 50 μL of sterile nuclease-free water. Wait for 5 min and centrifuge at 6,000 × g (8,000 rpm) for 1 min.
Elute the remaining DNA by adding another 50 μL of sterile nuclease-free water. Wait for 5 min and centrifuge at 6,000 × g (8,000 rpm) for 1 min. Pool together the DNA eluted from steps A15 and A16.
Note: Work under low luminosity throughout as much as possible (by turning off the lights and covering the windows with a blackout panel in the laboratory), as we experienced that light might reduce the yield of 8-oxodG IP. Also, add PBN (stock 28 mM in nuclease-free water; working 0.07 mM) to each solution indicated in steps A4–A16 just before use to preserve the oxidized state of DNA extracted from cells and to prevent the introduction of possible new nonspecific 8-oxodGs during DNA handling. Moreover, it is suggested reading the recommendations and troubleshooting guide displayed in the DNeasy Blood & Tissue kit handbook.
Sonication of genomic DNA
Note: Make sure that the Bioruptor is cold.
Quantify the eluted DNA using the Nanodrop.
Transfer 5 μg of quantified DNA, in a final volume of 100 μL of TE buffer (see Recipe 3), to a new 1.5 mL tube.
Sonicate using 15–20 cycles (30 s ON and 30 s OFF) in high power mode.
Note: The optimal number of cycles must be determined experimentally.
Use 400 ng of sonicated DNA to test the sonication on agarose gel (1.5%); a good sonication should produce fragments ranging in size between 200 and 800 bp. Keep samples on ice before and after sonication.
Note: Work under low luminosity throughout as much as possible and add PBN (stock 28 mM in nuclease-free water; working 0.07 mM) to each solution indicated in steps B1–B3.
Immunoprecipitation of 8-oxodG-containg ssDNA
Dilute 4 μg of sonicated DNA in 500 μL of IP buffer (see Recipe 6).
Take 3% as Input (15 μL), collect it in a new tube, and store it at -20 °C until step D7.
Denature the samples for 5 min at 95 °C on the thermo-shaker (except for the Input that must be stored at -20 °C). Then transfer to ice for 15 min.
Add 4 μL of polyclonal antibody against 8-hydroxydeoxyguanosine to the sample and incubate overnight at 4 °C under constant rotation.
Block magnetic beads with BSA as follow:
Put 30 μL/sample in a new 1.5 mL microcentrifuge tube.
Wash the magnetic beads with 1 mL of IP buffer.
Resuspend the magnetic beads in 1 mL IP buffer + BSA (0.5 mg/mL) and incubate overnight at 4 °C under constant rotation.
Day IIWash the magnetic beads with 1 mL of cold IP buffer twice.
Resuspend the beads in a final volume of 30 μL/sample of cold IP buffer.
Add 30 μL of BSA-blocked magnetic beads to the sample of step C4 and incubate under constant rotation for 3 h in a cold room (at 4 °C).
Note: Work under low luminosity throughout as much as possible for step C1–C6 and add PBN (stock 28 mM in nuclease-free water; working 0.07 mM) to each solution indicated in steps C1–C5.
Wash the beads–antibody–ssDNA complex with 1 mL of washing buffer (see Recipe 7) as follows:
Put the sample on the magnetic rack for 5 min.
Discard the flow-through.
Add 1 mL of washing buffer.
Take the sample under constant rotation for 10 min in a cold room (at 4 °C).
Repeat step 7 four times.
Note: From step 7 onwards, work under normal light condition in the laboratory.
Purification of 8-oxodG-containg ssDNA and Input DNA
When washes are finished, discard the flow-through and resuspend the beads with 250 μL of elution buffer (see Recipe 8).
Incubate O/N at 37 °C on the thermo-shaker (800 rpm).
Day IIIIncrease temperature to 52 °C and leave for 1 extra hour.
Spin the sample for 3 min at 13,000 rpm, capture beads on magnetic rack, and transfer supernatant to new 2 mL tubes.
Resuspend beads in 100 μL of elution buffer (see Recipe 8) again and re-incubate at 52 °C for 10 min on the thermo-shaker (800 rpm).
Spin the sample for 3 min at 13,000 rpm, capture beads on magnetic rack, and transfer supernatant into the same 2 mL tubes previously used in step D4.
Note: Elution is performed in one tube per sample to have 8-oxodG-containing ssDNA sample (IP) in a total 350 μL volume.
Thaw Input DNA sample (previously stored at -20 °C, step C2) and add 160 μL of elution buffer to reach the total volume of 175 μL.
Purify both IP and Input samples using Qiagen mini elute PCR purification kit:
Add 1,750 μL and 875 μL of PB buffer (without pH indicator, see Qiagen mini elute PCR purification kit) to IP and Input, respectively.
Vortex and spin briefly.
Transfer the samples to MiniElute columns (see Qiagen mini elute PCR purification kit).
Note: Only for the IP sample, as its volume is higher than what can be loaded on a single MiniElute column, split IP sample into two MiniElute columns (1,050 μL each) by loading 525 μL of IP sample on the same column twice.
Spin samples for 1 min at 13,000 rpm.
Discard the flow-through.
Wash the columns with 750 μL of PE buffer (wash buffer containing ethanol, see Qiagen mini elute PCR purification kit).
Spin samples for 1 min at 13,000 rpm.
Discard the flow-through and spin again for 1 min to dry the columns as much as possible.
Elute the DNA in a total volume of 30 μL of nuclease-free water by performing two elutions with 15 μL for each column.
Note: For IP sample, collect the eluted DNA from the two MiniElute columns in one 1.5 mL tube (so the final volume of each IP sample is 60 μL); for Input sample, add further 30 μL nuclease-free water to reach the same IP final volume (60 μL).
Optional: qPCR quality test of UV- and NAC-treated MCF10A samples
Design DNA oligonucleotides using IDT (Integrated DNA Technologies) platform considering both positive and negative regions (see Table 1).
Table 1. Primers for qPCR
Name Primer Positive control
(qPCR probe #6, Amente et al., 2019)FW_pos_ctrl CCAACATCTTAAATTTGTCAACTCTC RV_pos_ctrl TGCTGGCAGAAGTGTGATTT Negative control
(qPCR probe C1, Amente et al., 2019)FW_neg_ctrl AGACACAGCCTGGGAAACC RV_neg_ctrl CATCCGTCGTGCAGACCT Load 17 μL of master mix (see Table 2) and 3 μL of sample in a 96-well plate.
Notes:
Perform each reaction three times to obtain a statistically significant value.
We recommended StepOneTM Real-Time PCR system instrument and the proper thermocycling protocol.
Table 2. Master mix
Master mix components Final Concentration Amount SYBER GREEN (Luna universal) 1× 10 μL Oligos 0.2 μM 0.8 μL Nuclease-free water Up to 17 μL
Double stranding (using the Invitrogen random labelling kit)
Note: Only before NGS library prep. Not required if samples will be analyzed by qPCR.
Vacuum dry each IP sample to a final volume of 30 μL by using Speed-vac at RT.
Boil for 5 min at 95 °C and transfer to ice for 1 min.
Make the following double-stranding (D-S) master mix (1×) using reagents provided by the Invitrogen random labelling kit:
Reagent Amount dATP solution 0.5 mM 2.0 μL dCTP solution 0.5 mM 2.0 μL dGTP solution 0.5 mM 2.0 μL dTTP solution 0.5 mM 2.0 μL Random primers buffer mixture 1.5 μL Klenow fragment 1.0 μL dH2O 9.5 μL Total 20 μL Add 20 μL of D-S master mix to IP sample of step F2.
Incubate for 3 h at 25 °C with gently mixing (usually 450 rpm).
Add 5 μL of stop buffer (see Invitrogen random labelling kit) and put on ice.
Purify using Mini Elute PCR purification kit following manufacturer’s instructions.
Quantify dsDNA using Qubit dsDNA HS assay kit. Briefly, mix 1 μL of purified DNA (from step F7) and 199 μL of Qubit® working solution (Qubit dsDNA HS reagent 200× and Qubit dsDNA HS buffer) in a clean Qubit assay tube. Incubate at RT for 2 min and then measure the concentration in a Qubit 2.0 fluorometer. Usually, the Qubit dsDNA HS assay requires two standards. Each standard tube requires 10 µL of each Qubit standard and 190 µL of Qubit working solution.
The assay is highly selective for double-stranded DNA (dsDNA) and is accurate for initial sample concentrations from 10 pg/µL to 100 ng/µL.
Library preparation and sequencing
Prepare NGS Library using 1–2 ng of IP or Input DNA with TruSeq ChIP sample prep kit. Perform sequencing with a throughput of 40–50 million of 50 bp single-end reads per sample using Illumina platform according to standard operating procedures.
Data analysis
Analyze the OxiDIP-Seq (Figure 2) as indicated in Amente et al. (2019), Gorini et al. (2020), and Scala et al. (2022).
Briefly:
Necessary files
GRCh37 (hg19) reference fasta file
GRCh37 (hg19) bwa index
Filtering and mapping
The following steps need to be executed for each sequenced (IP and Input) sample (Figure 2A).
Perform reads quality check using FastQC.
Perform reads trimming using Trimmomatic with the following parameters: -phread33 ILLUMINACLIP:TruSeq3-SE.fa:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:30.
Perform quality check of trimmed reads using FastQC.
Map reads to GRCh37 (hg19) with BWA-backtrack method using “bwa aln” with default parameters.
Perform SAM to BAM format conversion with SAM-tools view module with default parameters.
Sort BAM file using SAM-tools sort with default parameters.
Index BAM file using SAM-tools index with default parameters.
Filter out unmapped reads and alignments with MPAQ smaller than 1 using SAM-tools view with the following parameters: -F 4 -q 1 -b.
Filter standard chromosomes using SAM-tools view with standard chromosomes id list as input.
Remove PCR duplicates using SAM-tools rmdup with default parameters.
Sort BAM file using SAM-tools sort with default parameters.
Generate BAM file index using SAM-tools index with default parameters.
Peak calling
This step needs to be executed for each sample starting from the OxiDIP and the corresponding DNA Input alignments (Figure 2B):
Perform peak calling using MACS2 with default parameters.
Perform peak annotation starting from narrowpeak file (step 1) using HOMER annotatePeaks.pl and specifying hg19 as genome.
Genomic signal processing
Signals normalization and correlation analyses are performed for each sample using DeepTools suite (Figure 2C).
Estimate GC bias using DeepTools computeGCbias with default parameters on the IP and Input BAM file.
If the estimation performed in step D1 reveals the presence of bias, perform GC content bias correction using DeepTools correctGCbias with default parameters on the BAM files to produce a GC corrected BAM file and proceed to step D3.
Generate normalized 8-oxodG signal over the corresponding genomic input (log2 8-oxodG/Input ratio) using the DeepTools bamCompare with SES method as scaling factor and visualize the output bigwig file on a genome browser.
Additionally, generate the bigwig files containing normalized reads count signal for IP and Input samples using DeepTools bamCoverage on the BAM file and choosing CPM (count per millions) as normalization method, and visualize the two signals (IP and Input) on a genome browser.
Perform metagene analysis using the DeepTools computeMatrix on the bigwig files produced in steps D3–D4 with default parameters. Generate metagene heatmap using DeepTools plotHeatmap on the matrices computed in the previous step with default parameters.
Only if you performed the optional steps D1 and D2 for NAC- and UV-treated cells:Perform a correlation analysis between the samples in the two experimental conditions (UV/NAC and Untreated) and between biological replicates using the DeepTools multiBamSummary command on and choosing “bins” as sub-command.
Plot the correlation between the samples in the two experimental conditions (UV/NAC and Untreated) and between biological replicates using the DeepTools plotCorrelation command.
Generate the bigwig-containing signal comparison between 8-oxodG signals in UV- or NAC- treated MCF10A cells using DeepTools bamCompare on the corresponding BAM files with exactScaling method as scaling factor and visualize the track on a genome browser.
Perform metagene analysis using the DeepTools computeMatrix on the bigwig files produced in step D7 with default parameters.
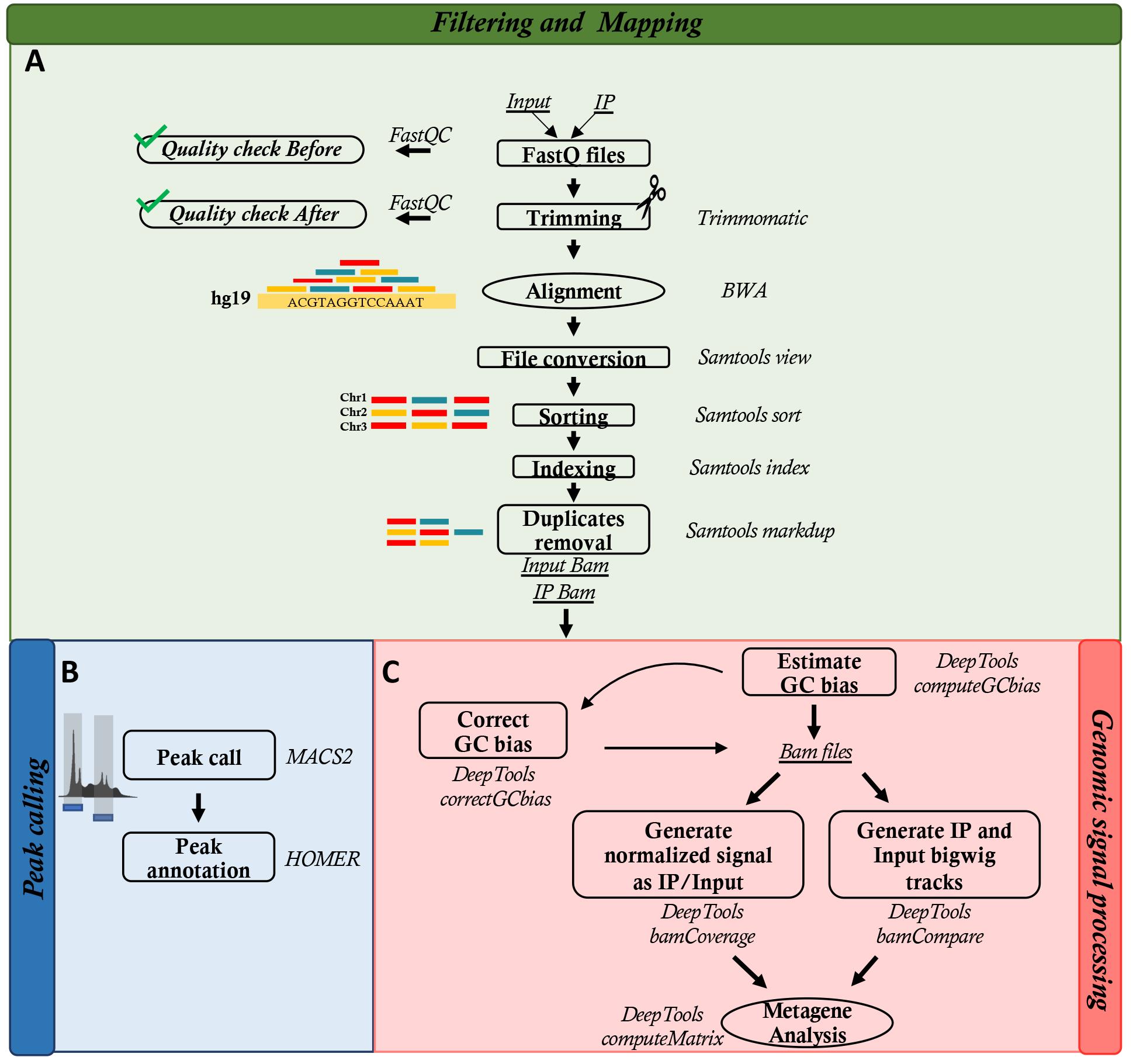
Figure 2. Workflow of bioinformatics analysis. The scheme represents sequential steps of data analysis.
Recipes
Growth medium for MCF10A cells
Reagent Final concentration Amount DMEM/HAM’S F12
Horse serum
Recombinant human EGF (100 μg/mL in sterile H2O)
Hydrocortisone (10 mg/mL in ethanol)
Cholera toxin
(1 mg/mL in sterile H2O)
n/a
n/a
n/a
n/a
n/a
95 mL
5 mL
20 μL
50 μL
10 μL
Insulin (10 mg/mL in sterile H2O with 1% glacial acetic acid) n/a 100 μL N-acetyl cysteine
Reagent Final concentration Amount N-acetyl cysteine n/a 100 mg Nuclease-free water n/a 1 mL TE buffer
Reagent Final concentration Amount Tris-HCl (1 M, pH 8)
EDTA (0.5 M, pH 8)
10 mM
1 mM
500 μL
100 μL
Nuclease-free water n/a 49.4 mL Total n/a 50 mL NaPi buffer (1 M, pH 7.4)
Reagent Final concentration Amount NaH2PO4 (1 M) n/a 43 mL Na2HPO4 (1 M) n/a 57 mL Total n/a 100 mL Adjust buffer
Reagent Final concentration Amount NaPi buffer (1 M, pH 7.4) 110 mM 110 μL NaCl (5 M)
Triton X-100 (20%)
Nuclease-free water
1,540 mM
0.5%
n/a
308 μL
25 μL
557 μL
Total n/a 1 mL IP buffer
Reagent Final concentration Amount Adjust buffer
TE buffer
n/a
n/a
1 mL
9 mL
Total n/a 10 mL Washing buffer
Reagent Final concentration Amount NaPi buffer (1 M, pH 7.4)
NaCl (5 M)
Triton X-100 (20%)
10 mM
150 mM
0.05%
100 μL
300 μL
25 μL
Nuclease-free water n/a 9.575 mL Total n/a 10 mL Elution buffer
Reagent Final concentration Amount Tris-HCl (1 M, pH 8)
EDTA (0.5 M, pH 8)
SDS (10%)
PK (20 mg/mL)
50 mM
10 mM
1%
0.5 mg/mL
250 μL
100 μL
500 μL
125 μL
Nuclease-free water n/a 4,025 μL Total n/a 5,000 μL
Acknowledgments
Funding: POR Campania FESR 2014-2020 “SATIN” grant to S.A. AIRC IG23066 grant to B.M. GS acknowledges support from PON AIM 2014–2020 E69F19000070001. The original papers have been published in Amente et al. (2019) (DOI: 10.1093/nar/gky1152), Gorini et al. (2020) (DOI: 10.1093/nar/gkaa175), and Scala et al. (20220) (DOI: 10.1093/nar/gkac143).
Competing interests
The authors declare that they have no other competing interests.
References
- Amente, S., Di Palo, G., Scala, G., Castrignano, T., Gorini, F., Cocozza, S., Moresano, A., Pucci, P., Ma, B., Stepanov, I., et al. (2019). Genome-wide mapping of 8-oxo-7,8-dihydro-2'-deoxyguanosine reveals accumulation of oxidatively-generated damage at DNA replication origins within transcribed long genes of mammalian cells. Nucleic Acids Res 47(1): 221-236.
- Baik, M.-H., Silverman, J. S., Yang, I. V., Ropp, P. A., Szalai, V. A., Yang, W. and Thorp, H. H. (2001). Using Density Functional Theory To Design DNA Base Analogues with Low Oxidation Potentials. J Physical Chemistry B 105(27): 6437-6444.
- Batra, V. K., Beard, W. A., Hou, E. W., Pedersen, L. C., Prasad, R. and Wilson, S. H. (2010). Mutagenic conformation of 8-oxo-7,8-dihydro-2'-dGTP in the confines of a DNA polymerase active site. Nat Struct Mol Biol 17(7): 889-890.
- Beckman, K. B. and Ames, B. N. (1998). The free radical theory of aging matures. Physiol Rev 78(2): 547-581.
- Boiteux, S., Coste, F. and Castaing, B. (2017). Repair of 8-oxo-7,8-dihydroguanine in prokaryotic and eukaryotic cells: Properties and biological roles of the Fpg and OGG1 DNA N-glycosylases. Free Radic Biol Med 107: 179-201.
- Cao, B., Wu, X., Zhou, J., Wu, H., Liu, L., Zhang, Q., DeMott, M. S., Gu, C., Wang, L., You, D. et al. (2020). Nick-seq for single-nucleotide resolution genomic maps of DNA modifications and damage. Nucleic Acids Res 48(12): 6715-6725.
- Cooke, M. S., Evans, M. D., Dizdaroglu, M. and Lunec, J. (2003). Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J 17(10): 1195-1214.
- Ding, Y., Fleming, A. M. and Burrows, C. J. (2017). Sequencing the Mouse Genome for the Oxidatively Modified Base 8-Oxo-7,8-dihydroguanine by OG-Seq. J Am Chem Soc 139(7): 2569-2572.
- Evans, M. D. and Cooke, M. S. (2004). Factors contributing to the outcome of oxidative damage to nucleic acids. Bioessays 26(5): 533-542.
- Fang, Y. and Zou, P. (2020). Genome-Wide Mapping of Oxidative DNA Damage via Engineering of 8-Oxoguanine DNA Glycosylase. Biochemistry 59(1): 85-89.
- Fortini, P., Parlanti, E., Sidorkina, O. M., Laval, J. and Dogliotti, E. (1999). The type of DNA glycosylase determines the base excision repair pathway in mammalian cells. J Biol Chem 274(21): 15230-15236.
- Frosina, G., Fortini, P., Rossi, O., Carrozzino, F., Raspaglio, G., Cox, L. S., Lane, D. P., Abbondandolo, A. and Dogliotti, E. (1996). Two pathways for base excision repair in mammalian cells. J Biol Chem 271(16): 9573-9578.
- Gorini, F., Scala, G., Di Palo, G., Dellino, G. I., Cocozza, S., Pelicci, P. G., Lania, L., Majello, B. and Amente, S. (2020). The genomic landscape of 8-oxodG reveals enrichment at specific inherently fragile promoters. Nucleic Acids Res 48(8): 4309-4324.
- Kim, G. H., Kim, J. E., Rhie, S. J. and Yoon, S. (2015). The Role of Oxidative Stress in Neurodegenerative Diseases. Exp Neurobiol 24(4): 325-340.
- Klaunig, J. E. and Kamendulis, L. M. (2004). The role of oxidative stress in carcinogenesis. Annu Rev Pharmacol Toxicol 44: 239-267.
- Koga, Y., Taniguchi, Y. and Sasaki, S. (2013). Synthesis of the oligoribonucleotides incorporating 8-oxo-guanosine and evaluation of their base pairing properties. Nucleosides Nucleotides Nucleic Acids 32(3): 124-136.
- Lindahl, T. (1993). Instability and decay of the primary structure of DNA. Nature 362(6422): 709-715.
- Lindahl, T. (1990). Repair of intrinsic DNA lesions. Mutat Res 238(3): 305-311.
- Lindahl, T. and Barnes, D. E. (2000). Repair of endogenous DNA damage. Cold Spring Harb Symp Quant Biol 65: 127-133.
- Liu, Z. J., Martinez Cuesta, S., van Delft, P. and Balasubramanian, S. (2019). Sequencing abasic sites in DNA at single-nucleotide resolution. Nat Chem 11(7): 629-637.
- Maga, G., Villani, G., Crespan, E., Wimmer, U., Ferrari, E., Bertocci, B. and Hubscher, U. (2007). 8-oxo-guanine bypass by human DNA polymerases in the presence of auxiliary proteins. Nature 447(7144): 606-608.
- Poetsch, A. R. (2020). The genomics of oxidative DNA damage, repair, and resulting mutagenesis. Comput Struct Biotechnol J 18: 207-219.
- Poetsch, A. R., Boulton, S. J. and Luscombe, N. M. (2018). Genomic landscape of oxidative DNA damage and repair reveals regioselective protection from mutagenesis. Genome Biol 19(1): 215.
- Scala, G., Gorini, F., Ambrosio, S., Chiariello, A. M., Nicodemi, M., Lania, L., Majello, B. and Amente, S. (2022). 8-oxodG accumulation within super-enhancers marks fragile CTCF-mediated chromatin loops. Nucleic Acids Res 50(6): 3292-3306.
- Shibutani, S., Takeshita, M. and Grollman, A. P. (1991). Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature 349(6308): 431-434.
- Steenken, S. and Jovanovic, S. V. (1997). How Easily Oxidizable Is DNA? One-Electron Reduction Potentials of Adenosine and Guanosine Radicals in Aqueous Solution. J Am Chem Soc 119(3): 617-618.
- van Loon, B., Markkanen, E. and Hubscher, U. (2010). Oxygen as a friend and enemy: How to combat the mutational potential of 8-oxo-guanine. DNA Repair (Amst) 9(6): 604-616.
- Wu, J., McKeague, M. and Sturla, S. J. (2018). Nucleotide-Resolution Genome-Wide Mapping of Oxidative DNA Damage by Click-Code-Seq. J Am Chem Soc 140(31): 9783-9787.
Article Information
Copyright
© 2022 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Gorini, F., Scala, G., Ambrosio, S., Majello, B. and Amente, S. (2022). OxiDIP-Seq for Genome-wide Mapping of Damaged DNA Containing 8-Oxo-2'-Deoxyguanosine. Bio-protocol 12(21): e4540. DOI: 10.21769/BioProtoc.4540.
Category
Cancer Biology > Genome instability & mutation > Genetics
Molecular Biology > DNA > DNA damage and repair
Molecular Biology > DNA > Chromatin accessibility
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.