- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

A New Tool for the Flexible Genetic Manipulation of Geobacillus kaustophilus
(*contributed equally to this work) Published: Vol 12, Iss 17, Sep 5, 2022 DOI: 10.21769/BioProtoc.4502 Views: 2080
Reviewed by: Alessandro DidonnaAksiniya AsenovaAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Mar 2022
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Geobacillus kaustophilus, a thermophilic Gram-positive bacterium, is an attractive host for the development of high-temperature bioprocesses. However, its reluctance against genetic manipulation by standard methodologies hampers its exploitation. Here, we describe a simple methodology in which an artificial DNA segment on the chromosome of Bacillus subtilis can be transferred via pLS20-mediated conjugation resulting in subsequent integration in the genome of G. kaustophilus. Therefore, we have developed a transformation strategy to design an artificial DNA segment on the chromosome of B. subtilis and introduce it into G. kaustophilus. The artificial DNA segment can be freely designed by taking advantage of the plasticity of the B. subtilis genome and combined with the simplicity of pLS20 conjugation transfer. This transformation strategy would adapt to various Gram-positive bacteria other than G. kaustophilus.
Graphical abstract:
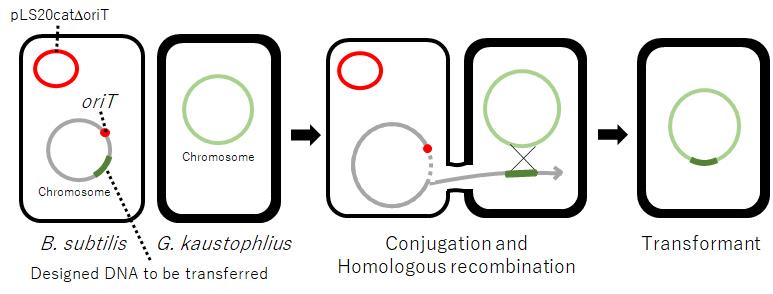
Background
Geobacillus kaustophilus HTA426 is a thermophilic Gram-positive bacterium isolated from sediments of the Mariana Trench (Nazina et al., 2001; Takami et al., 2004). It grows at temperatures ranging from 48 to 74 °C (optimal temperature of 60 °C) and is a promising chassis for high-temperature bioprocessing with the advantages of preventing contamination by mesophilic bacteria and lowering the cost of controlling fermentation heat. However, this bacterium is difficult to transform by natural competence or electroporation, and an approach based on a conjugation system for Gram-negative bacteria has been developed (Suzuki and Yoshida, 2012). This method has the disadvantage of not being able to select transformants that acquired mutations with adverse effects on growth. Therefore, better techniques for manipulating the genome of G. kaustophilus are needed.
Conjugation is one of the three main mechanisms for horizontal gene transfer in bacteria. Conjugative elements, which often are located on plasmids, encode all the proteins required for their transfer from a donor cell to a recipient cell. Conjugative elements encode adhesins or organelles facilitating interaction with recipient cells. In almost all conjugative systems only a single strand of the DNA (ssDNA) is transferred. Generation of the ssDNA is done by a relaxase, often assisted by one or two auxiliary proteins, that introduces a site- and strand-specific nick in a region of the plasmid named origin of transfer (oriT). After nicking, the relaxase remains covalently attached to the 5′ end of the DNA, and the generated 3′ end is used as a primer for DNA synthesis resulting in a rolling-circle mode of replication, thereby generating the ssDNA molecule that is destined for transfer into the recipient cell. Conjugative elements encode a sophisticated transferosome (type IV secretion system) connecting the donor and recipient cells. The relaxosome complex, which is composed of a number of proteins including the relaxase, is recruited to the cytosolic side of the transferosome in the donor cell, after which it guides the transfer of the ssDNA into the recipient cell (Christie et al., 2014; Christie, 2016; Waksman, 2019).
pLS20 is a 65 kb conjugative plasmid isolated from Bacillus subtilis var. natto (Tanaka and Koshikawa, 1977); it can transfer itself among various Bacillus species (Koehler and Thorne, 1987). Contrary to most conjugative systems that require prolonged interactions between donor and recipient cells on solid medium, efficient conjugation of pLS20 is achieved by mixing donor and recipient cells in liquid medium (Tanaka and Koshikawa, 1977; Singh et al., 2013; Miyano et al., 2018). Genes involved in pLS20 conjugation (genes 28–74) form a large operon controlled by a strong promoter named Pc. Regulation of pLS20 conjugation genes has been studied in detail (for a review, see Meijer et al., 2021). Gene 27 encodes RcopLS20, which is the master regulator of the conjugation operon. In its default state, conjugation is suppressed due to binding of RcopLS20 to its two operator sites within the Pc region, strictly repressing the Pc promoter. Gene 25 encodes an antirepressor named RappLS20, which activates conjugation by relieving repression from RcopLS20 and forming a Rap/Rco complex. Gene 26 encodes a quorum sensing peptide, Phr*pLS20, that regulates the activity of RappLS20. phrpLS20 synthesizes a small pre-proprotein. After being secreted, PhrpLS20 is proteolytically cleaved, generating the mature Phr*pLS20 peptide corresponding to its 5 C-terminal residues. When taken up by donor cells, Phr*pLS20 inactivates RappLS20 by changing its conformation upon binding to it, returning the system to its default repressed state (Singh et al., 2013). Ectopic overproduction of RappLS20 increased the efficiency of conjugation by weakening the effect of Phr*pLS20 (Singh et al., 2013).
A derivative of pLS20 lacking oriTpLS20, pLS20catΔoriT, is defective in conjugation (Miyano et al., 2018). However, we have recently shown that pLS20catΔoriT can mediate the transfer of large regions of the bacterial chromosome between B. subtilis cells when a copy of oriTpLS20 is present on the chromosome in the donor cell (Miyano et al., 2018). Inspired by this, we have developed a strategy to transform G. kaustophilus that allows for flexible manipulation of its chromosome (Mori et al., 2022). Here, we present the protocol to transform G. kaustophilus by pLS20-mediated conjugative transfer of a chromosomal DNA of B. subtilis designed to function in G. kaustophilus.
Materials and Reagents
Note: All reagents can be stored at room temperature unless otherwise specified.
Construction of the donor strain
B. subtilis 168 (Mori et al., 2022)
B. subtilis YNB211 (Mori et al., 2022)
G. kaustophilus MK244 (Mori et al., 2022)
pUCG18T (Mori et al., 2022)
LB medium (2% agar) (BD Difco, catalog number: 240230)
10% SDS (Nacalai Tesque, catalog number: 30562-04)
Lysozyme (Wako, catalog number: 129-06723)
Phenol/Chloroform/Isoamyl alcohol (25:24:1) (Wako, catalog number: 311-90151), store at 4 °C
70% Ethanol
99.5% Ethanol
KOD -Plus- Neo polymerase (Toyobo, catalog number: KOD-401), store at -20 °C
Agarose for electrophoresis gel (ME, Nacalai Tesque, catalog number: 01133-24)
In silico Molecular Cloning software for designing PCR primers (In Silico Biology, Inc., catalog number: IMCGF01)
Primer pairs used for the PCR reaction of five fragments
Fragment A1:
aprE-U-f1 ccggtacttgccaccacatcataac
aprE-U-r cagtaacctcatcaagccaagctacctctcgctatttccgtagagactcg
Fragment A2:
aprE-D-f2 gacagaggaattagatacattcgcgttaatcaacgtacaagcagctgcac
aprE-D-r ggccgagcagtattcgaatgtcaag
Fragment B1:
degA-U-f gtagcttggcttgatgaggttactg
degA-U-r2 catcggtcataaaatccgtatccttggttactttcatcgctcatcattc
Fragment B2:
degA-D-f2 ctgcaaggcgattaagttgggtaacagtgaaatcgtaaggatgtgagcag
degA-D-r cgcgaatgtatctaattcctctgtc
Fragment C1:
kan-f aaggatacggattttatgaccgatg
kan-r2 gttacccaacttaatcgccttgcag
Primers used for the recombinant PCR reactions that ligate the five fragments.
aprE-U-f3-nested caccgagctcatagcttgtcgcgatcacctcatcc
aprE-D-r2-nested tgctttcgctgattacaacattggtgacgctgcct
DNA purification kit (Wizard® SV Gel and PCR Clean-Up System, Promega, catalog number: A9281)
7.5 mg/mL Kanamycin sulfate stock solution in water (Wako, catalog number: 113-00343)
Solution A (see Recipes)
Solution B (see Recipes)
100× Trace element (see Recipes)
TKE buffer (see Recipes)
50× TAE buffer (see Recipes)
C-1 and C-2 medium for transformation (see Recipes)
Conjugation and selection of transconjugant (transformant)
7.5 mg/mL Kanamycin sulfate stock solution in water (Wako, catalog number: 113-00343)
10 mg/mL Chloramphenicol stock solution in 70% ethanol (Wako, catalog number: 030-19452)
100 mM IPTG stock solution in water (Nacalai Tesque, catalog number: 367-93-1)
LB medium (see Recipes)
LBMSM medium (see Recipes)
Southern blotting analysis
Primers used for amplification of a template DNA for synthesizing RNA probe
Km-probe-f taatacgactcactatagggtatggctctcttggtcgtc
Km-probe-r tctgattccacctgagatgc
Agarose for electrophoresis gel (SP, Nacalai Tesque, catalog number: 01163-76)
DNA Molecular Weight Marker II, DIG-labeled (Roche, catalog number: 11218590910)
DNA purification kit (Wizard® SV Gel and PCR Clean-Up System, Promega, catalog number: A9281)
T7 RNA polymerase (Roche, catalog number: 10881767001), store at -20 °C
DIG RNA Labeling Mix, 10× conc. (Roche, catalog number: 11277073910), store at -20 °C
Restriction enzymes
Cla I (Takara, catalog number: 1034A), store at -20 °C
Sac II (Takara, catalog number: 1079A), store at -20 °C
DIG Easy Hyb (Roche, catalog number: 11603558001), store at 4 °C
Amersham Hybond-N+ (GE Healthcare, catalog number: RPN82B)
Hybridization bag (Hybridization Bags Hybri-Bag Hard, COSMO BIO, catalog number: S-1001)
Blocking Reagent (Roche, catalog number: 11096176001)
Anti-Digoxigenin-AP, Fab fragment (Roche, catalog number: 11093274910), store at 4 °C
CSPD, ready-to-use (Roche, number: 11755633001), store at 4 °C
20× SSC buffer (see Recipes)
Maleic acid buffer (see Recipes)
Denaturing solution (see Recipes)
Neutralizing solution (see Recipes)
High-stringency wash solution (see Recipes)
Low-stringency wash solution (see Recipes)
Detection buffer (see Recipes)
Equipment
Construction of the donor strain
Thermal cycler (for example, Takara Bio, Thermal Cycler Dice Touch TP350, or equivalent)
Centrifuge (for example, TOMY, MX-307, or equivalent)
Spectrophotometer (for example, GE Healthcare, NanoVue Plus, or equivalent)
Agarose gel electrophoresis apparatus (for example, Takara Bio, Mupid®-2plus, or equivalent)
Incubator (for example, PHCbi, MIR-H163, or equivalent)
Conjugation and selection of transconjugant (transformant)
Two incubator shakers, one set at 37 °C and the other at 60 °C (TAITEC, Bioshaker BR-43FM-MR, or equivalent)
Centrifuge (TOMY, MX-307, or equivalent)
Two incubators, one is set at 37 °C and the other 60 °C (PHCbi, MIR-H163, or equivalents)
Spectrophotometer (SHIMADZU, UV-1800, or equivalent)
300 mL baffled Erlenmeyer flask
Southern blotting (or: Southern blot analysis)
Thermal cycler (Takara Bio, Thermal Cycler Dice Touch TP350, or equivalent)
Spectrophotometer (GE Healthcare, NanoVue Plus, or equivalent)
Agarose gel electrophoresis apparatus (Bio-Rad, Sub-Cell GT Cell, or equivalent)
Transfer apparatus (Bio Craft, Vacuum Transfer BS-31, or equivalent)
UV crosslinker (UVP, CL-1000, or equivalent)
Shaker (Bio Craft, Labo Shaker BC-730, or equivalent)
Chemiluminescence imager (Bio-Rad, ChemiDoc XRS+, or equivalent)
Procedure
Construction of the donor strain
Isolation of DNA from bacterial strains
Grow bacterial cells in 5 mL of LB medium at 37 °C overnight with shaking.
Collect bacterial cells of 1.5 mL of culture in a test tube by centrifugation at 8,152 × g for 3 min at room temperature.
Suspend the cells in 500 µL of TKE buffer containing 1 mg of freshly added lysozyme.
Incubate the tube for 30 min at 37 °C.
Add 50 µL of 10% SDS and mix thoroughly.
Add 500 µL of phenol/chloroform/isoamyl alcohol (25:24:1) and mix vigorously.
Centrifuge the tube at 20,380 × g for 10 min at 4 °C, and then transfer 200 µL of supernatant to a new tube; avoid transferring the white interface.
Add 500 µL of 99.5% ethanol to the tube, mix and stand on ice for 10 min, and centrifuge at 20,380 × g for 5 min. Discard the supernatant.
Wash the DNA pellet carefully with 0.7 mL of 70% ethanol and place the sample 10 min at 37 °C to evaporate remains of ethanol.
Add 100 µL of distilled water and place 10 min at 37 °C to dissolve the DNA gently.
Measure the concentration of DNA using a spectrophotometer such as NanoVue Plus.
Setting up recombinant PCR reaction to generate the “gene cassette”
The schematic organization of the ΔiolQGK::kan cassette, replacing the iolQGK coding region of G. kaustophilus with a thermostable kanamycin-resistance gene, is used in this protocol as an example (Figure 1). This gene cassette was generated by recombinant PCR, ligating the five PCR fragments as follows.
Amplify the five fragments individually (A1, A2, B1, B2, and C1) by PCR using KOD -Plus- neo polymerase. The specific primer pairs used to amplify each of the fragments are listed above in the Materials and Reagents section. It is strongly recommended to check the correct amplification of each PCR fragment by agarose gel electrophoresis. The PCR reactions are composed and carried out as follows:
PCR reaction:
Components Volume Final Concentration 10× PCR Buffer 5 μL 1× 2 mM dNTPs 5 μL 0.2 mM each 25 mM MgSO4 3 μL 1.5 mM Primer (10 μM each) 0.75–1.5 μL 0.15–0.3 μM each Template DNA variable Plasmid/PCR fragment DNA ~50 ng/50 μL Genomic DNA ~200 ng/50 μL KOD -Plus- Neo (1 U/μL) 1 μL 1 U/50 μL H2O variable Total 50 μL Thermal cycler conditions:
Initial Denaturation 98 °C 2 min 30 cycles 98 °C 10 s 68 °C 30 s/kb Hold 4 °C Purify the PCR reactions using a DNA purification kit and determine the concentration of the purified DNA by measuring OD260nm.
Mix all the amplified and purified fragments in a 1:1:1:1:1 molar ratio.
Run the recombinant PCR for the recombination of the five fragments using primers aprED-r2-nested and aprEU-f3-nested. The PCR reaction is the same as described above, but set the thermal cycler conditions as follows:
Thermal cycler conditions:
Initial Denaturation 98 °C 2 min 30 cycles 98 °C 10 s 68 °C 2.5 min Hold 4 °C
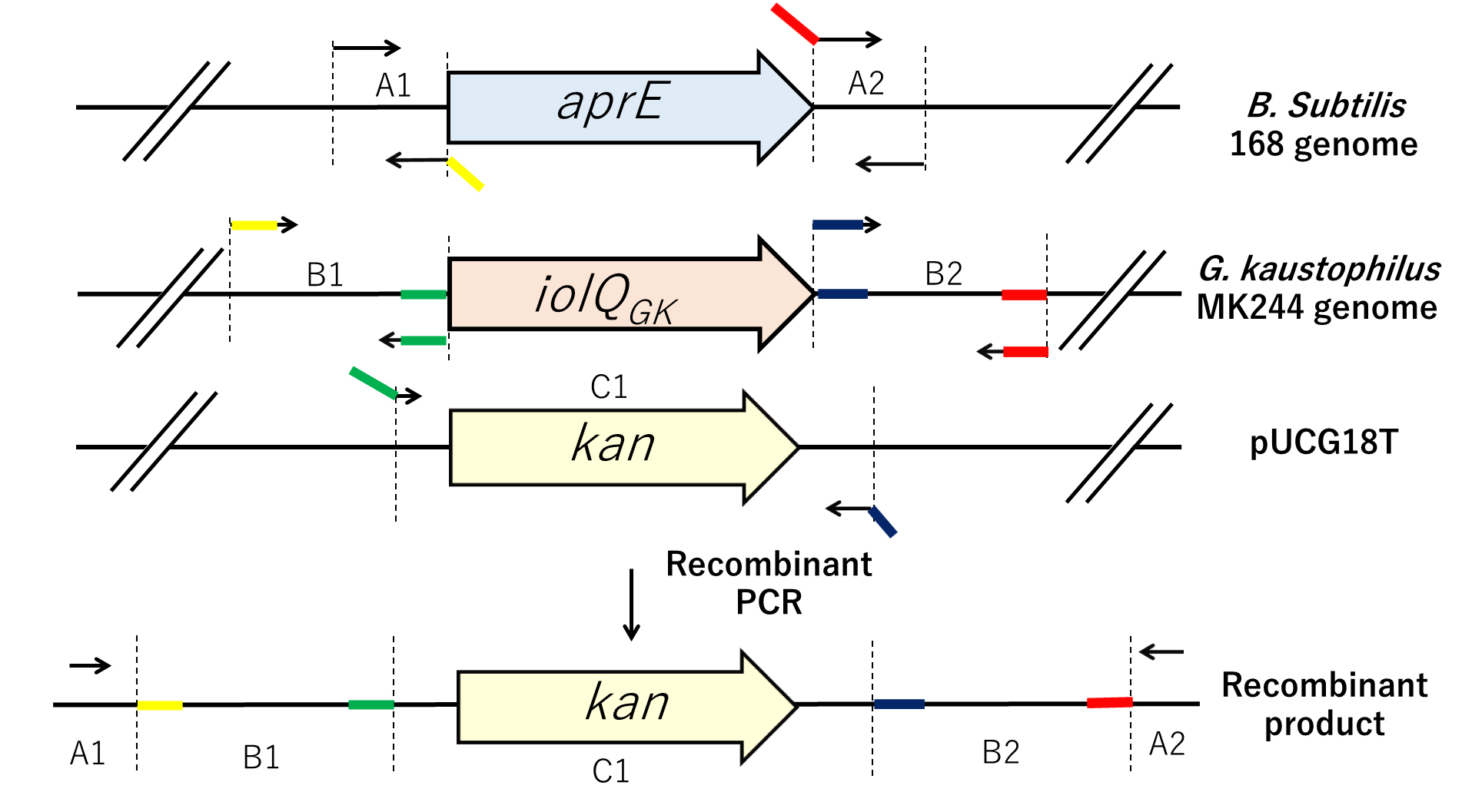
Figure 1. Strategy to generate the ΔiolQGK::kan cassette (recombinant product). Five fragments are amplified by PCR using the primer pairs mentioned in A.14. Two fragments correspond to “upstream” and “downstream” regions of the B. subtilis aprE locus and are named A1 and A2, respectively. Two other fragments corresponding to sequences located upstream and downstream of G. kaustophilus MK244 iolQGK are named B1 and B2, respectively; the DNA fragment containing the kanamycin-resistance gene of pUCG18T is named C1. Fragments A1 and A2 are designed to be approximately 1 kb long, and fragments B1 and B2 are approximately 3 kb. The specific primers, indicated as arrows, are listed in the Materials and Reagents section. Colored regions of bent arrows correspond to 5′ extensions designed to create overlap with a flanking region in the final recombinant product. Overlapping sequences are indicated using different colors. The five individual PCR fragments are linked together by PCR to form one long “gene cassette” fragment.Transformation
The presence of pLS20 lowers competence levels (Singh et al., 2012). Therefore, it is recommended to generate the final B. subtilis donor YNB213 in two steps. First, the five-fragment recombinant product generated by PCR is used to transform competent B. subtilis 168 cells. Second, chromosomal DNA isolated from a kanamycin-resistant transformant of this transformation is then used to transform competent B. subtilis YNB211 cells harboring pLS20catΔoriT and containing both the oriTpLS20 and the IPTG-inducible rappLS20 present on the chromosome at the yhfK and amyE loci, respectively (Miyano et al., 2018; Mori et al., 2022). Transformation of B. subtilis is performed as follows:
Grow a strain of B. subtilis to be transformed overnight (~16 h) on an LB 2% agar plate at 30 °C to form colonies.
Pick up a fresh colony and use it to inoculate 5 mL of C-1 medium in a test tube to allow the cells to grow at 37 °C with vigorous shaking (at least 200 rpm) for 2.5 h.
Collect the cells by centrifugation at 6,000 rpm at room temperature for 3 min.
Suspend the cell pellet in 10 mL of C-2 medium in a new test tube and incubate the tube with shaking at 37 °C for 40 min.
Transfer 1 mL of the C-2 medium culture into a new tube containing less than 100 µL of DNA solution (1–10 µg/mL).
Incubate the tube with shaking at 37 °C for 2 h.
Spread the culture onto plates containing appropriate antibiotics to select transformants.
Conjugation and selection of transconjugant (Figure 2)
Based on the previous findings that the addition of sorbitol (Kananavičiūtė et al., 2015), MgCl2, and maleic acid-NaOH buffer (Wyrick and Rogers, 1973) was effective in increasing the efficiency of transformation by electroporation and stabilizing L-form cells, we developed a modified medium, named LBMSM, suitable for elevating the efficiency of this transformation of G. kaustophilus mediated by conjugation.
Preparation of donor culture
Grow B. subtilis YNB213 overnight on an LB 2% agar plate containing 10 µg/mL chloramphenicol at 37 °C to form colonies.
Pick up a fresh colony and use it to inoculate 5 mL of LB liquid medium containing 10 µg/mL chloramphenicol, pre-warmed at 37 °C in a test tube.
Grow overnight at 37 °C with shaking at 200 rpm.
In a 300 mL baffled Erlenmeyer flask pre-warmed at 37 °C, dilute the culture in 10 mL of LBMSM medium containing 1.0 mM IPTG to reach an OD600 of 0.05 (check using a spectrophotometer).
Grow the cells at 37 °C with shaking at 200 rpm until OD600 reaches 0.6–0.8, and use this as the donor culture.
Preparation of recipient culture
Grow G. kaustophilus MK244 overnight on an LB 2% agar plate at 60 °C to form colonies.
Pick up a fresh colony and inoculate it into 10 mL of LB liquid medium pre-warmed to 60 °C in a 300 mL Erlenmeyer flask.
Grow the cells at 60 °C for 2 h with shaking at 200 rpm.
Dilute the culture (to make OD600 adjusted to 0.01 using a spectrophotometer) in 10 mL of LBMSM medium in a 300 mL baffled Erlenmeyer flask pre-warmed at 60 °C.
Allow the cells to grow at 60 °C with shaking at 200 rpm until OD600 reaches 0.6–0.8 measured with a spectrophotometer, and use this as the recipient culture.
Mating
Mix 2 mL of the donor culture and 8 mL of the recipient culture in a sterilized 300 mL Erlenmeyer flask.
Stand the mixed culture at 37 °C for 90 min, and then allow the cells to grow at 60 °C for 180 min with shaking at 200 rpm.
Colony formation of transconjugant
Transfer the mating culture into a test tube and centrifuge at 8,152 × g for 2 min and resuspend the pellet in 1.2 mL of LB liquid medium pre-warmed at 60 °C in a 1.5 mL tube.
Spread the suspension onto LB 2% agar plates containing 7.5 µg/mL of kanamycin pre-warmed at 60 °C (200 µL on each Petri dish 9 cm in diameter).
As negative control experiments, spread separately donor and recipient cells onto LB 2% agar plates containing 7.5 µg/mL of kanamycin pre-warmed at 60 °C.
Incubate the plates at 60 °C overnight and score the number of colonies.
Colony formation of recipient
Sample a small portion (~100 µL) of the mating culture, dilute it 107-fold, and spread 100 µL of the dilution on LB 2% agar plates.
Incubate the plates at 60 °C overnight and score the number of colonies.
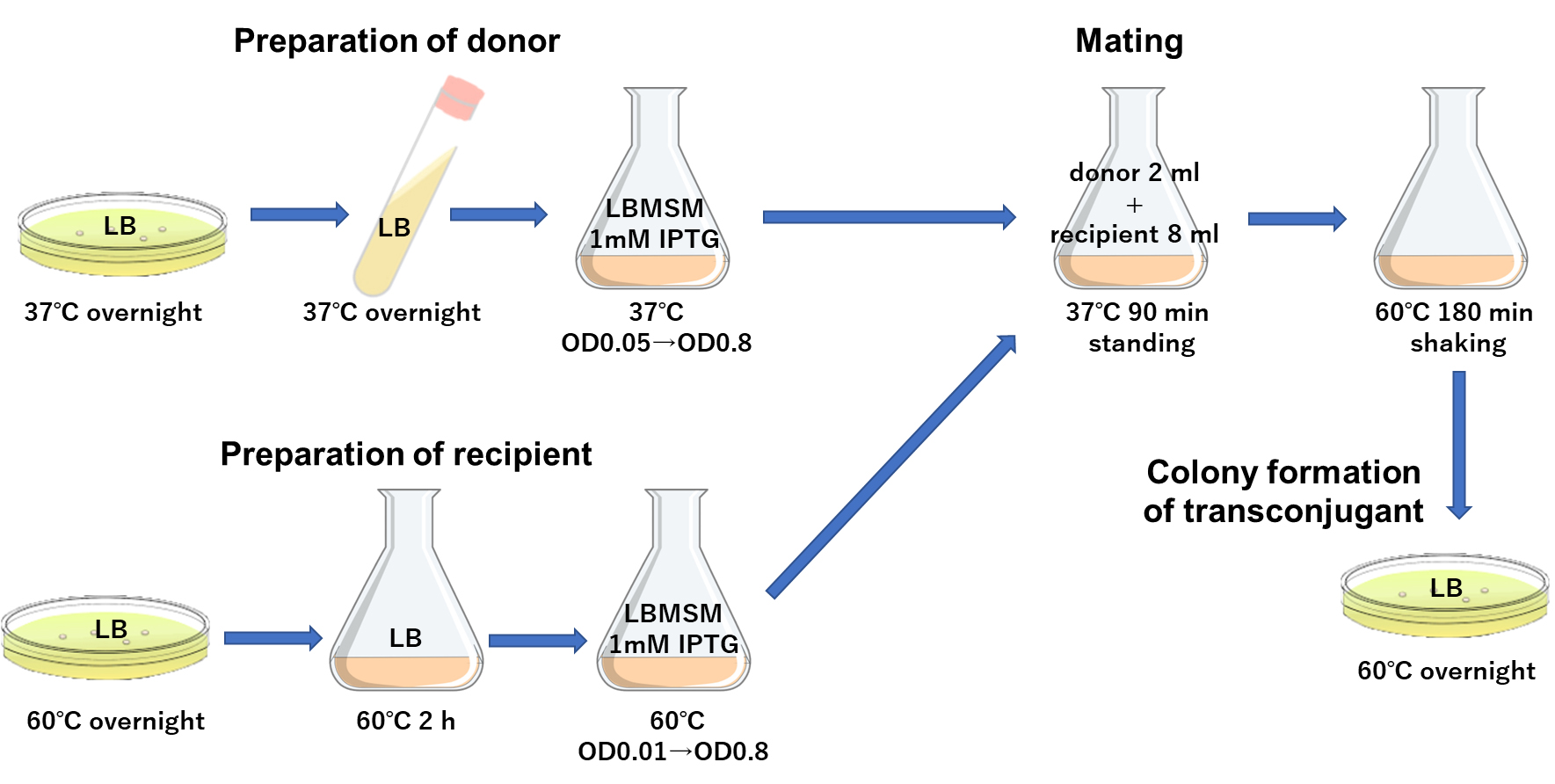
Figure 2. A schematic presentation of the conjugation procedure. Donor strain B. subtilis YNB213 and recipient strain G. kaustophilus MK244 were grown independently. Mating was performed by mixing two cultures in a 1 to 4 ratio of donor and recipient cells, followed by further incubation to enhance colony formation.Southern blot analysis
Southern blot analysis allows us to detect a specific DNA sequence in chromosomal DNA samples, which usually involves four successive steps, including (i) Fragmentation of chromosomal DNA with restriction enzymes, (ii) Separation of DNA fragments by electrophoresis, (iii) Transfer of the DNA onto the membrane, and (iv) Detection of DNA on the membrane using the probe. It is important to choose appropriate restriction enzymes for the fragmentation of chromosomal DNA. In the case of this protocol, we decided on Cla I and Sac II (Figure 3). For the detection of DNA on the membrane, a fluorescence-based nonradioactive technology with a DIG-labeled RNA probe is used for the detection of target DNA.
Preparation of DIG-labeled RNA
Run PCR to amplify the stretch corresponding to the kanamycin-resistance gene using KOD -Plus- neo polymerase and the primer pairs Km-probe-f and Km-probe-r. The PCR reactions are composed and carried out as follows:
PCR reaction:
Components Volume Final Concentration 10× PCR Buffer 5 μL 1× 2 mM dNTPs 5 μL 0.2 mM each 25 mM MgSO4 3 μL 1.5 mM Primer (10 μM each) 0.75–1.5 μL 0.15–0.3 μM each pUCG18T DNA variable ~50 ng/50 μL KOD -Plus- Neo (1 U/μL) 1 μL 1 U/50 μL H2O variable Total 50 μL Thermal cycler condition:
Initial Denaturation 98 °C 2 min 30 cycles 98 °C 10 s 68 °C 30 s/kb Hold 4 °C Using the DNA purification kit, purify the amplified DNA as the template for in vitro transcription.
Perform the in vitro transcription of the template DNA using T7 RNA polymerase and DIG RNA Labeling Mix, following the protocol provided by the manufacturer. This step generates a DIG-labeled RNA probe.
Store the DIG-labeled RNA probe at -80 °C until being used.
Digestion and electrophoresis of the genomic DNA
Extract genomic DNAs and measure OD260 to determine their concentration.
Digest 3 µg of genomic DNAs with appropriate restriction enzymes at 37 °C for at least 1 h to complete the digestion.
Subject the digested DNAs in parallel with the size markers (DNA Molecular Weight Marker II, DIG-labeled) to 1.0% agarose gel electrophoresis.
Run the electrophoresis for 1 h at 100 V.
Blotting, hybridization, and detection
Soak the agarose gel successively in 150 mL of (i) 0.25 M HCl (30 min), (ii) denaturing solution (30 min), and (iii) neutralizing solution (30 min).
Transfer DNAs in the gel onto a positively charged nylon membrane (Amersham Hybond-N+), using the transfer apparatus (Vacuum Transfer BS-31) as instructed by the supplier.
Crosslink the transferred DNAs to the membrane with UV crosslinker L-1000 with accumulative radiation set to 0.07 J/cm2.
Transfer the membrane into a hybridization bag, and fill up the bag with 30 mL of DIG Easy Hyb at 50 °C for at least 10 min for pre-hybridization.
Denature the DIG-labeled RNA probe by boiling the water solution containing the probe for 10 min and then snap-cool by placing the sample on ice.
Soak the membrane in the bag filled with 30 mL of DIG Easy Hyb containing 100 ng/mL of denatured DIG-labeled RNA probe at 42 °C overnight with gentle shaking.
Wash the membrane twice for 5 min with 150 mL of low-stringency wash solution at room temperature with gentle shaking.
Wash the membrane twice for 15 min with 150 mL of pre-warmed high-stringency wash solution at 65 °C with gentle shaking.
Wash the membrane briefly with 150 mL of maleic acid buffer with gentle shaking.
Soak the membrane in blocking reagent diluted in 150 mL of maleic buffer for 30 min at room temperature to prevent non-specific binding of antibodies.
Transfer the membrane into a hybridization bag and incubate for 30 min with 30 mL of blocking reagent containing 1/104 volume of Anti-Digoxigenin-AP, Fab fragment (20 mL of blocking reagent per 100 cm2 of membrane should be used).
Wash the membrane twice with 150 mL of wash buffer for 15 min with gentle shaking.
Transfer the membrane into a new hybridization bag and equilibrate with 30 mL of detection buffer for 3 min.
Discard the detection buffer and incubate the membrane with 30 mL of detection buffer containing CSPD at 37 °C for 5–10 min (20 mL of detection buffer should be used per 100 cm2 of membrane).
Detect the chemiluminescence signal using ChemiDoc XRS+ following the instructions of the supplier.
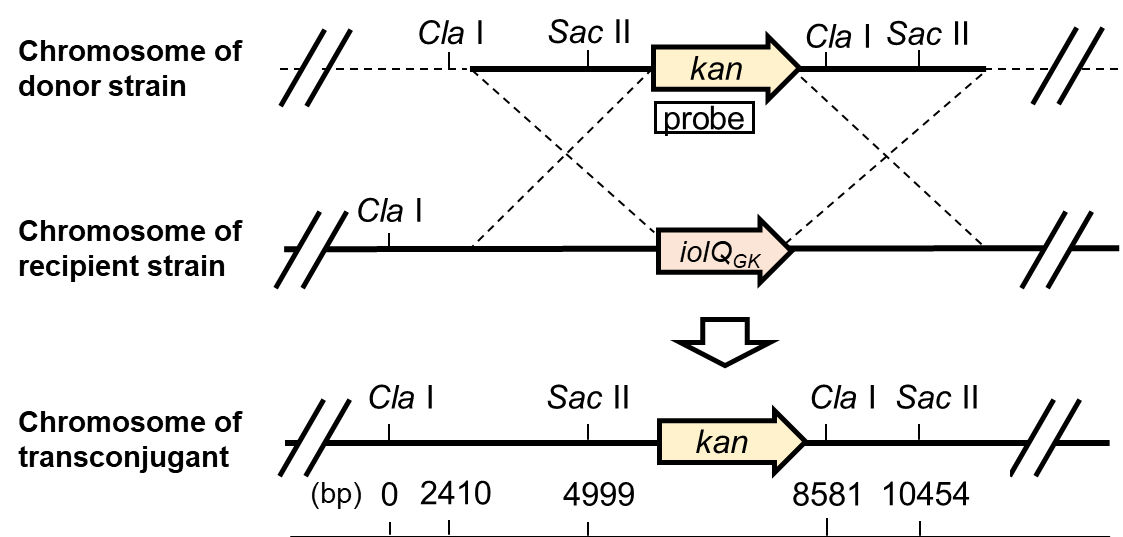
Figure 3. Physical maps of the relevant regions of the chromosomes of donor, recipient, and transconjugant strains.
Data analysis
Conjugation and selection of transconjugant
Evaluate the efficiency judging from the value of CFUtransconjugant/CFUrecipient, which usually is approximately 1.0 × 10-9.
Count the number of colonies that appear on the plates containing kanamycin incubated at 60 °C overnight. Calculate CFUtransconjugant as follows:
CFUtransconjugant = Mean ± SD of colony numbers per plate × 1.2 mL/200 μL/10 mL
For example, when 3, 3, and 5 colonies were formed on three plates, respectively:
CFUtransconjugant = 3.7 ± 1.2 ×1.2/0.2/10 = 2.2 ± 0.7
Count the number of colonies that appear on the plates without kanamycin incubated at 60 °C overnight. Calculate CFUrecipient as follows:
CFUrecipient = Mean ± SD of colony numbers per plate × 107 × 1 mL/100 μL
For example, when 18, 13, and 11 colonies were formed on three plates, respectively:
CFUrecipient = 14.0 ± 3.6 × 107 × 1.0 /0.1 = 1.4 ± 0.36 × 109
Southern blot analysis
Take a picture of the chemiluminescence signal on the membrane to visualize the bands of DNA fragments containing the kanamycin-resistance gene (Figure 4).
Estimate the length of each band in relation to the positions of the size marker DNAs electrophoresed in parallel with the samples.
Check against the physical maps of the chromosome to ensure that the kanamycin-resistance gene has been inserted at the desired location. Proper insertion of the kanamycin-resistance gene, in this case, is confirmed by the appearance of a specific combination of bands on the membrane: a 5,456 bp band from both transconjugant and donor for SacII digestion, an 8,581 bp band from transconjugant or a 6,172 bp band from a donor for ClaI digestion, and a 3,583 bp band from both transconjugant and donor for SacII and ClaI double digestion.
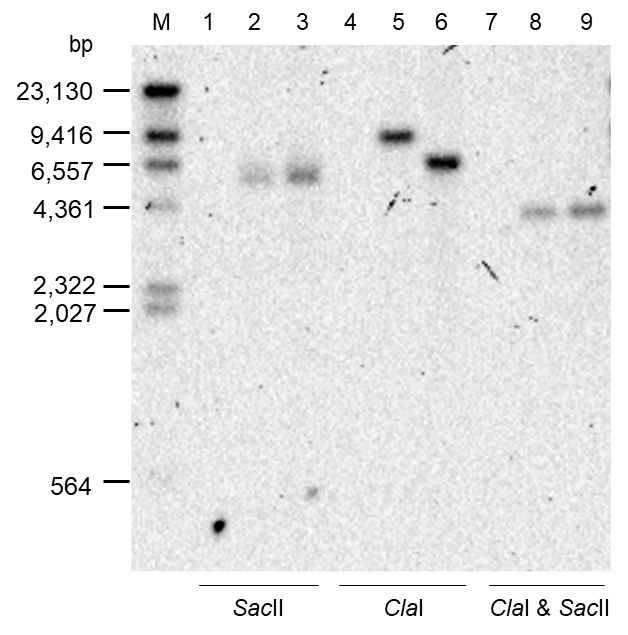
Figure 4. Results of Southern blot analysis. Chromosomal DNAs of the Recipient (lanes 1, 4, and 7), Transconjugant (lanes 2, 5, and 8), and Donor (lanes 3, 6, and 9) strains digested with Sac II (lanes 1–3), Cla I (lanes 4–6), and Cla I & Sac II (lanes 7–9) were subjected to agarose gel electrophoresis in parallel with size marker DNA fragments (M) followed by Southern blot analysis probing for the kanamycin-resistance gene.
Recipes
LB medium
Reagent Final concentration Amount LB (Difco Lennox) 2% 20 g H2O Total 1,000 mL Autoclave at 121 °C for 20 min.
LBMSM
Reagent Final concentration Amount LB (Difco Lennox) 2% 20 g H2O 500 mL After autoclaving the above at 121 °C for 20 min, add the following solutions sterilized separately. 1.25 M sorbitol 0.5 M 400 mL 0.4 M MgCl2 0.02 M 50 mL 0.4 M maleic acid-NaOH buffer (pH 7.0) 0.02 M 50 mL Total 1,000 mL Solution A
Reagent Final concentration Amount K2HPO4 0.16 M 5.6 g KH2PO4 0.088 M 2.4 g (NH3)2SO4 0.03 M 0.8 g Sodium citrate·2H2O 0.0068 M 0.4 g H2O Total 200 mL Autoclave at 121 °C for 20 min.
Solution B
Reagent Final concentration Amount MgSO4·7H2O 0.010 M 0.5 g Glucose 0.056 M 2 g H2O After autoclaving the above, add the following solutions sterilized separately. 2 mg/mL FeCl3·4H2O 4.02 × 10-6 M 80 µL 0.1 mg/mL MnSO4·5H2O 1.66 × 10-7 M 80 µL 100× trace element × 0.2 400 µL Total 200 mL 100× Trace element
Reagent Final concentration Amount CaCl2·2H2O 4.97 × 10-3 M 0.73 g ZnCl2 1.25 × 10-3 M 0.17 g CuCl2·2H2O 2.52 × 10-4 M 0.043 g CoCl2·6H2O 2.52 × 10-4 M 0.06 g Na2MoO4·2H2O 2.48 × 10-3 M 0.06 g H2O Total 1,000 mL Autoclave at 121 °C for 20 min.
C-1 medium
Reagent Final concentration Amount Solution A 2.5 mL Solution B 2.5 mL After autoclaving the above at 121 °C for 20 min, add the following solutions sterilized separately. 5% yeast extract 0.05% 50 µL 10% casamino acids 0.10% 10 µL 5 mg/mL L-Trp 50 µg/mL 50 µL Total 5 mL C-2 medium
Reagent Final concentration Amount Solution A 5.0 mL Solution B 5.0 mL After autoclaving the above at 121 °C for 20 min, add the following solutions sterilized separately. 5% yeast extract 0.05% 10 µL 10% casamino acids 0.10% 10 µL 5 mg/mL L-Trp 5 µg/mL 10 µL Total 10 mL 50× TAE buffer
Reagent Final concentration Amount Tris (hydroxymethyl) aminomethane 2 M 242 g Acetic acid 1 M 57.1 mL Na2EDTA·2H2O 0.5 M 18.6 g H2O Total 1,000 mL Autoclave at 121 °C for 20 min.
TKE buffer
Reagent Final concentration Amount 1 M Tris-HCl buffer (pH 8.0) 0.1 M 10 mL 0.5 M Na2EDTA (pH 8.0) 0.05 M 10 mL 1 M KCl 0.1 M 10 mL H2O 70 mL Total 100 mL Autoclave at 121 °C for 20 min.
20× SSC
Reagent Final concentration Amount Sodium citrate·2H2O 0.3 M 88.2 g NaCl 3 M 175.3 g H2O Total 1,000 mL Adjust pH to 7 with 14 N HCl.
Autoclave at 121 °C for 20 min.
Maleic acid buffer
Reagent Final concentration Amount Maleic acid 0.1 M 11.61 g NaCl 0.15 M 8.77 g NaOH 0.18 M 7.2 g H2O Total 1,000 mL Adjust pH to 7.5.
Autoclave at 121 °C for 20 min.
Denaturing solution
Reagent Final concentration Amount NaCl 1.5 M 87.6 g NaOH 0.5 M 20 g H2O Total 1,000 mL Neutralizing solution
Reagent Final concentration Amount 1 M Tris-HCl buffer (pH 7.5) 0.5 M 500 mL NaCl 1.5 M 87.6 g H2O Total 1,000 mL Low-stringency wash solution
Reagent Final concentration Amount 20× SSC 0.1× SSC 30 mL 10% SDS 0.2% 12 mL H2O Total 600 mL High-stringency wash solution
Reagent Final concentration Amount 20× SSC 2× SSC 180 mL 10% SDS 0.2% 12 mL H2O Total 600 mL Detection buffer
Reagent Final concentration Amount Tris 0.1 M 12.1 g NaCl 0.1 M 5.84 g MgCl2 0.05 M 4.76 g H2O Total 1,000 mL Adjust pH to 9.5.
Acknowledgments
The authors thank Valeria Verrone and Anil Wipat for their indispensable contribution to the original research paper where this protocol was derived from Mori et al. (2022). This study was supported by KAKENHI 18H02128.
Competing interests
This protocol is related to a patent and is only permitted for use in academic research. In particular, the distribution of the strains YNB211 and YNB213 requires the conclusion of an appropriate material transfer agreement.
References
- Christie, P. J., Whitaker, N. and Gonzalez-Rivera, C. (2014). Mechanism and structure of the bacterial type IV secretion systems. Biochim Biophys Acta 1843(8): 1578-1591.
- Christie, P. J. (2016). The Mosaic Type IV Secretion Systems. EcoSal Plus 7(1).
- Kananavičiūtė, R., Kanišauskaitė, I., Novickij, V. and Čitavičius, D. J. (2015). Geobacillus stearothermophilus NUB3621R genetic transformation by electroporation. Biologija61(3-4): 101-108.
- Koehler, T. M. and Thorne, C. B. (1987). Bacillus subtilis (natto) plasmid pLS20 mediates interspecies plasmid transfer. J Bacteriol 169(11): 5271-5278.
- Meijer, W. J. J., Boer, D. R., Ares, S., Alfonso, C., Rojo, F., Luque-Ortega, J. R. and Wu, L. J. (2021). Multiple Layered Control of the Conjugation Process of the Bacillus subtilis Plasmid pLS20. Front Mol Biosci 8: 648468.
- Miyano, M., Tanaka, K., Ishikawa, S., Takenaka, S., Miguel-Arribas, A., Meijer, W. J. J. and Yoshida, K. I. (2018). Rapid conjugative mobilization of a 100 kb segment of Bacillus subtilis chromosomal DNA is mediated by a helper plasmid with no ability for self-transfer. Microb Cell Fact 17(1): 13.
- Mori, K., Fukui, K., Amatsu, R., Ishikawa, S., Verrone, V., Wipat, A., Meijer, W. J. J. and Yoshida, K. I. (2022). A novel method for transforming Geobacillus kaustophilus with a chromosomal segment of Bacillus subtilis transferred via pLS20-dependent conjugation. Microb Cell Fact 21(1): 34.
- Nazina, T. N., Tourova, T. P., Poltaraus, A. B., Novikova, E. V., Grigoryan, A. A., Ivanova, A. E., Lysenko, A. M., Petrunyaka, V. V., Osipov, G. A., Belyaev, S. S., et al. (2001). Taxonomic study of aerobic thermophilic bacilli: descriptions of Geobacillus subterraneus gen. nov., sp. nov. and Geobacillus uzenensis sp. nov. from petroleum reservoirs and transfer of Bacillus stearothermophilus, Bacillus thermocatenulatus, Bacillus thermoleovorans, Bacillus kaustophilus, Bacillus thermodenitrificans to Geobacillus as the new combinations G. stearothermophilus, G. th. Int J Syst Evol Microbiol 51(Pt 2): 433-446.
- Singh, P. K., Ramachandran, G., Duran-Alcalde, L., Alonso, C., Wu, L. J. and Meijer, W. J. (2012). Inhibition of Bacillus subtilis natural competence by a native, conjugative plasmid-encoded comK repressor protein. Environ Microbiol 14(10): 2812-2825.
- Singh, P. K., Ramachandran, G., Ramos-Ruiz, R., Peiró-Pastor, R., Abia, D., Wu, L. J. and Meijer, W. J. (2013). Mobility of the native Bacillus subtilis conjugative plasmid pLS20 is regulated by intercellular signaling. PLoS Genet 9(10): e1003892.
- Suzuki, H. and Yoshida, K. (2012). Genetic transformation of Geobacillus kaustophilus HTA426 by conjugative transfer of host-mimicking plasmids. J Microbiol Biotechnol 22(9): 1279-1287.
- Takami, H., Takaki, Y., Chee, G. J., Nishi, S., Shimamura, S., Suzuki, H., Matsui, S. and Uchiyama, I. (2004). Thermoadaptation trait revealed by the genome sequence of thermophilic Geobacillus kaustophilus. Nucleic Acids Res 32(21): 6292-6303.
- Tanaka, T. and Koshikawa, T. (1977). Isolation and characterization of four types of plasmids from Bacillus subtilis (natto). J Bacteriol 131(2): 699-701.
- Waksman, G. (2019). From conjugation to T4S systems in Gram-negative bacteria: a mechanistic biology perspective. EMBO Rep 20(2).
- Wyrick, P. B. and Rogers, H. J. (1973). Isolation and characterization of cell wall-defective variants of Bacillus subtilis and Bacillus licheniformis. J Bacteriol 116(1): 456-465.
Article Information
Copyright
© 2022 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Amatsu, R., Mori, K., Ishikawa, S., Meijer, W. J. J. and Yoshida, K. (2022). A New Tool for the Flexible Genetic Manipulation of Geobacillus kaustophilus. Bio-protocol 12(17): e4502. DOI: 10.21769/BioProtoc.4502.
Category
Microbiology > Microbial genetics > Transformation
Molecular Biology > DNA > Transfection
Molecular Biology > DNA > DNA recombination
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.