- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

RNA Interference Method for Gene Function Analysis in the Japanese Rhinoceros Beetle Trypoxylus dichotomus
(*contributed equally to this work) Published: Vol 12, Iss 8, Apr 20, 2022 DOI: 10.21769/BioProtoc.4396 Views: 2753
Reviewed by: Teiya KijimotoLeonardo Gaston GuilgurAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Oct 2020

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
In the Japanese rhinoceros beetle Trypoxylus dichotomus, various candidate genes required for a specific phenotype of interest are listed by next-generation sequencing analysis. Their functions were investigated using RNA interference (RNAi) method, the only gene function analysis tool for T. dichotomus developed so far. The summarized procedure for the RNAi method used for T. dichotomus is to synthesize double-stranded RNA (dsRNA), and inject it in larvae or pupae of T. dichotomus. Although some dedicated materials or equipment are generally required to inject dsRNA in insects, the advantage of the protocol described here is that it is possible to inject dsRNA in T. dichotomus with one syringe.
Keywords: RNA interferenceBackground
The Japanese rhinoceros beetle Trypoxylus dichotomus is useful from different perspectives, such as in developmental biology, morphology, ethology, ecology, biomimetics, aerodynamics, and drug discovery. For instance, Siva-Jothy (1987) revealed the feeding time of T. dichotomus from ecological research, Miyanoshita et al. (1996) isolated a new antibacterial peptide from larvae of T. dichotomus, Hongo (2007) clarified the relationship between horn length and body size in T. dichotomus, Emlen et al. (2012) showed that signaling through the insulin receptor is involved in T. dichotomus horn development, Chen et al. (2017) clarified that the elytron plate of T. dichotomus has a mechanism of high energy absorption, and Ohde et al. (2018) identified several horn formation genes using RNA-sequencing analysis and RNAi techniques.
Recently, the genome of T. dichotomus was clarified (Ogata, 2021; Morita et al., 2022), and various candidate genes required for a specific phenotype of interest were listed by RNA-sequencing analysis (Ohde et al., 2018; Zinna et al., 2018). Furthermore, the functions of the candidate genes were investigated using RNA interference (RNAi) method, the only gene function analysis tool for T. dichotomus developed so far (Emlen et al., 2012; Ito et al., 2013; Gotoh et al., 2015; Adachi et al., 2018, 2020; Ohde et al., 2018; Morita et al., 2019).
RNAi method for small insects generally requires specialized setups for microinjection, such as those for preparing glass capillaries, loading double-stranded RNA (dsRNA) into microcapillaries, injecting dsRNA into insects using high pressure, and manipulating the needle for injection under a stereomicroscope (e.g., Kim et al., 2004; Niimi et al., 2005; Masumoto et al., 2009; Posnien et al., 2009). The RNAi method in T. dichotomus does not require most of the above setups, it only requires a 1-mL syringe for dsRNA injection, since the beetle’s larval body size is as large as a mouse. This study provides a detailed protocol for the RNAi method in the third instar larvae of T. dichotomus (Figure 1). This method can also be applied to the first and second instar larvae and pupae of T. dichotomus.
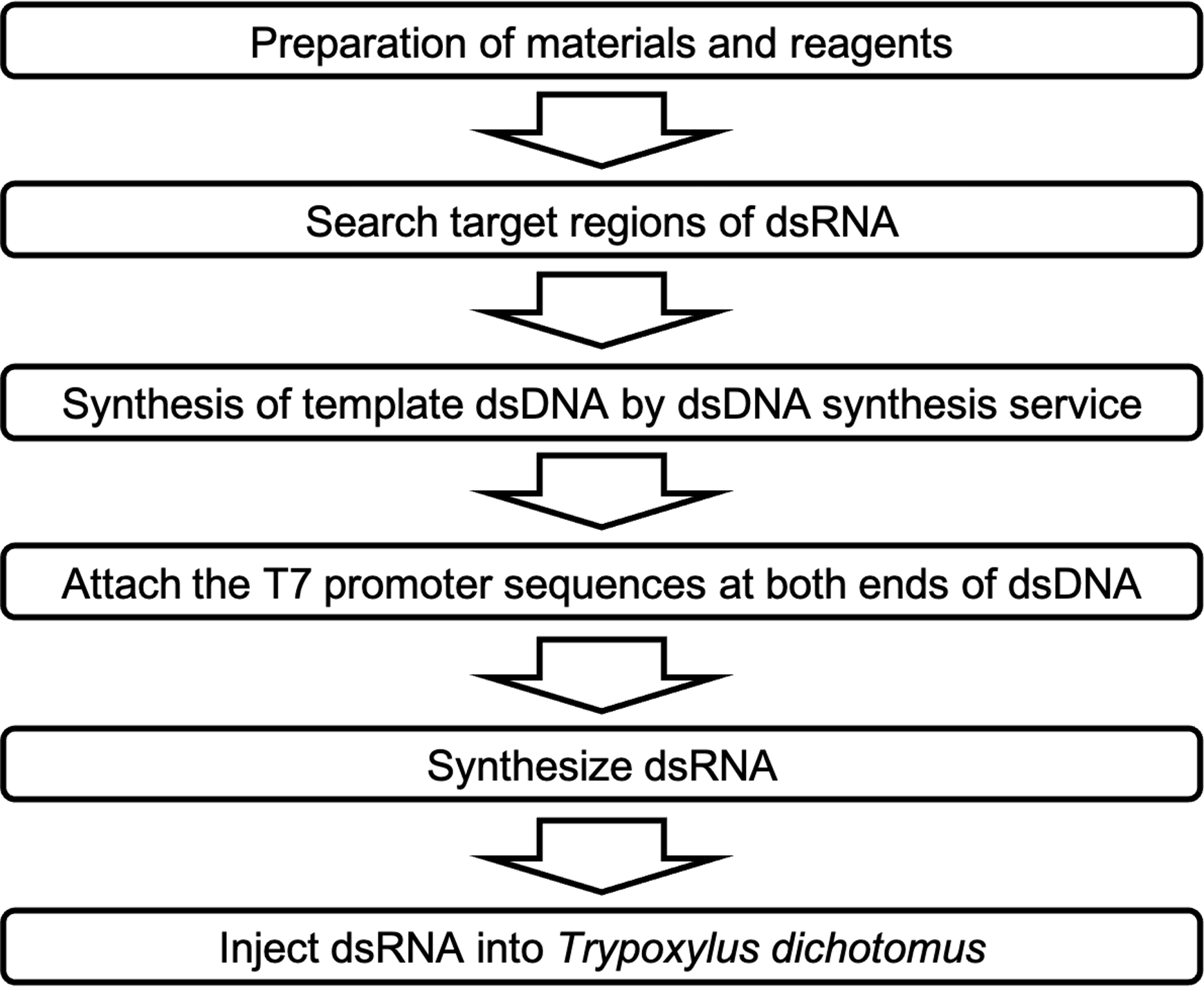
Figure 1. Overview of RNA interference method procedure in Trypoxylus dichotomus.
Materials and Reagents
Filter Pipette tips (RNase/DNase-free) (Labcon, catalog numbers: 1057-965-018-9 [P1000], 1059-965-008-9 [P200], 1055-965-018-9 [P40]; and QSP, catalog number: TF102-10-Q [P2])
Disposable needles No.30 (30-gauge) (Dentronics, No.30)
0.2 mL Flat PCR Tube 8-Cap Strips (INA OPTIKA, catalog number: 3247-00)
1.5 mL microtubes, flat-bottom, DNase/RNase-free (Watson, catalog number: 131-415C)
Plastic bottles (Hobby Club, blow container 750)
Lavender Nitrile Powder-free Exam Gloves (Kimberly-Clark, catalog number: 52818)
Styrofoam container
Aluminum foil
Third instar larvae of Trypoxylus dichotomus (Loiinne, third instar larva)
UltraPureTM DNase/RNase-Free Distilled Water (UPW) (Invitrogen, catalog number: 10977023)
Loading Dye (New England Biolabs, catalog number: B7024S)
100 bp DNA Ladder (New England Biolabs, catalog number: N3231L)
TaKaRa Ex Taq (TaKaRa, catalog number: RR001B)
AmpliScribeTM T7-FlashTM Transcription Kit (Epicentre Technologies, catalog number: ASF3257)
Rnase AWAY (Thermo Fisher Scientific, catalog number: 7002)
Monarch PCR & DNA Cleanup Kit (5 μg) (BioLabs, catalog number: T1030)
Terumo Syringe 1 mL for Tuberculin Slip Tip White (Terumo, catalog number: SS-01P)
Ethanol (FUJIFILM Wako Chemicals, catalog number: 057-00456)
Phenol/Chloroform/Isoamyl alcohol (25:24:1) (PCI) (NIPPON GENE, catalog number: 311-90151)
Chloroform/Isoamyl alcohol (24:1) (CIA) (Sigma-Aldrich, catalog number: C0549)
3 M Sodium Acetate (pH 5.2) (NIPPON GENE, catalog number: 316-90081)
Agarose S (NIPPON GENE, catalog number: 318-01195)
10× TAE Buffer (NIPPON GENE, catalog number: 318-90301)
Ethidium Bromide Powder (SIGMA, catalog number: E7637-1G)
Humus (Dorcus Owner’s shop, Osaka, Japan)
PCR Reaction mixture (see Recipes)
Solution for 1% agarose gel (see Recipes)
Equipment
Thermal cycler (Bio-Rad Laboratories, model: T100)
Pipetman (GILSON, catalog numbers: F123602 [P1000], F123601 [P200], F123600 [P20], and F144801 [P2])
Spectrophotometer (DeNovix, model: DS-11)
Electrophoresis chamber (TaKaRa, model: AD115)
Microwave oven (Panasonic, model: NE-MS261)
Gel imaging device (ATTO, model: WSE-6100)
Centrifuge (TOMY, model: MX-307)
Heat block (Major Science, model: MD-MINI)
Vortex mixer (LMS, model: VTX-3000L)
Procedure
Search target regions of dsRNA
Search target mRNA in RNA-seq database of Trypoxylus dichotomus (Ohde et al., 2018; Zinna et al., 2018).
Search for target regions of dsRNA.
Note: The lengths of target regions range from 150 bp to 600 bp. The target region should have low homology with any other T. dichotomus gene to avoid off-target effects. Therefore, it is desirable to not include any domains in the target design. Furthermore, the target region is checked for homology with other T. dichotomus genes, by performing a blastn search in the T. dichotomus database. dsRNA should be redesigned in other regions, if consecutive regions of 25-bp or more homological with other T. dichotomus genes are included.
Add the adapter sequence 5’-AACGAATTCGCCCTT-3’ at the 5’ end of the dsDNA, and the sequence 5’-AAGGGCGAATTCGCG-3’ at the 3’ end of dsDNA.
Note: The final sequence is as follows: 5’-AACGAATTCGCCCTT-[the sequence in the target region of dsRNA]-AAGGGCGAATTCGCG-3’. The adapter sequences are designed to be homologous with both ends of the pCR 4-TOPO vector (Invitrogen, catalog number: K457502).
Synthesize the above sequences using a dsDNA synthesis service (e.g., IDT eBlocks).
Synthesize dsDNA
Add the T7 promoter sequences at both ends of dsDNA, by performing PCR using the following primers: 5’-taatacgactcactatagggAACGAATTCGCCCTT-3’ [melting temperature (Tm) = 74°C] and 5’-taatacgactcactatagggCGCGAATTCGCCCTT-3’ (Tm = 78°C).
Note: T7 promoter sequences are written in lowercase and underlined. The primers can also be used to attach T7 promoter sequences to inserts that are subcloned into the pCR 4-TOPO vector.
Perform PCR with a reaction mixture in which dsDNA (10–100 ng) synthesized in step A-4 is added as a template.
Note: The following steps constitute the PCR reaction: the initial denaturing step is 98°C for 30 s, followed by 30 cycles of denaturing at 98°C for 10 s, annealing at 55°C for 30 s, and an extension of for 60 s per 1 kb at 72°C, with an additional step of 72°C for 5 min. React about four 50 µL-reaction mixture tubes per target gene, to obtain more than 1 µg of dsDNA.
Collect PCR products from step B-2 into a 1.5-mL microtube (50 µL × 4 = total of 200 µL).
Add a sufficient amount of 1× TAE and 1 % agarose gel into the electrophoresis chamber.
Load a 100-bp DNA Ladder into the 1 % agarose gel well.
Load 5 μL of PCR product mixed (step B-3) with 1 μL of 6× DNA loading dye into a well.
Perform electrophoresis on full power (100 V) for 30 min.
Using a gel imaging device, check whether the PCR product size corresponds to the expected size.
Purify the PCR product from step B-3 with Monarch PCR & DNA Cleanup Kit (5 μg), according to the manufacturer’s instructions.
Note: dsDNA is eluted with 100 µL of elution buffer.
Make dsDNA solution RNase-free.
Note: Lay aluminum foil on the laboratory table, and make the workplace and the experimental materials and equipment RNase-free with RNase AWAY. Wear rubber gloves.
Add 100 μL (equal to the volume of dsDNA solution) of Phenol/Chloroform/Isoamyl alcohol (PCI) to the result from step B-9 and vortex.
Centrifuge at 13,000 × g and 25°C for 15 min.
Transfer upper layer to new 1.5-mL RNase-free microtube.
Add 100 μL (equal to the volume of dsDNA solution) of chloroform, and vortex.
Centrifuge at 13,000 × g and 25°C for 5 min.
Transfer upper layer to new 1.5-mL RNase-free microtube.
Add 250 μL (2.5-fold the volume of dsDNA solution) of ethanol, and 10 μL (0.1-fold the volume of dsDNA solution) of 3 M Sodium Acetate (pH 5.2), and vortex.
Centrifuge at 20,400 × g and 4°C for 15 min .
Discard the supernatant.
Add 250 μL of 70% ethanol solution, made with UltraPure water (UPW), and vortex.
Centrifuge at 20,400 × g and 4°C for 5 min.
Discard the supernatant.
Open the lid of the tube, and dry for 5 min.
Add <7.3 µL of UPW.
Mix 1 µL of dsDNA solution from step B-10-n, and 9 µL of UPW, to make a 10-fold dilution.
Measure the concentration of the 10-fold diluted dsDNA solution from step B-10-o with the spectrophotometer.
Synthesize dsRNA
Synthesize dsRNA with 1 µg of dsDNA and AmpliScribeTM T7-FlashTM Transcription Kit according to the manufacturer’s instructions.
Purify dsRNA
Note: Lay aluminum foil on the laboratory table, and make the workplace and the experimental materials and equipment RNase-free with RNase AWAY. Wear rubber gloves.
Add 80 μL of UPW to the result from step C-1, for a total volume of 100 µL.
Add 100 μL (equal to the volume of dsRNA solution) of PCI, and vortex for 30 s.
Centrifuge at 13,000 × g and 25°C for 15 min.
Transfer the upper layer to a new 1.5-mL RNase-free microtube.
Add 100 μL (equal to the volume of dsRNA solution) of chloroform, and vortex for 30 s.
Centrifuge at 13,000 × g and 25°C for 5 min.
Transfer the upper layer to a new 1.5-mL RNase-free microtube.
Add 250 μL (2.5-fold the volume of dsRNA solution) of ethanol, and 10 μL (0.1-fold the volume of dsRNA solution) of 3 M Sodium Acetate (pH 5.2), and vortex for 30 s.
Centrifuge at 20,400 × g and 4°C for 15 min.
Discard the supernatant.
Add 250 μL of 70% ethanol solution made with UPW, and vortex for 30 s.
Centrifuge at 20,400 × g and 4°C for 5 min.
Discard the supernatant.
Open the lid of the tube, and dry for <5 min.
Note: Do not dry RNA pellets for too long.
Add 20 μL of UPW.
Incubate the tube containing dsRNA solution from step C-2-o in a heat block at 65°C for 10 min.
Transfer the aluminum block of the heat block into a Styrofoam™ container, and cool it to room temperature, to anneal the dsRNA (Figure 2A).
Take an aliquot of the annealed dsRNA, and dilute it with UPW for quantification. We routinely dilute 1 µL of the dsRNA solution from step C-2-o with 9 µL of UPW.
Measure the concentration of 1 µL of 10-fold diluted dsRNA solution from step C-2-r with the spectrophotometer.
Add a sufficient amount of 1× TAE and 1% agarose gel into the electrophoresis chamber.
Load 100-bp DNA Ladder into the 1% agarose gel well.
Load 9 μL of 10-fold diluted dsRNA solution from step C-2-s mixed with 2 μL of 6× DNA loading dye into a well.
Perform electrophoresis on full power (100 V) for 30 min.
Check the concentration, and whether the dsRNA product size is the same as expected.
Add UPW to dsRNA solution.
Note: It is helpful to adjust the final concentration of the dsRNA solution to 5 µg/µL.
Store at −30°C until use.
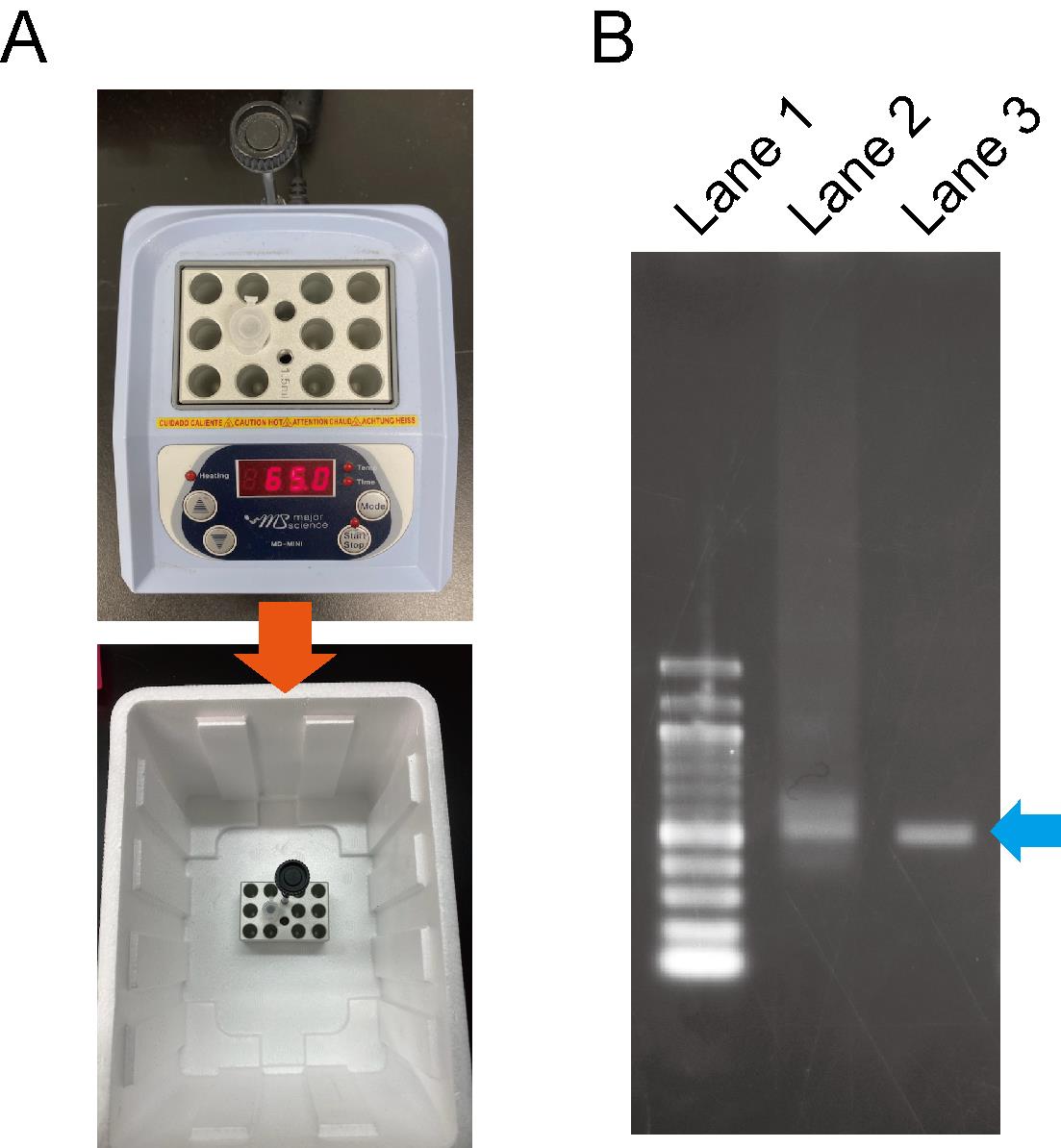
Figure 2. dsRNA annealing process. A. The annealing process of dsRNA. B. Evaluation of dsRNA patterns via the annealing process. Lane 1; 100 bp DNA ladder. Lane 2; dsRNA before the annealing process looks smeared. Lane 3; dsRNA after the annealing process resembles a sharp band.
Inject dsRNA in Trypoxylus dichotomus
Note: EGFP is used as negative control.
Mix dsRNA (50–100 µg) and UPW.
Note: The final volume of dsRNA solution should be 50 µL.
Wear rubber gloves.
Softly hold the larva of T. dichotomus (Figure 3A).
Suck dsRNA aqueous solution using a 1-mL syringe with a 30-gauge needle.
Pierce the needle at the lateral body wall of the first thoracic segment (T1) of a larva, and inject the solution into the hemocoel, just beneath the epidermis (Figure 3B and C).
Note: It is possible for bubbles to be injected in T. dichotomus to a small extent.
Place the larva in a bottle filled with humus, and incubate it at room temperature (24–28°C) until the larva develops into the pupa or the adult.
Observe the phenotype in the pupa and/or the adult.
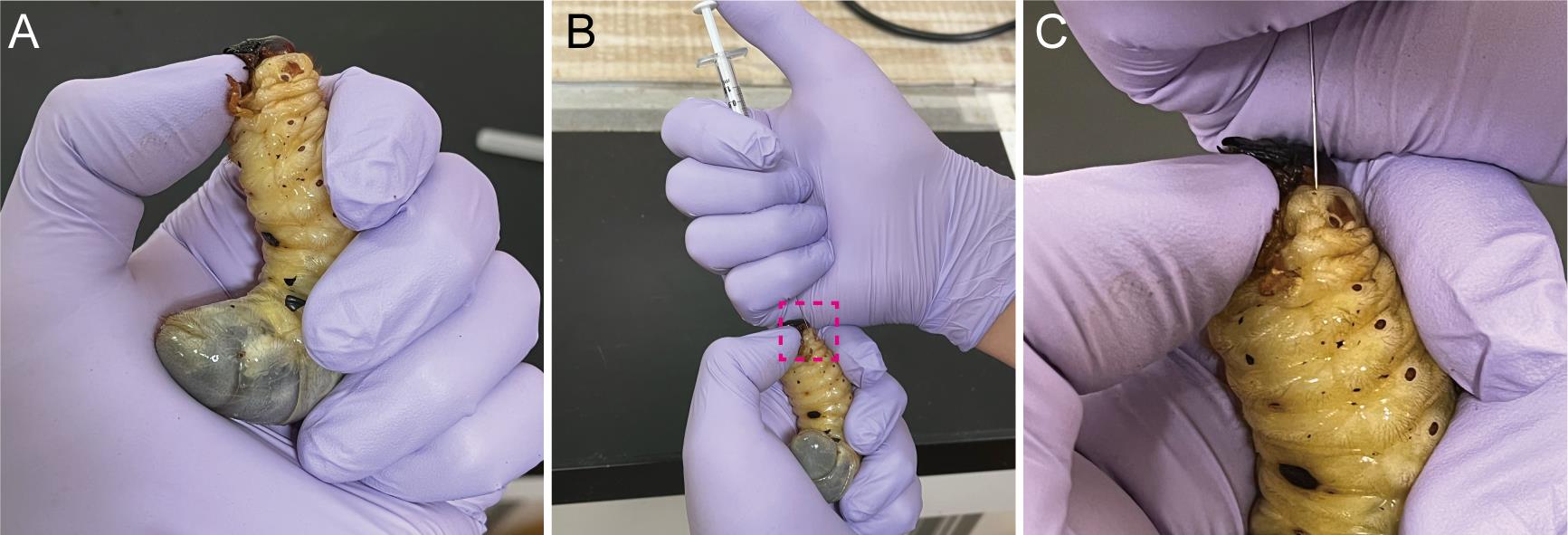
Figure 3. Injection of dsRNA into third instar larvae of Trypoxylus dichotomus. A. How to hold a larva during the injection. To prevent a larva from moving its head during injection, place your thumbnail under the mandible and push the larval head upward. B. Injection of dsRNA into third instar larvae. C. A highly magnified view of the injection point in the first thoracic (T1) body segment. dsRNA is injected into the T1 of a larva. The needle should be inserted about 2 mm beneath the epidermis. The needle should be held in place for 10 sec after dsRNA injection, to prevent the backflow of dsRNA.
Recipes
PCR Reaction mixture
5 μL of 10× Ex taq Buffer
4 μL of dNTP Mixture (each 2.5 mM)
2 μL of Primer (10 mM) (1 μL of forward, and 1 μL of reverse)
0.5 μL of Ex Taq
38.5 μL of UPW (the amount of DNA template solution)
Solution for 1% agarose gel
150 mL of TAE (1×)
1.5 g Agarose S
Note: Dissolve the above solution in a microwave oven. Next, add 20 μL of 10 mg/mL ethidium bromide to the solution. Allow the solution to polymerize at room temperature.
Acknowledgments
We thank Toshiya Ando and Taro Nakamura for critical reading, and Glen Cowan for linguistic corrections, the Model Plant ResearchFacility/NIBB BioResource Center, the Data Integration and Analysis Facility/NIBB for technical assistance. This protocol was adapted from Ito et al. (2013), Ohde et al. (2018), and Morita et al. (2019). This work was supported by MEXT KAKENHI Grant Numbers 20H05944, 20H04933, 18H04766, 16H01452, 23128505, and 25128706 (to T. N.), and JSPS KAKENHI Grant Numbers 19K16181 and 21K15135 (to S. M.).
Competing interests
No conflict of interest is declared regarding this article.
References
- Adachi, H., Matsuda, K., Niimi, T., Inoue, Y., Kondo, S., Gotoh, H. (2018). Anisotropy of cell division and epithelial sheet bending via apical constriction shape the complex folding pattern of beetle horn primordia. Mech Dev 152: 32–37.
- Adachi, H., Matsuda, K., Niimi, T., Kondo, S., Gotoh, H. (2020). Genetical control of 2D pattern and depth of the primordial furrow that prefigures 3D shape of the rhinoceros beetle horn. Sci Rep 10: 18687.
- Emlen, D. J., Warren, I. A., Johns, A., Dworkin, I. and Lavine, L. C. (2012). A mechanism of extreme growth and reliable signaling in sexually selected ornaments and weapons. Science 337(6096): 860–864.
- Gotoh, H., Hust, J. A., Miura, T., Niimi, T., Emlen, D. J. and Lavine, L. C. (2015). The Fat/Hippo signaling pathway links within-disc morphogen patterning to whole-animal signals during phenotypically plastic growth in insects. Dev Dyn 244: 1039–1045.
- Hongo, Y. (2007). Evolution of male dimorphic allometry in a population of the Japanese horned beetle Trypoxylus dichotomus septentrionalis. Behav Ecol Sociobiol 62(2): 245–253.
- Ito, Y., Harigai, A., Nakata, M., Hosoya, T., Araya, K., Oba, Y., Ito, A., Ohde, T., Yaginuma, T. and Niimi, T. (2013). The role of doublesex in the evolution of exaggerated horns in the Japanese rhinoceros beetle. EMBO Rep 14(6): 561–567.
- Chen, J., Zhang, X., Okabe, Y., Saito, K., Guo, Z., and Pan, L. (2017). The deformation mode and strengthening mechanism of compression in the beetle elytron plate. Mater Des 131(5): 481–486.
- Miyanoshita, A., Hara, S., Sugiyama, M., Asaoka, A., Taniai, K., Yukuhiro, F. and Yamakawa, M. (1996). Isolation and characterization of a new member of the insect defensin family from a beetle, Allomyrina dichotoma. Biochem Biophys Res Commun 220(3): 526–531.
- Kim, Y.-O., Park, S.-J., Balaban, R. S., Nirenberg, M. and Kim, Y. (2004). A functional genomic screen for cardiogenic genes using RNA interference in developing Drosophila embryos. Proc Nati Acad Sci 101(1): 159–164.
- Masumoto, M., Yaginuma, T. and Niimi, T. (2009). Functional analysis of Ultrabithorax in the silkworm, Bombyx mori, using RNAi. Dev Genes Evol 219(9-10): 437–444.
- Morita, S., Ando, T., Maeno, A., Mizutani, T., Mase, M., Shigenobu, S. and Niimi, T. (2019). Precise staging of beetle horn formation in Trypoxylus dichotomus reveals the pleiotropic roles of doublesex depending on the spatiotemporal developmental contexts. PLOS Genet 15(4): e1008063.
- Morita, S., Shibata, T., Nishiyama, T., Kobayashi, Y., Yamaguchi, K., Toga, K., Ohde, T., Gotoh, H., Kojima, T., Weber, J., et al. (2022). The draft genome sequence of Japanese rhinoceros beetle Trypoxylus dichotomus. bioRxiv https://doi.org/10.1101/2022.01.10.475740
- Niimi, T., Kuwayama, H. and Yaginuma, T. (2005). Larval RNAi Applied to the Analysis of Postembryonic Development in the Ladybird Beetle, Harmonia axyridis. J Insect Biotechnol Sericology 74(3): 95–102.
- Ogata, N. (2021). Whole-Genome Sequence of the Trypoxylus dichotomus Japanese rhinoceros beetle. MicroPubl Biol 2021.
- Ohde, T., Morita, S., Shigenobu, S., Morita, J., Mizutani, T., Gotoh, H., Zinna, R. A., Nakata, M., Ito, Y., Wada, K., et al. (2018). Rhinoceros beetle horn development reveals deep parallels with dung beetles. PLOS Genet 14(10): e1007651.
- Posnien, N., Schinko, J., Grossmann, D., Shippy, T. D., Konopova, B. and Bucher, G. (2009). RNAi in the red flour beetle (Tribolium). Cold Spring Harb Protoc 2009(8): pdb prot5256.
- Siva-Jothy, M. T. (1987). Mate securing tactics and the cost of fighting in the Japanese horned beetle, Allomyrina dichotoma L. (Scarabaeidae). J Ethol 5(2): 165–172.
- Zinna, R., Emlen, D., Lavine, L. C., Johns, A., Gotoh, H., Niimi, T. and Dworkin, I. (2018). Sexual dimorphism and heightened conditional expression in a sexually selected weapon in the Asian rhinoceros beetle. Mol Ecol 27(24): 5049–5072.
Article Information
Copyright
© 2022 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Sakura, K., Morita, S. and Niimi, T. (2022). RNA Interference Method for Gene Function Analysis in the Japanese Rhinoceros Beetle Trypoxylus dichotomus. Bio-protocol 12(8): e4396. DOI: 10.21769/BioProtoc.4396.
Category
Developmental Biology > Morphogenesis > Metamorphosis
Molecular Biology > RNA > RNA interference
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.