- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

RNA-mediated in vivo Directed Evolution in Yeast
Published: Vol 12, Iss 5, Mar 5, 2022 DOI: 10.21769/BioProtoc.4346 Views: 3908
Reviewed by: Alessandro DidonnaAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Sep 2021
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Directed evolution is a powerful approach to obtain genetically-encoded sought-for traits. Compared to the prolonged adaptation regimes to mutations occurring under natural selection, directed evolution unlocks rapid screening and selection of mutants with improved traits from vast mutated sequence spaces. Many systems have been developed to search variant landscapes based on ex vivo or in vivo mutagenesis, to identify and select new-to-nature and optimized properties in biomolecules. Yet, the majority of such systems rely on tedious iterations of library preparation, propagation, and selection steps. Furthermore, among the relatively few in vivo directed evolution systems developed to mitigate handling of repetitive ex vivo steps, directed evolution of DNA is the standard approach. Here, we present the protocol for designing the transfer of genetic information from evolving RNA donors to DNA in baker’s yeast, using CRISPR- and RNA-assisted in vivo directed evolution (CRAIDE). We use mutant T7 RNA polymerase to introduce mutations in RNA donors, while incorporation into DNA is directed by CRISPR/Cas9. As such, CRAIDE offers an opportunity to study fundamental questions, such as RNA’s contribution to the evolution of DNA-based life on Earth.
Graphic abstract:
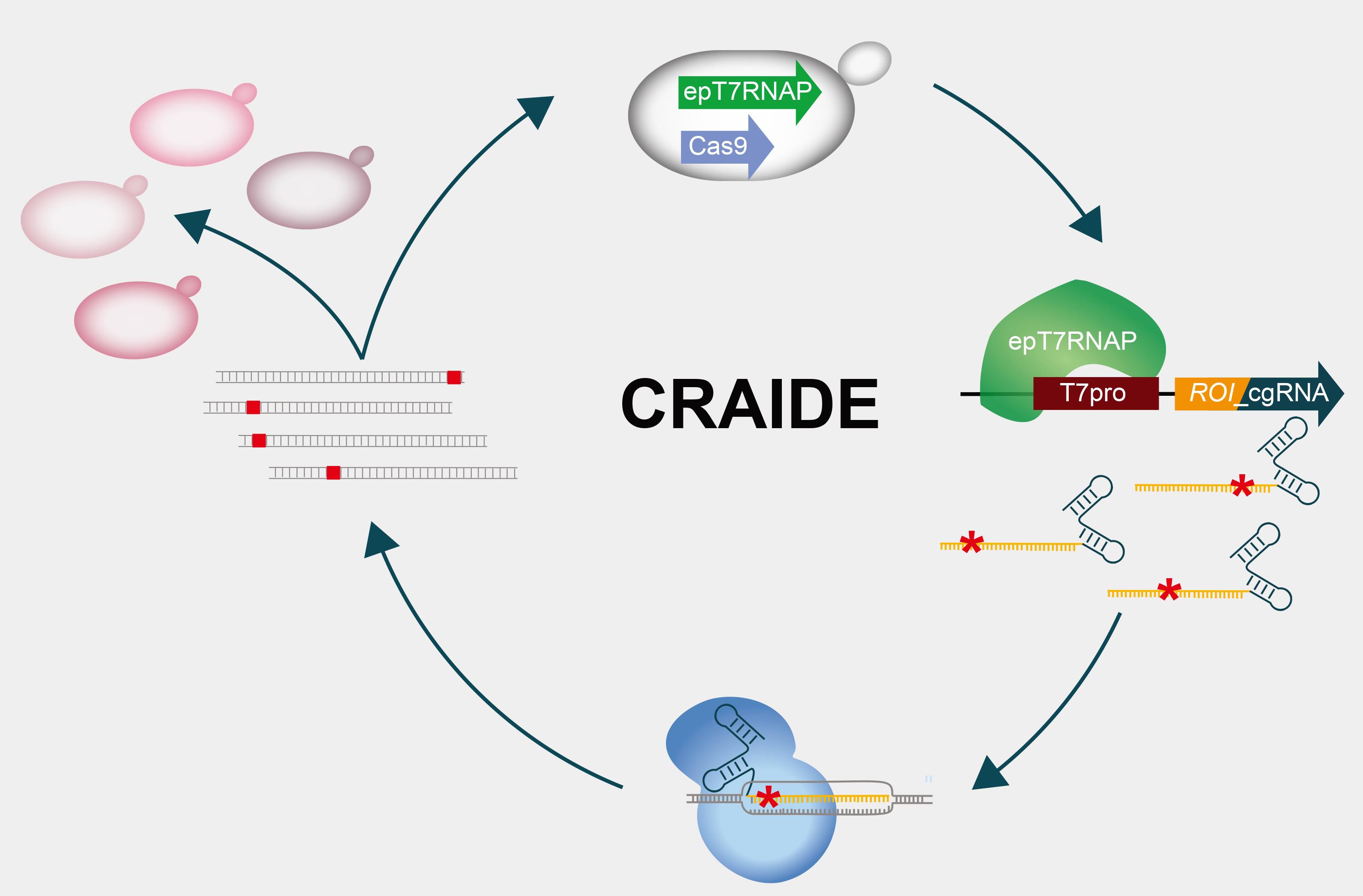
CRISPR- and RNA-assisted in vivo directed evolution (CRAIDE).
Background
The contribution of RNA to the evolution of DNA-based life is poorly understood. Detailed understanding of fundamental mechanisms and circumstances that facilitate information transfer from RNA to DNA are key to elucidate RNA’s role in evolution (Morris, 2015). Current technologies used in the laboratory to study evolution in vivo often focus on fixation of mutations directly into DNA by the use of error-prone and engineered polymerases (Ravikumar et al., 2014; Badran and Liu 2015; Moore et al., 2018; Halperin et al., 2018), and do not support studies on the possibility and frequency of evolving RNA molecules as templates for plasmid and genome editing. Here, we designed a synthetic genetically-encoded system to introduce evolving RNA donors into Cas9-targeted DNA loci in baker’s yeast, serving as a tool for investigating RNA-mediated evolution on plasmids and genomic contexts. This system called CRISPR- and RNA-assisted in vivo directed evolution (CRAIDE) builds upon previous reports, showing that transcripts delivered in vivo can support RNA-DNA repair of induced DNA double-strand breaks (DSBs) in yeast RNase H knock-out strains (Keskin et al., 2014 and 2016). Extending from these seminal findings, CRAIDE makes use of an orthogonal error-prone T7 RNA polymerase to control the expression of chimeric guide RNAs (cgRNAs), which encode both a seed sequence and an evolved template for repair at user-defined loci targeted by Cas9 (Jensen et al., 2021). We envision CRAIDE to be useful for i) studies on the basics of RNA-templated DNA repair during evolution, ii) the development and further optimization of design principles for targeted and efficient genome engineering, as well as iii) in vivo directed evolution by use of a simple set of plasmid-based designs.
Materials and Reagents
1.5 mL safe-lock tubes (Sigma-Aldrich, catalog number: 0030 120.086)
Petri dishes (TH. Geyer, Labsolute, catalog number: 7696405)
14 mL culture tubes (Fisher Scientific, Greiner Bio-One, catalog number: 07-000-212)
PCR tubes (Sarstedt Inc., 0.2 mL PCR tube multiply μstrip 8 neutral PCR perf, catalog number: 72.985.002)
Inoculation loops (Deltalab S.L., 10 μL, catalog number: 302704)
Cuvettes (BrandTech Scientific Inc., catalog number: 7590 15)
Whatman paper (Sigma-Aldrich, catalog number: WHA10300045)
Replication block (Sigma-Aldrich, catalog number: Z363391-1EA)
50 mL syringe (VWR, catalog number: 89215-240)
0.2 μm single use filter unit (TH. Greyer, Labsolute, catalog number: 7699822)
Yeast (CEN.PK2-1C, EUROSCARF) (see Table 1)
Lithium acetate (Sigma-Aldrich, catalog number: 517992)
PEG 3350 (Sigma-Aldrich, catalog number: 1546547)
ssDNA (Sigma-Aldrich, single stranded deoxyribonucleic acid from salmon testes, catalog number: D9156)
Sodium acetate, 3 M, pH 5.2 (Merck Millipore, catalog number: 567422)
100% ethanol (VWR, catalog number: 20821.310)
70% ethanol (VWR, catalog number: 83801.360)
NucleoSpin gel and PCR clean-up kit (Macherey-Nagel, catalog number: 740609.50)
Yeast synthetic drop-out medium supplements without leucine, tryptophan, and uracil (Sigma-Aldrich, catalog number: Y1771)
Yeast synthetic drop-out medium supplements without histidine (Sigma-Aldrich, catalog number: Y1751)
Yeast synthetic drop-out medium supplements without leucine (Sigma-Aldrich, catalog number: Y1376)
YPD Broth (ThermoFisher Scientific, catalog number: A1374501)
D-(+)-Galactose (Sigma-Aldrich, catalog number: G0625)
D-(+)-Glucose (Sigma-Aldrich, catalog number: G8270)
Yeast nitrogen base without amino acids (Sigma-Aldrich, catalog number: Y0626)
Yeast nitrogen base without amino acids and ammonium sulfate (Sigma-Aldrich, catalog number: Y1251)
L-Glutamic acid monosodium salt monohydrate (Sigma-Aldrich, catalog number: 49621)
Agar (Merck Millipore, catalog number: 05040)
Nourseothricin (Jena Bioscience, catalog number: AB-102XL)
GeneRuler 1 kb DNA ladder (Thermo Scientific, catalog number: SM0311)
Agarose (Sigma-Aldrich, catalog number: A4718)
RedSafe (iNtRON Biotechnology, catalog number: 21141)
DNA gel loading dye 6× (Thermo Scientific, catalog number: R0611)
OneTaq 2× Master Mix with standard buffer (New England BioLabs, catalog number: M0482S)
Oligonucleotides (IDT) (see Table 3)
Plasmids (see Table 2)
FastDigest NotI (ThermoFisher Scientific, catalog number: FD0593)
FastDigest buffer (ThermoFisher Scientific, catalog number: B64)
TAE Buffer (Tris-Acetate-EDTA) 50× (ThermoFisher Scientific, catalog number: B49)
200 mg/mL nourseothricin solution (see Recipes)
Synthetic complete drop-out media (see Recipes)
20% (w/v) glucose and galactose solutions (see Recipes)
Table 1. Yeast strains.
Strain Genotype Plasmids Parental strain Reference CEN.PK2-1C MATa ura3-52 his3∆1 leu2-3_112 trp1-289 MAL2-8c SUC2 N/A N/A EUROSCARF Sc35 MATa ura3-52 his3∆1 leu2-3_112 trp1-289 MAL2-8c SUC2 pEDJ391 CEN.PK2-1C Jensen et al., 2021 Sc36 MATa ura3-52 his3∆1 leu2-3_112 trp1-289 MAL2-8c SUC2 rnh1∆ rnh201∆ N/A CEN.PK2-1C Jensen et al., 2021 Sc43 MATa ura3-52 leu2-3_112 trp1-289 MAL2-8c SUC2 rnh1∆ rnh201∆ his3∆ pEDJ391 Sc40 Jensen et al., 2021 Sc138 MATa ura3-52 leu2-3_112 trp1-289 MAL2-8c SUC2 rnh1∆ rnh201∆ his3∆ HIS3_23∆29-XII-5 N/A Sc43 Jensen et al., 2021 Sc146 MATa ura3-52 leu2-3_112 trp1-289 MAL2-8c SUC2 rnh1∆ rnh201∆ his3∆ HIS3_23∆29-XII-5 pEDJ333; pMLB10; pEDJ508 Sc138 Jensen et al., 2021 Sc147 MATa ura3-52 leu2-3_112 trp1-289 MAL2-8c SUC2 rnh1∆ rnh201∆ his3∆ HIS3_23∆29-XII-5 pEDJ333; pMLB10; pEDJ400 Sc138 Jensen et al., 2021 Table 2. Plasmids.
Plasmid Contents Genetic marker Antibiotic marker Origin of replication Addgene ID pEDJ8 SNR52p:sgRNA_RNH1∆:SUP4t_SNR52:sgRNA_RNH201∆:SUP4t URA3 Ampicillin 2μ 177271 pEDJ332 SNR52p:sgRNA_HIS3-KO:SUP4t Nourseothricin resistance Ampicillin 2μ N/A pEDJ333 GAL1p:Cas9:CYC1t LEU2 Ampicillin Cen/ARS 177272 pEDJ391 TEF1p:Cas9:CYC1t LEU2 Ampicillin Cen/ARS N/A pEDJ400 USER cloning site URA3 Ampicillin 2μ 177269 pEDJ437 USER cloning site HIS3 Ampicillin 2μ 177270 pEDJ506 ADH1t:HIS3_23∆29-XII-5:<-PGK1p N/A (no marker) Ampicillin 2μ 177274 pEDJ508 ADH1t:T7p:cgRNA_HIS3_stop:tZ URA3 Ampicillin 2μ 177275 pMLB10 GAL1p:T7RNAP(F11L;T613A):CYC1t TRP1 Ampicillin 2μ 177273 pCfB3050 SNR52p:sgRNA_XII-5:SUP4t Nourseothricin resistance Ampicillin 2μ 73292 Table 3. Oligos.
Oligo Sequence F_RNH1_KO CCTACTGTATTGTACTTGAAACAGG R_RNH1_KO GCCACTCCGAAGGTTTGAAAGTCG F_RNH201_KO GTACCCCCCACGGTAGAAGCATC R_RNH201_KO CAATTATCTAGGGTTCTCAGC F_HIS3_KO ACTCTTGGCCTCCTCTAGTACAC R_HIS3_KO CATGTATATATATCGTATGCTGCAG F_Seq CAAGTTCTTAGATGCTTTC R_Seq CTACATAAGAACACCTTTGGTG RNH1_90-mer AAAGTGTCACTCCTTGCTTATCGAAGGAACTATCGATTCCTAATTTGCTGTTTTTTGCTTGGCTTCTTACTGGACCTGTTGTACCTGTAA RNH201_90-mer AAAAACCTTGAAAACAACTACTGCACACCAAATTGATACGATTAATCACCTCCAGTTTGCACATACTATATATCTTCATGTAATACTACA HIS3_90-mer AAGAATATACTAAAAAATGAGCAGGCAAGATAAACGAAGGCAAAGTGACACCGATTATTTAAAGCTGCAGCATACGATATATATACATGT
Equipment
-80°C freezer (Thermo Scientific, Forma 88000 Series, catalog number: 88-500A)
30°C shaking incubator (Thermo Scientific, MaxQ 8000, model: SHKE8000)
Stationary incubator (Thermo Scientific, Heratherm, catalog number: 51028112)
Safety cabinet (Thermo Scientific, Safe 2020, catalog number: 51026637)
Autoclave (CertoClav, CertoClav Multicontrol, catalog number: 8502312A)
Centrifuge (Thermo Scientific, Multifuge X1 Centrifuge, catalog number: 75004211)
Benchtop centrifuge (VWR, Micro Star 17, catalog number: 521-1646)
Refrigerated centrifuge (VWR, Micro Star 17R, catalog number: 521-1647)
Spectrophotometer (IMPLEN, model: NanoPhotometer P300)
Thermomixer (VWR, model thermomixer comfort)
Vortexer (Scientific Industries, model: Vortex-Genie 2)
Water purification system (VWR, PURELAB flex 2, catalog number: 89204-092)
Molecular Imager Gel Doc (Bio-Rad Laboratories, catalog number: 1708195)
DNA electrophoresis chamber (Bio-Rad Laboratories, catalog number: 1704487EDU)
Fixed-Height Comb (Bio-Rad Laboratories, catalog number: 1704465EDU)
Sub-Cell GT UV-Transparent Mini-Gel tray (Bio-Rad Laboratories, catalog number: 1704435EDU)
Mini-Gel caster (Bio-Rad Laboratories, catalog number: 1704422EDU)
PowerPac basic power supply (Bio-Rad Laboratories, catalog number: 1645050)
Thermal cycler (Bio-Rad Laboratories, S1000, catalog number: 1852148)
Procedure
This procedure describes plasmid construction, strain engineering, and gain-of-function analysis of cgRNA-DNA repair in yeast. The purpose of this method is to confer random gain-of-function mutagenesis in an artificially engineered STOP codon on the cgRNA delivery plasmid (pEDJ508) into a dysfunctional ectopic his3 cassette in the pEDJ506 integration plasmid (PGK1pro-HIS3_23Δ29-XII-5-ADH1t). Strains must be RNase H knock-out mutants for the system to function. Successful CRAIDE mutants are functionally screened on selective dropout plates following system induction (Figure 1).
CRAIDE target sequence and plasmid designThe pEDJ506 plasmid contains an integration cassette for chromosome XII with PGK1pro-HIS3_23Δ29-XII-5-ADH1t in reverse orientation. Here, the native HIS3 sequence is disrupted by a 29 bp deletion and an insertion of 23 bases comprising the gRNA seed sequence and protospacer-adjacent motif (PAM). The HIS3_23Δ29-XII-5 sequence is given below, with Δ29 depicted in orange, PAM in blue, the gRNA seed sequence in bold, and the start codon (ATG) in green:
CTACATAAGAACACCTTTGGTGGAGGGAACATCGTTGGTACCATTGGGCGAGGTGGCTTCTCTTATGGCAACCGCAAGAGCCTTGAACGCACTCTCACTACGGTGATGATCATTCTTGCCTCGCAGACAATCAACGTGGAGGGTAATTCTGCTAGCCTCTGCAAAGCTTTCAAGAAAATGCGGGATCATCTCGCAAGAGAGATCTCCTACTTTCTCCCTTTGCAAACCAAGTTCGACAACTGCGTACGGCCTGTTCGAAAGATCTACCACCGCTCTGGAAAGTGCCTCATCCAAAGGCGCAAATCCTGATCCAAACCTTTTTACTCCACGCACGGCCCCTAGGGCCTCTTTAAAAGCTTGACCGAGAGCAATCCCGCAGTCTTCAGTGGTGTGATGGTCGTCTATGTGTAAGTCACCAATGCACTC-∆29-CCGTCGAGGAATTTTTACTAACACCAGAGCATGTATCATATGGTCCAGAAACCCTATACCTGTGTGGACGTTAATCACTTGCGATTGTGTGGCCTGTTCTGCTACTGCTTCTGCCTCTTTTTCTGGGAAGATCGAGTGCTCTATCGCTAGGGGACCACCCTTTAAAGAGATCGCAATCTGAATCTTGGTTTCATTTGTAATACGCTTTACTAGGGCTTTCTGCTCTGT CAT
The pEDJ508 plasmid is engineered to express his3 with an engineered STOP-codon (inside the Δ29 missing from pEDJ506) and a frame-shifting artificial intron (AI). This his3 sequence is fused to a gRNA to make a chimeric gRNA (cgRNA) for targeting the HIS3_23Δ29-XII-5 sequence from pEDJ506 in the genome. The error-prone T7 RNA polymerase transcribes the cgRNA from which the AI is spliced out, to give the complete open reading frame of his3, still maintaining the STOP-codon, including Δ29 missing from pEDJ506, and yet excluding the 23 bp insertion in that plasmid (this removal also terminates Cas9 targeting in HIS3_23Δ29-XII-5). The STOP-codon is subject to random mutagenesis introduced with error-prone T7 RNA polymerase, to create permissive mutations that can be introduced into HIS3_23Δ29-XII-5 from cgRNA-DNA double-strand break repair. The full expression cassette in pEDJ508 is given below, with all components depicted: insulator (ADH1 terminator—grey), T7 promoter (pink), STOP-codon (red), AI (yellow), gRNA seed sequence (bold), gRNA structural sequence (purple), and the T7 terminator (cyan). The start codon (ATG) is omitted in this design.
Note: Any portion of a target gene/sequence can be selected for mutagenesis, but homology arms should be as long as possible and at least 200 bp on either side of the Cas9 binding site matching the cgRNA seed sequence. The homologous sequence must contain a mutation that disrupts Cas9 targeting to protect the plasmid, and later to protect the engineered target gene/sequence. With pEDJ508, 29 bp are reintroduced into the target sequence, within which a STOP-codon has been artificially engineered. This design ensures that only evolving cgRNA can be used for templating homology-directed repair of the targeted gene and restoring its function (HIS3).
CGTTGGTAGATACGTTGTTGACACTTCTAAATAAGCGAATTTCTTATGATTTATGATTTTTATTATTAAATAAGTTATAAAAAAAATAAGTGTATACAAATTTTAAAGTGACTCTTAGGTTTTAAAACGAAAATTCTTATTCTTGAGTAACTCTTTCCTGTAGGTCAGGTTGCTTTCTCAGGTATAGCATGAGGTCGCTCGTTAATTAAGAGCTCGAATGCagggaacaaaagctggagctTAATACGACTCACTATAGGGAGACTACATAAGAACACCTTTGGTGGAGGGAACATCGTTGGTACCATTGGGCGAGGTGGCTTCTCTTATGGCAACCGCAAGAGCCTTGAACGCACTCTCACTACGGTGATGATCATTCTTGCCTCGCAGACAATCAACGTGGAGGGTAATTCTGCTAGCCTCTGCAAAGCTTTCAAGAAAATGCGGGATCATCTCGCAAGAGAGATCTCCTACTTTCTCCCTTTGCAAACCAAGTTCGACAACTGCGTACGGCCTGTTCGAAAGATCTACCACCGCTCTGGAAAGTGCCTCATCCAAAGGCGCAAATCCTGATCCAAACCTTTTTACTCCACGCACGGCCCCTAGGGCCTCTTTAAAAGCTTGACCGAGAGCAATCCCGCAGTCTTCAGTGGTGTGATGGTCGTCTATGTGTAAGTCACCAATGCACTCAACGATTAGCGACCAGCCGGAATGCTAGGGTATGTTAATATGGACTAAAGGAGGCTTTTCTGCAGGTCGACTCTAGAGGATCCCCGGGTACCGAGCTCGAATTTTTACTAACAAATGGTATTATTTATAACAGCCAGAGCATGTATCATATGGTCCAGAAACCCTATACCTGTGTGGACGTTAATCACTTGCGATTGTGTGGCCTGTTCTGCTACTGCTTCTGCCTCTTTTTCTGGGAAGATCGAGTGCTCTATCGCTAGGGGACCACCCTTTAAAGAGATCGCAATCTGAATCTTGGTTTCATTTGTAATACGCTTTACTAGGGCTTTCTGCTCTGTTGTTAGTAAAAATTCCTCGAGTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGGTGCCCTAGCATAACCCCGCGGGGCCTCTTCGGGGGTCTCGCGGGGTTTTTTGCTGAAAGAAGCTTCAAATAAAACGAAAGGCTCAGTCGAAAGACTGGGCCTTTCGTTTTATCTGTTGTTTGTCGCTGCGGCCGCACTCGAGCTCGACCTCGACCTCCACTGAGATCCGGCTGCTAACAAAGCCCGAAAGGAAGCTGAGTTGGCTGCTGCCACCGCTGAGCAATAACTAGCATAACCCCTTGGGGCCTCTAAACGGGTCTTGAGGGGTTTTTTGCTGAAAG
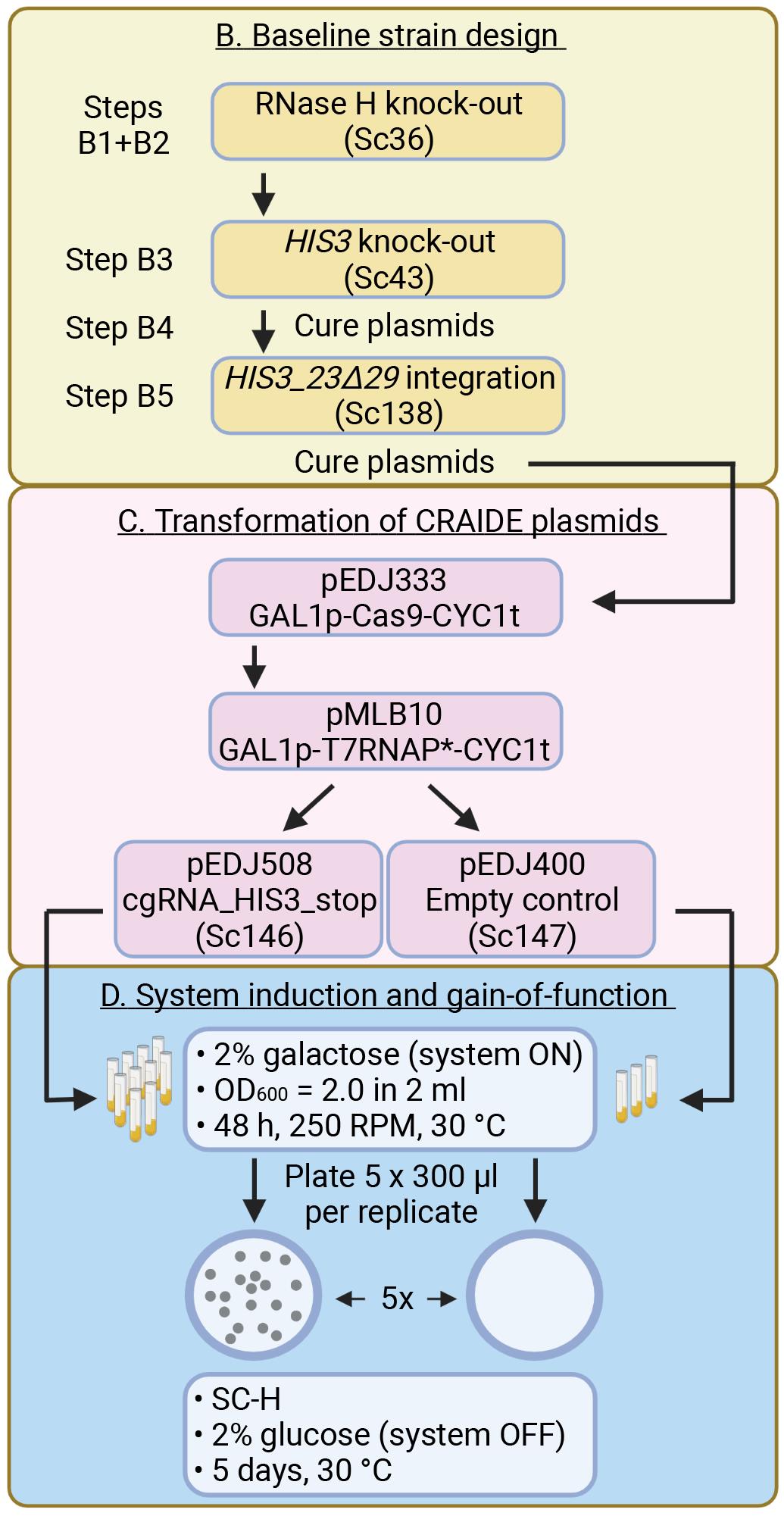
Figure 1. CRAIDE flow chart referring to main text illustrating B. Baseline strain design, C. Transformation of CRAIDE plasmids, and D. System induction and gain-of-function. Individual steps 1-5 are shown for panel B, and resulting strains are shown in parentheses in panels B and C. Plasmid expression cassettes are described for each plasmid in panel C with T7RNAP* referring to the T7RNAP(F11L;T613A) variant. Plasmids pEDJ333 and pMLB10 are inducible with galactose, and pEDJ508 is transcribed by T7RNAP(F11L;T613A) for introducing random mutations into the resulting cgRNA_HIS3_stop transcript. cgRNA_HIS3_stop does not translate into a functional protein, and mutagenesis of the built-in stop-codon is necessary to functionally repair the Cas9-targeted HIS3_23Δ29-XII-5 expression cassette residing in the genome of Sc138. Panel D highlights relevant conditions in liquid cultures (indicated with vials) for inducing CRAIDE and for plate-based scoring of HIS3 gain-of-function mutants after a 48 h induction in strains expressing cgRNA_HIS3_stop (Sc146) or an empty control plasmid (Sc147).Baseline strain design
Cas9 transformation for strain engineering
Strains are overviewed in Table 3. First, a single colony of catalog strain CEN.PK2-1C is grown overnight in 0.5 mL of YPD in a 14 mL culture tube at 250 rpm and 30°C, and diluted 10-fold in fresh YPD the next morning. After 4 h of incubation under the same conditions, 100 μL of the culture is transferred to a 1.5 mL Eppendorf tube and pelleted at 13,000 × g for 30 s by centrifugation at room temperature. The supernatant is discarded by pipetting. To the cell pellet, add 1 μg of a centromeric plasmid constitutively expressing Cas9 with LEU2 as its marker (pEDJ391), 10 μL of ssDNA (boiled for 5 min), 36 μL of lithium acetate (1 M), 74 μL of MilliQ water minus the volume of added plasmid, and 240 μL PEG 3350 (50%). Resuspend the pellet in the transformation mix by pipetting up and down and then vortex it for a few seconds until a homogenous suspension occurs. Incubate the transformation mix at 42°C for 40 min and then centrifuge it at 13,000 × g for 30 s before removing the supernatant by pipetting. Resuspend the pellet in 100 μL of MilliQ water and plate it with glass beads on synthetic complete dropout medium without L-leucine (SC-L). Dry the plate with the lid off in a safety cabinet, before incubating it for 2 days in a stationary 30°C incubator, to obtain strain Sc35.
RNase H deletions
Inoculate a single colony of strain Sc35 in 0.5 mL of SC-L in a 14 mL culture tube and incubated overnight at 250 rpm and 30°C. The next morning, add 4.5 mL of fresh SC-L to the overnight culture and incubate for another 4 h before transformation. The transformation was done as before, but this time with 1 nmol each of two double-stranded 90-mer oligos targeting RNH1 and RNH201 for knock-out (45 bases homology immediately up- and downstream of the open reading frames) and 1 μg of gRNA plasmid pEDJ8 (2× gRNA plasmid targeting RNH1 and RNH201 with URA3 selection marker). Plate the transformed pellet on SC-LU and incubate for 3 days at 30°C, to obtain strain Sc36. In the resulting colonies, colony PCR (cPCR) with oligos interspaced by 500-1,000 bases following genetic deletion verified knock-out of RNH1 and RNH201. Perform cPCR by picking 12 different colonies with toothpicks and resuspending each colony in two PCR tubes, one for testing rnh1 and one for rnh201, with 8 μL of MilliQ water and 1 μL of each primer per knock-out. Heat the suspension in a Thermal Cycler at 98°C for 15 min and add 10 μL of OneTaq 2× MM to a final volume of 20 μL in each tube. Return the reactions to the Thermal Cycler and run the following program:
98°C for 30 s
40× (98°C for 30 s; 50°C for 20 s; 68°C for 1 min/kb)
10°C forever
Load the final reactions on a 1% agarose gel. The gel is made by dissolving 4 g of agarose in 400 mL of 1× TAE buffer by heating, and cast in a Sub-Cell GT UV-Transparent Mini-Gel tray with 20 μL RedSafe and Fixed-Height Combs in a Mini-Gel caster. Run the loaded gel with 10 μL of GeneRuler 1 kb DNA ladder at 90 V for 30 min, in a DNA electrophoresis chamber. The power is supplied from a PowerPac Basic Power Supply, and gel imaging is done with a Molecular Imager Gel Doc.
Note: Double knock-outs were infrequent using this approach, and sequentially knocking out RNH1 and RNH201 can be used as an alternate strategy. Please also note that rnh1 rnh201 double knock-out strains require an incubation time approximately 50% longer compared to wild-type.
HIS3 deletion
To completely remove the open reading frame of native HIS3, to avoid genetic crossover during downstream analyses, a single colony from strain Sc36 is prepared for transformation in SC-L, as before. Transform 1 μg of gRNA plasmid pEDJ332 (contains nourseothricin resistance marker) with 1 nmol of a double-stranded 90-mer oligo, designed as before to knock out HIS3. After heat shock at 42°C, resuspend the pellet in 500 μL of YPD and incubate it at 800 rpm and 30°C for 2 h. Centrifuge the culture at 13,000 × g for 30 s, discard the supernatant, resuspend the pellet in 100 μL of sterile MilliQ water, and then plate it out on SC-L containing nourseothricin. After 3 days at 30°C, screen the resulting colonies by cPCR, to verify the removal of the HIS3 open reading frame, resulting in strain Sc43.
Strain curing (plasmid removal)
Before proceeding to the next engineering step, the Sc43 strain must be cured for all other plasmids than pEDJ391, which contains Cas9. Inoculate a single colony of strain Sc43 in 1 mL of SC-L and incubate it over-night at 250 rpm and 30°C. Dilute 10 μL of the overnight culture in 990 μL of sterile MiiliQ water, from which another 10 μL is diluted into 990 μL of sterile MiiliQ water. Repeat this dilution strategy again, plate 100 μL from all dilutions on SC-L, and incubate this for 3 days at 30°C. From each plate, perform three replica-plates to selective plates: SC+nourseothricin, SC-U, and SC-H. Replica-plating was achieved on a replication block with Whatman paper. Pick a single colony that did not grow on any of the three replica plates from the original plate following another overnight incubation of all plates at 30°C.
Dysfunctional his3 design integration (HIS3_23Δ29-XII-5)
Inoculate the cured Sc43 in 0.5 mL of SC-L and allow to grow overnight at 250 rpm and 30°C. Prepare the strain for transformation as before. In the meantime, linearize pEDJ506, which contains HIS3_23Δ29-XII-5, by restriction digest with NotI by mixing 3 μg of pEDJ506, with 2 μL of FastDigest buffer, 2 μL of NotI (20 U), and MiiliQ water to a total volume of 20 μL. Incubate the digest at 37°C in a stationary incubator for 4 h, prior to column purification with a NucleoSpin Gel and PCR Clean-up kit. Add 130 μL of NTI buffer to the digest and transfer it to a spin column. Discard the flow-through following centrifugation at 13,000 × g for 30 s. Add 700 μL of NT3 buffer to the spin column and repeat the centrifugation. Discard the flow-through and centrifuge the spin column at 13,000 × g for 1 min to dry the membrane. Add 60 μL of sterile MiiliQ water to the spin column, transfer this to a sterile 1.5 mL Eppendorf tube, and centrifuge at 13,000 × g for 30 s. Use the entire eluate for co-transformation with 1 μg of gRNA plasmid pCfB3050, which contains the nourseothricin resistance marker, using the same protocol as before. Resuspend the transformed pellet in 500 μL of YPD and incubate it at 800 rpm and 30°C for 2 h. Centrifuge the culture at 13,000 × g for 30 s, discard the supernatant, resuspend the pellet in 100 μL of sterile MiiliQ water, and then plate it out on SC-L containing nourseothricin. Screen the resulting transformants after 3 days incubation at 30°C for integration by cPCR, resulting in strain Sc138. Cure Sc138 essentially as before, but pre-grow it in YPD instead of SC-L, to completely eliminate all plasmids before proceeding to the introduction of CRAIDE plasmids.
Transformation of CRAIDE plasmids into engineered yeast
Inoculate the Sc138 cured strain in 0.5 mL of YPD and prepare it for transformation as before. Sequentially, transform 1.5 μg of plasmid pEDJ333 (GAL1p-Cas9-CYC1t, LEU2) with Cas9, and 1.5 μg of plasmid pMLB10 (GAL1p-T7RNAP(F11L;T613A)-CYC1t, TRP1) with error-prone T7 RNA polymerase, as before. After 3 days at 30°C on SC-LW, inoculate a single transformant in 0.5 mL of SC-LW and prepare it for transformation, as before. Transform 1.5 μg of pEDJ508 (cgRNA_HIS3_stop, URA3) or pEDJ400 (control, URA3), and incubate it on SC-LWU as before, to obtain strains Sc146 and Sc147, respectively.
Note: Cas9 and error-prone T7 RNA polymerase are both under galactose-inducible control from the GAL1 promoter.
System induction and screen for gain-of-function mutants (HIS3+)
Workflow preparations
Prepare at least thirteen 14 mL cultivation tubes, to contain 5 mL of SC-LWU with 2% glucose.
Strain inoculations
Use an inoculation loop to inoculate each cultivation tube with a single colony from the transformation plates. Inoculate at least 10 colonies for strain Sc146, and at least three colonies for control strain Sc147. Incubate overnight (20 h) at 250 rpm and 30°C (system OFF).
System induction
Pellet overnight cultures by centrifugation (2,000 rpm for 5 min), discard the supernatant, and wash once in 5 mL of sterile MiiliQ water. Resuspend pellets in 2 mL of SC-LWU with 2% galactose (system ON) and measure OD600 by spectrophotometry on 1 mL of culture per cuvette. Adjust to OD600 = 2.0 in 2 mL total volume, using SC-LWU with 2% galactose in new cultivation tubes. Incubate cultivation tubes for 48 h at 250 rpm and 30°C.
Screen for gain-of-function mutants (plates)
Measure the final OD600 by spectrophotometry and plate 5 × 300 μL from each culture on five synthetic dropout plates without histidine (SC-H) containing 2% glucose (system OFF). In addition, dilute 10 μL of the cultures in 990 μL of MiiliQ water and, as before, plate a 10× dilution series with 3 × 100 μL per dilution on non-selective plates (SC-LWU). Incubate all plates in a stationary incubator at 30 °C for 5 days, and score the resulting colonies.
Note: The resulting colonies have acquired HIS3+ at the targeted locus and an equal the number of successful functional cgRNA-DNA repair events.
Screen for gain-of-function mutants and sequencing (liquid cultures)
Optional: Add 5 mL of SC-H with 2% glucose to cultivation tubes with the culture remnants (~190 μL per tube) and incubate at 250 rpm and 30°C for an additional 72 h. Transfer and harvest 500 μL from each culture tube in 1.5 mL Eppendorf tubes, by centrifugation and resuspension of pellets in 400 μL of 400 mM lithium acetate. Incubate at 70°C for 15 min, pellet the remnants by centrifugation, and transfer 300 μL of the supernatant to a fresh 1.5 mL Eppendorf tube. Add 30 μL of 3 M sodium acetate and 900 μL of 100% ethanol, and incubate at -80°C for 1 h. Spin down in a pre-cooled centrifuge (4°C) at full speed for 20 min, discard the supernatant, and wash once with 70% ethanol. Centrifuge at full speed for 5 min at room temperature, and discard the supernatant. Dry the pellets at 60°C for 5-10 min, and resuspend in 100 μL of MiiliQ water by gentle pipetting. Use 100-400 ng as template for PCR with OneTaq, 2× MM, and oligos: F_Seq (CAAGTTCTTAGATGCTTTC) and R_Seq (CTACATAAGAACACCTTTGGTG). Allow elongation to occur for 2 min, to amplify the targeted locus (HIS3+). Sanger sequencing of the amplification product can be done to verify mutagenesis in the region of interest.
Data analysis
Count colonies from step D4 above from (mutant) selective SC-H plates and from non-selective SC-LWU plates after 5 days of incubation at 30°C.
Divide the number of mutants by the number of viable cells per volume determined from non-selective plates, to obtain the mutation frequency.
Determine the number of undergone generations during system induction with galactose (48 h) from starting and final OD600 measurements, using the formula: LN(OD600)end/LN(OD600)start - 1.
Determine the mutation rate per-base by first dividing the mutation frequency by the number of generations, and then by the sequence space (three bases = the artificial STOP codon) that may allow for permissive mutations.
Recipes
200 mg/mL nourseothricin solution
Dissolve 5 g nourseothricin in 25 mL of MiiliQ water (200 mg/mL) and sterile filter the solution with a 0.2 μm single use filter unit and a 50 mL syringe.
20% (w/v) glucose and galactose solutions
Dissolve 20 g of glucose or galactose in 80 mL of MiiliQ water and then brought to 100 mL total volume. Autoclave the final solution at 121°C for 15 min.
Synthetic complete drop-out media
Yeast synthetic drop-out medium supplements with appropriate drop-outs are weighed as follows:
Without leucine: 1.62 g/L
Without histidine: 1.92 g/L
Without uracil, leucine and tryptophan: 1.46 g/L
Bring the synthetic drop-outs to solution in 880 mL of MiiliQ water with i) 6.7 g/L yeast nitrogen base without amino acids or (standard media), ii) 1.7 g/L yeast nitrogen base without amino acids and ammonium sulfate, and iii) 1.1 g/L L-glutamic acid monosodium salt monohydrate (for media containing nourseothricin).
Adjust pH to 5.6 and then the volume to 900 mL with MiiliQ water. Add 20 g agar (for plates) before sterilization at 121°C for 15 min in an autoclave.
After heat treatment, let the media cool to ~60°C and add 100 mL of 20% (w/v) glucose or 20% (w/v) galactose and mix by stirring for 20 s. Add also 0.5 mL of 200 mg/mL nourseothricin solution for selective plates from point ii) above before stirring.
Pour plates in a safety cabinet and let the plates solidify for ~30 min.
Acknowledgments
This work was financially supported by the Novo Nordisk Foundation (NNF20CC0035580), and the Villum Foundation (grant #28309). This protocol has been adapted from Jensen et al.( 2021).
Competing interests
The authors declare there are no potential conflicts of interest.
References
- Badran, A. H. and Liu, D. R. (2015). Development of potent in vivo mutagenesis plasmids with broad mutational spectra. Nat Commun 6: 8425.
- Halperin, S. O., Tou, C. J., Wong, E. B., Modavi, C., Schaffer, D. V. and Dueber, J. E. (2018). CRISPR-guided DNA polymerases enable diversification of all nucleotides in a tunable window. Nature 560(7717): 248-252.
- Jensen, E. D., Laloux, M., Lehka, B. J., Pedersen, L. E., Jakociunas, T., Jensen, M. K. and Keasling, J. D. (2021). A synthetic RNA-mediated evolution system in yeast. Nucleic Acids Res 49(15): e88.
- Keskin, H., Shen, Y., Huang, F., Patel, M., Yang, T., Ashley, K., Mazin, A. V. and Storici, F. (2014). Transcript-RNA-templated DNA recombination and repair. Nature 515(7527): 436-439.
- Keskin, H., Meers, C. and Storici, F. (2016). Transcript RNA supports precise repair of its own DNA gene. RNA Biol 13(2): 157-165.
- Morris, K. V., (2015). The theory of RNA-mediated gene evolution. Epigenetics 10(1): 1-5.
- Moore, C. L., Papa, L. J. and Shoulders, M. D. (2018). A Processive Protein Chimera Introduces Mutations across Defined DNA Regions In Vivo. J Am Chem Soc 140(37): 11560-11564.
- Ravikumar, A., Arrieta, A. and Liu, C. C. (2014). An orthogonal DNA replication system in yeast. Nat Chem Biol 10(3): 175-177.
Article Information
Copyright
© 2022 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Jensen, E. D. and Jensen, M. K. (2022). RNA-mediated in vivo Directed Evolution in Yeast. Bio-protocol 12(5): e4346. DOI: 10.21769/BioProtoc.4346.
Category
Microbiology > Microbial genetics > CRISPR-Cas9
Cell Biology > Cell engineering > CRISPR-cas9
Molecular Biology > DNA > DNA damage and repair
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.