- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

An in vitro Assay of mRNA 3’ end Using the E. coli Cell-free Expression System
Published: Vol 12, Iss 4, Feb 20, 2022 DOI: 10.21769/BioProtoc.4333 Views: 3909
Reviewed by: Emilia KrypotouSrujana Samhita YadavalliAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Mar 2019
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
At the end of about 80% of the operon in Escherichia coli, translation termination decouples transcription, leading to Rho-dependent transcription termination (RDT). However, no in vitro or in vivo assay system has proven to be good enough to see the 3’ end of the mRNA generated by RDT. Here, we present a cell-free assay system that could provide detailed information on the 3’ end of a transcript RNA generated by RDT. Our protocol shows how to extract transcript RNA generated by transcription reactions from a cell-free extract, followed by an RNA oligomer ligation to the 3’ end of a transcript RNA of interest. The 3' end of the RNA is amplified using RT-PCR. Its genetic location can be determined using a gene-specific primer extension reaction. The 3’ ends of mRNA can be visualized and quantified by polyacrylamide gel electrophoresis. One significant advantage of a cell-free assay system is that factors involved in the generation of the 3' end, such as proteins and sRNA, can be directly assayed by exogenously adding factor(s) to the reaction.
Graphic abstract:
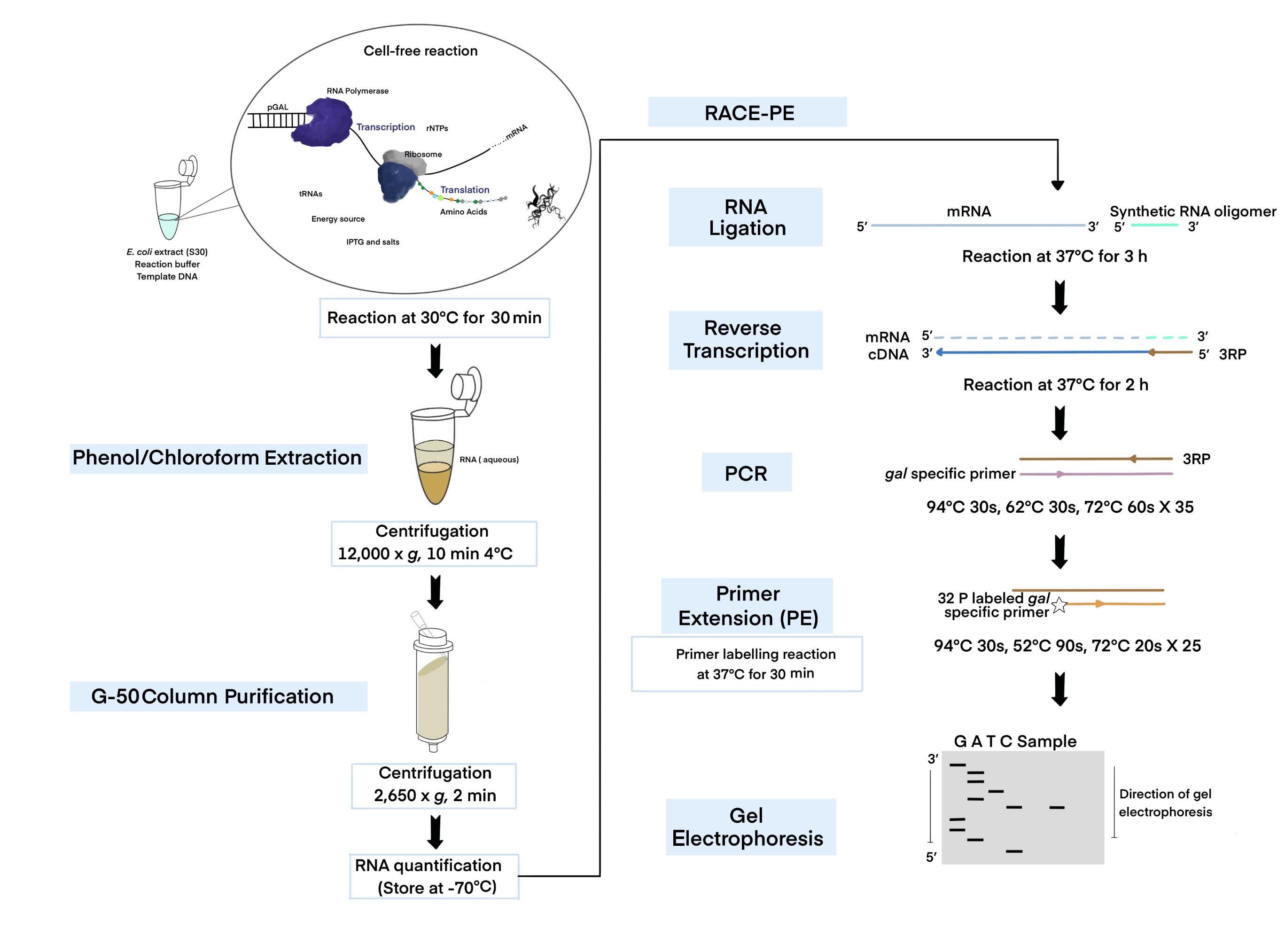
An illustration of the experimental methodology.
Background
In bacteria, there are two types of transcription termination: 1) intrinsic or Rho-independent termination (RIT), caused by the terminator hairpin formed on the transcript at the end of the last transcription unit, and 2) Rho-dependent termination (RDT) (Ray-Soni et al., 2016). In prokaryotes, transcription termination and RNA processing are required for mRNA 3' end production (Altman and Robertson, 1973; Zhao et al., 1999; Nudler and Gottesman, 2002). As a result, understanding the mechanisms for producing transcription termination requires an examination of the 3' end of an mRNA regarding its position and relative amount.
The 3’ end of a specific transcript could be identified and located using the 3’ random amplification of cDNA ends (RACE) assay (Sambrook and Russell, 2006; Wang, X. et al., 2018). This assay is based on the ligation of an RNA oligomer to the 3’ end of an RNA transcript, which is amplified using RT-PCR (Lee et al., 2008). Using gene-specific primer extension (PE) and DNA sequencing gel electrophoresis, the 3' end position is determined (Lee et al., 2008). The labeled primer embedded in PCR products is extended by a denaturant PAGE analysis, enabling the visualization and quantification of the 3' end. Using the 3’ RACE-PE method, the gal operon mRNA 3′ end has been previously identified in vivo and studied (Lee et al., 2008; Wang, X. et al., 2014, 2015, 2019; Jeon et al., 2021). The Escherichia coli gal operon has four structural genes, galE, galT, galK, and galM, which encode enzymes for catabolic and anabolic metabolism of D-galactose (Holden et al., 2003). Transcription initiated from the P1 and P2 promoters (Adhya and Miller, 1979) terminate at the terminator hairpin of the last structural gene galM (Wang, X. et al., 2019), generating the 3’ end of galETKM mRNA at 4,313 (from the transcription initiation site of the P1 promoter, +1).
In E. coli, transcription is tightly coupled to translation (Burmann et al., 2010; Chakrabarti and Gorini, 1975; Proshkin et al., 2010), mediating the translation of the nascent transcript (Demo et al., 2017; Fan et al., 2017; Kohler et al., 2017; Wang, C. et al., 2020; Webster et al., 2020). The RNA polymerase often transcribes open reading frames (ORFs) without a linked ribosome, resulting in ribosome-free mRNA (Chen and Fredrick, 2018; Zhu et al., 2019; Jeon et al., 2020). To understand the mechanism of transcription termination for the above-mentioned transcription states, we need specifically targeted tools and methodology. Cell-free expression or in vitro transcription-translation techniques can be utilized for studying functional protein synthesis without requiring a living cell (Silverman et al., 2020). The coupled transcription-translation system provides a time-saving alternative to conventional methods, by coupling both processes in a microcentrifuge tube (Figure 1). Unlike other in vitro approaches based on cell culture, the in vitro transcription-translation system does not require gene transfection, bacterial cell culture, or extensive purification (Silverman et al., 2019, 2020). Although various cell-free extracts have recently been created, E. coli extracts remain the most practical and best choice for protein production due to their high yields (Silverman et al., 2020). For the first time, we report the extraction of RNA from a cell-free system to investigate the mechanism of 3’ end generation in an in vitro system, where transcription is coupled with translation. The in vitro transcription-translation system is comprised of two basic components: (1) the template DNA containing RNA polymerase (RNAP) promoters, and (2) a reaction buffer supplemented with the necessary components for the transcription and translation processes (Kim et al., 2007).
Cell extracts contain all the essential molecules such as RNAP, ribosomes, tRNAs, amino acids, several other cofactors, and a source of energy for transcription and translation (Kim et al., 2007). Any template DNA with an E. coli holoenzyme or T7 RNA polymerase promoter, ribosome binding site (rbs), and terminator (Takahashi et al., 2015) can be used to study protein-protein, protein-nucleic acid interactions (Wong and Blobel, 2008; Tando et al., 2010; Muratore et al., 2012), or transcription termination (Ray-Soni et al., 2016). For instance, to examine Rho-dependent termination, cloning the transcription terminator rho gene in a template with the T7 promoter/terminator and an rbs can be employed. Although in vitro expression is impractical for extensive commercial production, it can support RNA/protein synthesis as a coupled reaction (transcription and translation), making it significantly useful and manageable for many research applications (Tinafar et al., 2019; Silverman et al., 2020).
So far, numerous methodologies have been described for cell-free protein expression (Cole et al., 2020). Here, we present an optimized protocol that combines a cell-free extract system and a 3’ RACE assay, to better understand mRNA generating mechanisms (Jeon et al., 2021). We investigated how transcription termination could be studied in a naturally occurring dual terminator system influenced by transcription-translation coupling. We also show how RNA extracted from a cell-free reaction contains pre-mRNAs previously unseen before processing and after RDT. We anticipate that our in vitro-based experimental approach will be relevant in future studies of gene regulation.
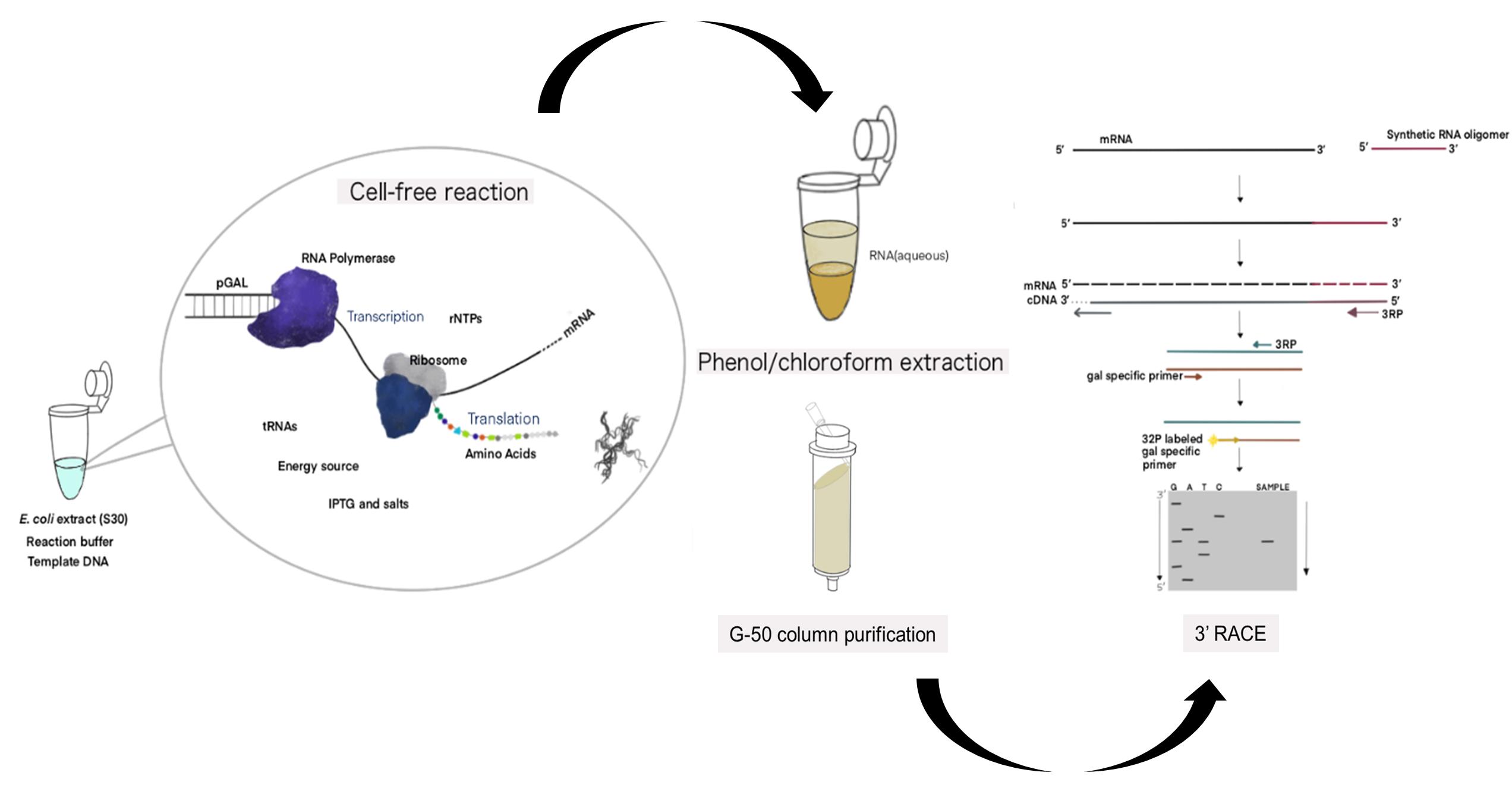
Figure 1. Schematic of the cell-free expression system (CFS) followed by the 3' RACE-PE (CFS-3’RACE-PE) assay. Target RNA is obtained from the template DNA (pGal) using the E. coli extract containing RNA polymerase, ribosome, rNTPs, tRNAs, amino acids, energy source, other components supplied, and a reaction buffer via a coupled transcription-translation reaction, followed by phenol-chloroform extraction and G-50 column purification to purify RNA for use in the RACE assay.
Materials and Reagents
Materials
Pipette (STARLAB International, catalog numbers: S7100-0510, S7100-2200, and S7110-1000)
Pipette tips (Starlabs., S Korea and Sorenson Bioscience, Inc., catalog numbers: S1120-3810, S1122-1830, 2937T, 17370-X)
1.5 mL microcentrifuge tubes (SPL Life Sciences, catalog number: SPL-060015)
1.5 mL microcentrifuge tubes, sterilized (Bio-Fact, catalog number: EMT-1530)
Plasmid DNA mini-prep kit (Bio-Medic, catalog number: BM-K110B)
PCR tubes (Corning Incorporated, catalog number: PCR-02-C)
PCR purification kit (Qiagen, catalog number: 28106)
Whatman 3MM paper (GE Healthcare, catalog number: 3017-915)
Kodak CL-Xposure Film (Thermo Scientific, catalog number: 34090)
Micro-flex Gloves (Ansell, catalog number: MF-300-L)
RNaseZap (RNase Decontamination Solution) (Thermo Scientific, catalog number: AM9780)
E. coli extract (S30) (Bioneer, catalog number: K-7250)
Reaction buffer for cell-free reaction (Bioneer, catalog number: K-7250)
DEPC water (Bioneer, catalog number: K-7250)
RNase-free water (Thermo Scientific, catalog number: 7732-18-5)
Galactose (Merck, catalog number: G5388)
DNase I (Thermo Fisher Scientific, Invitrogen, catalog number: AM2222)
T4 RNA ligase (Ambion, catalog number: AM2141)
RNasin Ribonuclease Inhibitors (Promega, catalog number: N2111)
PCI (Phenol:Chloroform:Isoamyl Alcohol) (Merck, catalog number: 77617)
G-50 column (Sephadex) (GE Healthcare, catalog number: 27-5330-01)
Omniscript Reverse Transcriptase (Qiagen, catalog number: 205111)
HotStarTaq Plus DNA Polymerase (Qiagen, catalog number: 203603)
dNTP mix (10 mM each) (ThermoFisher, catalog number: R0191)
T4 Polynucleotide Kinase (NEB, catalog number: M0201L)
T4 Polynucleotide Kinase 10× buffer (NEB, catalog number: M0201L)
ATP, [γ-32P]-6000Ci/mmol (PerkinElmer, catalog number: NEG002Z250UC)
5× TBE (Bioneer, catalog number: C-9002)
Urea (Merck, catalog number: U5128)
Acrylamide (Merck, catalog number: V900845)
Bis-acrylamide (Merck, catalog number: V900301)
Ammonium persulfate (Merck, catalog number: 248614)
TEMED (Merck, catalog number: T9281)
Formamide (Merck, catalog number: F9037)
Xylene cyanol (Sigma, catalog number: X4126)
Bromphenol blue (Sigma, catalog number: B0126)
Sigmacote (Sigma, catalog number: SL2)
5× Developer (Vivid, catalog number: 0514_00004)
5× Developer & Fixer (Vivid, catalog number: 0514_00003)
8% sequencing solution (see Recipes)
8% Urea-PAGE gel (see Recipes)
2× stop buffer (see Recipes)
Plasmids and primers (Table 1)
pGal plasmid (contains gal operon, Figure 3A) (Wang, X. et al., 2014)
pRho plasmid (contains rho gene under the T7 promotor control) (Jeon et al., 2021)
pRNaseIII (contains rnc gene under the T7 promoter control) (Jeon et al., 2021)
Table 1. Primers used in this study.
Primer name Primer sequence (5’-3’) Use Synthetic RNA UUCACUGUUCUUAGCGGCCGCAUGCUC RNA Oligomer ligation for 3′ RACE assay 3RP-R AGCATGCGGCCGCTAAGAAC RT and PCR for 3′ RACE assay M3-F TCCGCACGACGGCCTGAAAT
PCR for 3′ RACE assayT3-F ACGGTAGCCGTACCGTTGTC M4240-R CGAAGAGTATTCCAGCCTG
PE for 3’ RACE assayT8-F ATCCATTTTCGCGAATCCGGA
Equipment
30/37°C stationary incubator (SNT, model: MCL-20A)
Vortex Genie-2 (Scientific Industries, model: G560E)
37°C Heat block (FINEPCR, model: ALB64)
Centrifuge (4°C) (Hanil Scientific Inc., model: SU-R17)
NanoDrop 1000 (Thermo Fisher, model: ND-1000)
Thermo cycler machine (Bio-Rad, model: T100, catalog number: 1861096)
Gel sequencing apparatus (Labrepco, model: S2, catalog number: 21105036)
Automatic pipette (Thermo Scientific, USA, model: S1 pipette filler, catalog number: 9501)
Cassette (X-ray film) (Duksan (DS) Lab, model: L-07019-001)
Power supply unit (Bio-Rad, USA, model: PowerPac 3000, catalog number: 165-5057)
Procedure
Cell-free extract reaction (Time for Completion: 1 h)
Set-up a cell-free reaction in a 60 µL reaction volume with 10 nM of the DNA template, which should contain a promoter, the rbs followed by the inserted gene of interest, and the transcription terminator [a pGal plasmid is used for the reaction in Figure 3A (Wang, X. et al., 2014)], 25 µL of the reaction buffer (Bioneer), 14 µL of E. coli extract (S30) (Bioneer), 0.5% (w/v) galactose, and DEPC water up to the final volume.
Note: Dissolve 100 g galactose in 500 mL RNase-free water to make a 20% (w/v) galactose stock solution. Stir/mix thoroughly at room temperature to dissolve, then filter sterilize the solution. A final galactose concentration of 0.5% (w/v) is used for the reaction.
Incubate at 30°C in a stationary incubator for 30 min.
Note: We recommend setting up the cell-free reaction in a stationary incubator.
Add an equivalent amount of PCI (Phenol:Chloroform:Isoamyl Alcohol) and centrifuge at 12,000 × g and 4°C for 10 min, to stop the reaction.
Note: At this stage, three distinctive phases can be seen. The upper aqueous phase will retain the total RNA, whereas the yellowish-interphase and the lower organic phase contain the protein.
Carefully use a pipette to take 25 µL of the upper aqueous phase, which contains the RNA, without disturbing the inter/organic phase.
Apply the sample to the upper side of the G-50 resin column and centrifuge at 2,650 × g for 2 min. The purified RNA is collected at the bottom of the microcentrifuge tube.
Note: To prepare the G-50 column, vortex the resin to resuspend and centrifuge at 2,650 × g for 1 min. Place the column in a new sterile 1.5 mL microcentrifuge tube.
Quantify the purified RNA (usually 1.0–1.2 µg/µL) and store it at -70°C (Pause step).
CRITICAL STEP: Use RNase-free filter tips to avoid RNase contamination.
RNA ligation (Time for Completion: 3.5 h)
Set-up the RNA ligation reaction in 1.5 mL sterile microcentrifuge tubes to a volume of 25 μL, comprised of 2.5 µL of 10× RNA ligase buffer, 1 µL of 100 nM RNA oligo (27 mer) (Table 1), 2.5 µg RNA, 0.5 µL of 40 U/µL RNasin, 1.5 μL of 3U DNase I, and 2 μL of T4 5 U/µL RNA Ligase.
Note: If there is any DNA carryover from the previous step, DNase I ensures full eradication.
Incubate at 37°C in a stationary incubator for 3 h.
Clean the subsequent ligation mix using a G-50 column by centrifugation at 2,650 × g for 2 min.
Note: To prepare the G-50 column, vortex the resin to resuspend and centrifuge at 2,650 × g for 1 min. Place the column in a new sterile 1.5 mL microcentrifuge tube.
Reverse transcription (RT) (Time for Completion: 2.5 h)
Set-up a reverse transcription reaction in 1.5 mL sterile microcentrifuge tubes to a volume of 20 μL containing 2 µL of 10× RTase buffer, 1 µL of 25 µM 3RP-R primer (Table 1), 2 µL of 5 mM dNTP mix, 0.25 µL of 40 U/µL RNasin, 1 μL of 4 U/µL RTase, and 4 µg ligated RNA.
Incubate at 37°C in a stationary incubator for 2 h.
Note: cDNAs can be stored at -20°C until the next step (Pause step).
PCR (Time for Completion: 2 h)
Set-up a PCR reaction in 0.2 mL PCR tubes to a final volume of 50 μL, containing 5 µL of 10× PCR buffer, 1 µL of 10mM dNTP mix, 2.5 µL of 10 µM 3RP-R primer, 2.5 µL of 10 µM gene of interest (GOI) primer (Table 1), 0.5 µL of 5 U/µL HotStarTaq plus DNA polymerase, 2.5 µL of cDNA, and 36 µL of RNase-free water.
Move PCR tubes to a thermo-cycler machine. Pre-heat the block to 105°C.
Start the thermocycling process with the following parameters: initial denaturation at 94°C for 5 min, and 30 cycles of: denaturation at 94°C for 30 s, hybridization at 60°C for 30 s, and extension at 72°C for 60 s. Cool the samples down to 4°C.
Store the PCR tubes at -20°C (Pause step).
CRITICAL STEP: PCR purification ensures that the PCR reaction components are removed.
Primer end labeling (Time for Completion: 1.5 h)
Set-up a 20 µL reaction in 1.5 mL sterile microcentrifuge tubes as follows: 2 µL of 10× Kinase buffer, 2 µL of 10 µM gene-specific extension primer, 2 µL of [γ-32P] ATP, 1 µL of 10U/µL Kinase, and 13 µL of RNase-free water.
CRITICAL STEP: Use gloves and protective gear, as well as a plexiglass shield to avoid radiation.
Incubate at 37°C for 30 min in a heat block.
Note: The enzyme can be inactivated at 65°C for 10 min.
Dilute the reaction mixture with 30 µL of RNase-free water and purify the labeled primer using a G-50 column by centrifugation at 2,650 × g for 2 min. Store at 4°C (Pause step).
Primer extension (PE) (Time for Completion: 2 h)
Set-up a 20 µL PCR reaction in 0.2 mL PCR tubes, as follows: 2 µL of 10× PCR buffer, 0.3 µL of 10 mM dNTP mix, 0.75 µL of 5’ end-labeled gene-specific extension primer, 0.2 µL of 5 U/µL HotStarTaq plus DNA polymerase, 2 µL of DNA template, and 14.75 µL of RNase-free water.
CRITICAL STEP: Use gloves and protective gear, as well as a plexiglass shield to avoid radiation.
Run the thermocycling process with the following parameters: initial denaturation at 94°C for 5 min, and 25 cycles of: denaturation at 94°C for 30 s, hybridization at 52°C for 90 s, and extension at 72°C for 30 s. Cool the samples down to 4°C.
Note: Preheat the block to 105°C.
Store the PCR tubes at -20°C (pause step).
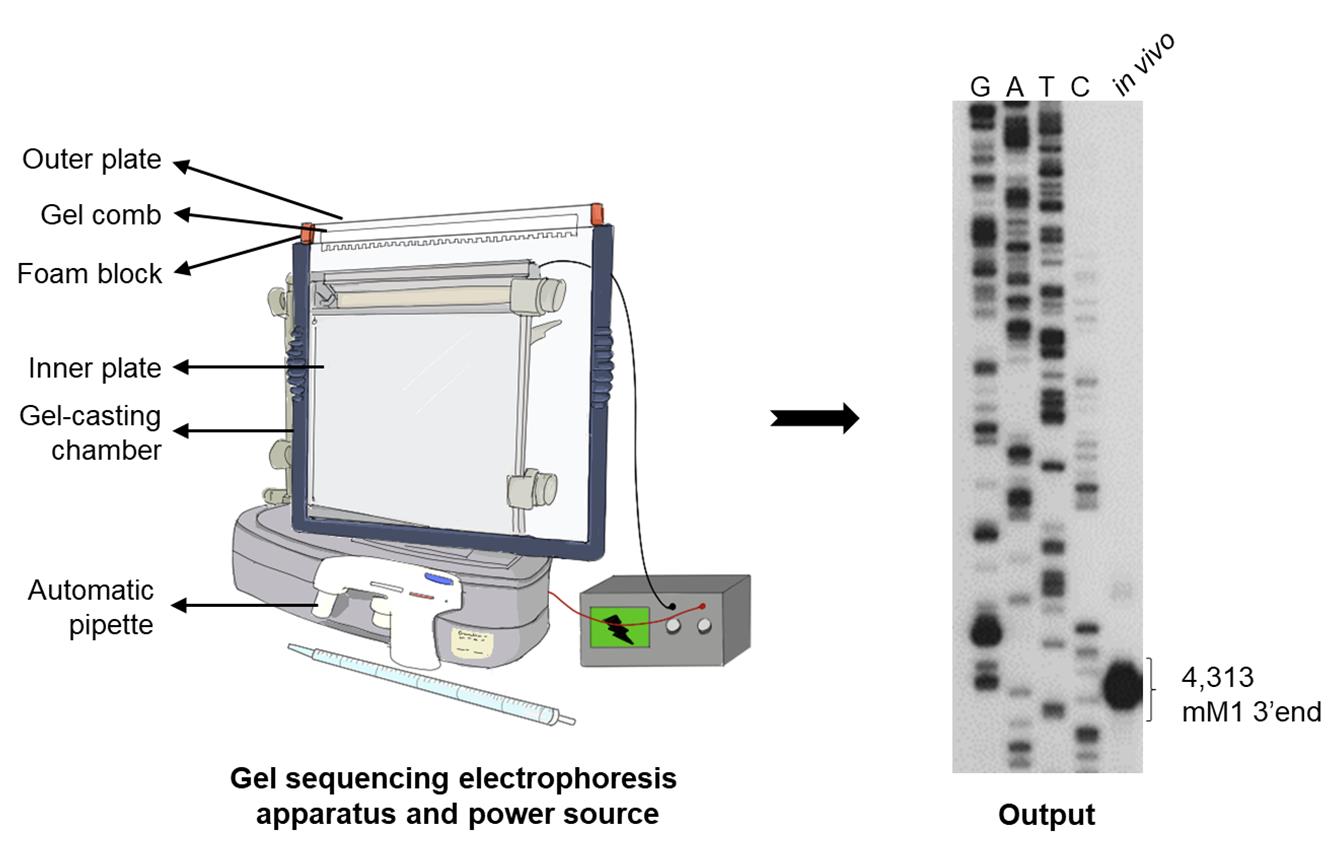
Figure 2. Schematic of the components of an assembled electrophoresis apparatus. The illustrated output of the 3′ RACE assay on gal transcripts generated in E. coli MG1655 wild-type (WT) cells showing the 3’ end of galETKM mRNA (previously called mM1) at 4,313 (the number represents the position of the nucleotide from the gal transcription initiation site); GATC: DNA sequencing ladder.
Gel electrophoresis and analysis (Time for Completion: 16 h)
Assemble spacers (0.4 mm), inner and outer glass plates (W × H: 31 × 38.5 cm) in the gel-casting chamber according to the manufacturer’s description (Labrepco) (Figure 2) (Video 1).
CRITICAL STEP: Coat the inner glass plate with a hydrophobic Sigmacote solution, to facilitate removing the gel from the outer glass plate after electrophoresis.
 Video 1. Gel-casting procedure.
Video 1. Gel-casting procedure.Prepare 50 mL of polyacrylamide solution from the 8% sequencing solution (see Materials and Reagents).
Add 300 µL of ammonium persulfate (10%) and 30 µL of TEMED to the polyacrylamide solution. Mix well by stirring.
Using an automated pipette, gently pipette the solution between glass plates, without introducing any air bubbles.
Allow 1 h after inserting the gel comb (4 × 0.4 mm, 14 cm), for the gel polymerization to occur.
Remove the polymerized gel from the gel-casting chamber and transfer it to the gel sequencing electrophoresis apparatus (Labrepco) (Figure 2).
Fill the bottom and upper buffer chambers with 1× TBE buffer (submerged by at least 2–3 cm).
Detach the comb from the gel and use a pipette to rinse the wells.
CRITICAL STEP: As urea gets seeped into the wells, pipette them thoroughly every 2 min.
Add 15 µL of 2× stop buffer (see Materials and Reagents) to the sample and denature at 95°C for 5 min.
Place the samples on ice for 2 min to cool.
Load 2.5 µL of the sample carefully, avoiding any air bubbles.
CRITICAL STEP: Always keep samples on ice.
Close the lid of the apparatus, attach the cords to a high-voltage power source, and run the gel at 60 W for 2–3 h.
Remove glass plates from the gel electrophoresis device by removing the clamps. Carefully disassemble the glass plates after they have cooled down completely.
Note: Check and wait for the glass plates to cool down.
Cut a piece of Whatman paper (3 mm) to the same size as the gel and carefully remove the gel after attaching the Whatman paper to the outer glass plate.
Cover the gel with saran wrap and seal it with scotch tape.
CRITICAL STEP: Check the radiation level often with a Geiger counter, focusing on the front of the protective screen and your gloves, in case of contamination.
Transfer the saran wrap-sealed gel to a gel cassette containing the Whatman paper, cover the gel with an X-ray film, and expose it at -80°C for 12–16 h.
Note: At -80°C, the film exposure period can be adjusted.
Scan the film once it has dried from development and fixing. ImageJ software can be used to calculate relative band intensities.
Data analysis
Analysis of DNA and RNA used in the CFS-3’RACE-PE assay
We present an efficient strategy for better understanding mRNA generating mechanisms and studying transcription termination procedures in the coupled and uncoupled transcription states, while limiting the experiment time to a minimum. Agarose gel electrophoresis was utilized to confirm the DNA template employed in the cell-free reaction (Figure 3A). The 23s and 16s rRNA bands of the RNA extracted after the cell-free reaction were visible on a denaturing gel (Figure 3B). The amount of protein synthesized after the reaction was sufficient to be detected with conventional Coomassie blue staining.
Data analysis of CFS-3’RACE-PE reaction
After the cell-free reaction only in the presence of pGal DNA template, the 3’ RACE produced no 3’ ends at 4,313 (Wang, X. et al., 2019) (Figure 3C, lane 2). However, the addition of galactose (0.5%) with different reaction times produced 3’ ends at 4,313 (Figure 3C, lanes 3–7), indicating that these bands were generated from the gal operon transcription, as shown in vivo (Figure 3C, lane 1).
Validation of CFS-3’RACE-PE
We also investigated the role of Spot 42 binding at the galT-galK junction in the formation of galET mRNA species, to validate the cell-free system (Jeon et al., 2021). The 3’ ends of galET mRNA are found at 2,125–2,129 (galET-short) and 2,164–2,167 (galET-long) downstream of the galT stop codon, respectively (Figure 3D, lane 1) (Wang, X. et al., 2015). Base-pairing of the two regions of Spot 42 with corresponding complementary regions of mRNA generated two different 3’ ends. The binding of Spot 42 resulted in a Rho-dependent termination (RDT) of galT transcription, resulting in the generation of galET mRNA (formerly called mT1) (Wang, X. et al., 2015).
Using the cell-free assay system, we investigated the role of Spot 42 and RNaseIII in RDT. In the presence of pGal DNA template and 0.5% galactose, the 3’ RACE assay produced weak 3’ ends (Figure 3D, lane 2). In the presence of plasmid pRho, where rho is controlled by the T7 promoter, the band at 2,184 increased five-fold. These findings indicate that the 2,184-band is due to RDT (Figure 3D, lane 3). In the cell-free reaction containing pGal DNA template and 0.5% galactose, we added 150 nM of Spot 42 RNA. The RNA with 3’ ends at 2,184 increased exponentially. A cluster of 3' ends at 2,125–2,129 also increased with Spot 42 RNA (Figure 3D, lane 4). These findings suggest that the 3’ end of the RDT terminated product at 2,184 is processed to the 3’ ends of galET-short, implying that Spot 42 can modulate RDT utilizing the endogenous Rho protein in the E. coli extract.
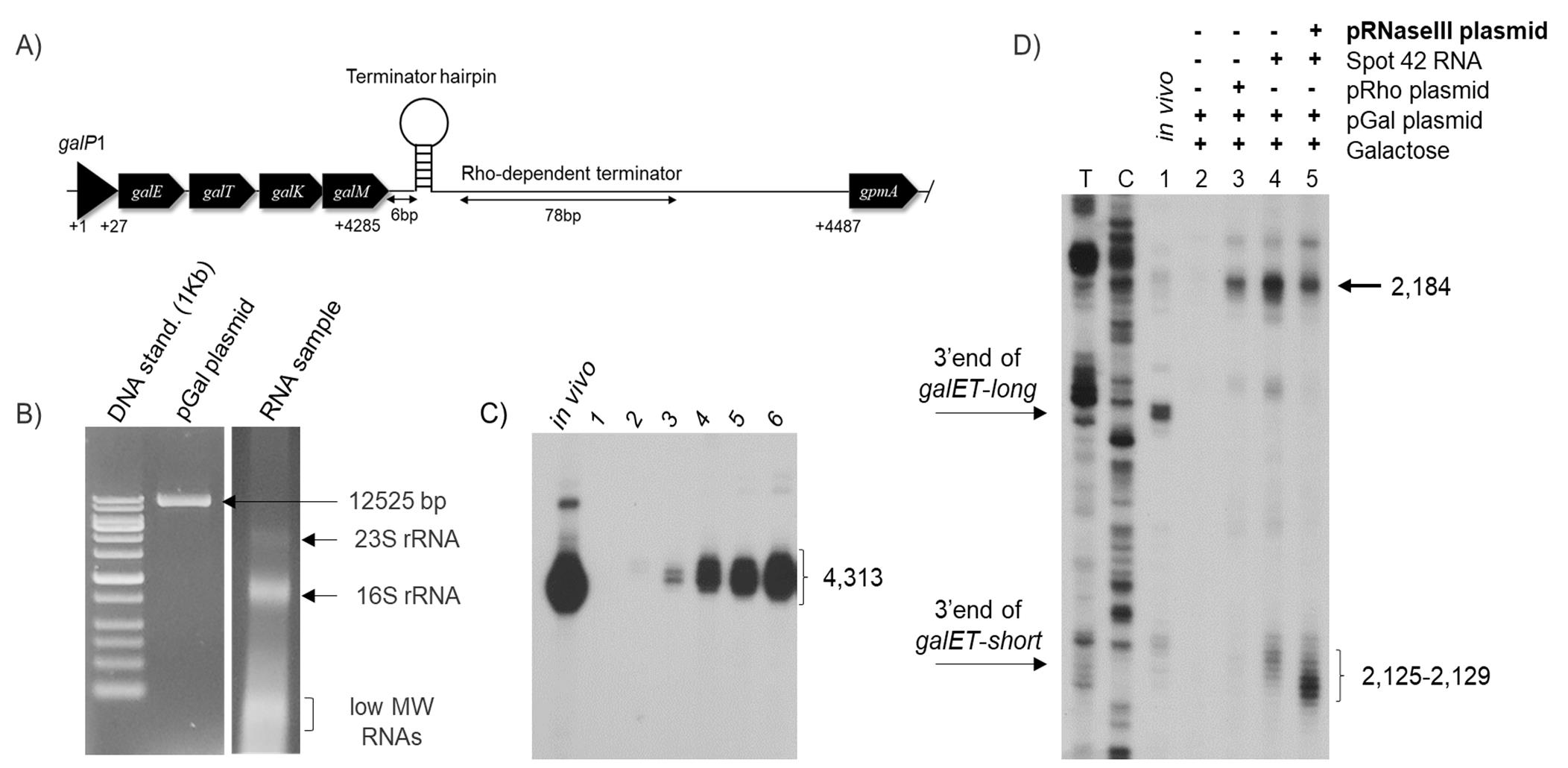
Figure 3. An overview of the cell-free reaction followed by the data output of the 3’ RACE assay. (A) A diagram of the pGal plasmid used as the template for this study. The transcription initiation site from the P1 promoter is marked as galP1. The galactose operon structural genes; galE, galT, galK, and galM followed by the two transcription terminations at the end of gal are shown. The Rho-independent termination signal (the terminator hairpin) is presented as a hairpin structure and the Rho-dependent termination signal (C-rich region) is depicted as a double-arrow. (B) Agarose gel electrophoresis of the pGal plasmid (12,525 bp) and RNA samples extracted from the cell-free reaction on a 1.2% agarose gel, showing 23S rRNA, 16S rRNA, and other low molecular weight RNAs. (C) The 3′ RACE assay on gal transcripts generated in E. coli MG1655 wild-type (WT) cells and in a cell-free reaction in the presence of pGal and 0.5% galactose. In vitro showing the 3’ end of galETKM mRNA (previously called mM1) at 4,313; lane 1: No DNA (Negative control); lane 2: DNA template pGal alone; lanes 3–6: pGal and 0.5% galactose with different reaction times; 30 min, 1 h, 2 h, and 3 h. (D) RNase III-mediated transcript cleavage using the cell-free system. 3′ RACE assay of the galET mRNA 3’ ends from MG1655 in vitro (lane 1) and the cell-free system in the presence of DNA template, pGal, and 0.5% galactose (lane 2); pGal, 0.5% galactose, and pRho plasmid (lane 3), where the gene for Rho (rho) is under control of the T7 promoter; pGal, 0.5% galactose, and Spot42 RNA (150nM) (lane 4) and pGal, 0.5% galactose, Spot42 RNA (150nM), and pRNaseIII plasmid (lane 5), where the gene for RNase III (rnc) is under control of the T7 promoter. 2,125–2,129: 3’ end of galET-short mRNA; 2,161–2,163: 3’ end galET-long mRNA. The numbers represent the position of the nucleotide from the gal transcription initiation site. TC: DNA sequencing ladder.Suspecting the involvement of an endoribonuclease in the RNA processing of the 3’ ends at 2,184 to 2,125–2,129, we added pRNaseIII plasmid (rnc gene controlled by the T7 promoter) into the cell-free reaction containing pGal DNA template, 0.5% galactose, and Spot 42 RNA. RNaseIII is an endoribonuclease known to cleave dsRNA (Robertson et al., 1968; Nashimoto and Uchida, 1985). In the presence of RNaseIII, the RNA with 3' ends at 2,184 was reduced to 50% (Figure 3D, lane 5). However, the galET-short bands at 2,121–2,129 increased three-fold compared to those in Figure 3D, lane 4.
These findings show that RDT is controlled by Spot 42 and that RDT of transcript 3’ end at 2,184 is processed to 2,121–2,129, which is cleaved by RNaseIII. The 3’ end at 2,184 is a terminated product completely dependent on Spot 42 and Rho for termination. Normally, this invisible ‘pre-mRNA’ species is not recorded in vitro, but can be seen in our optimized cell-free system. Our findings show that RNase III can cleave the transcript at 2,184, to generate galET-Short. The 3' end of 2,184 cannot be detected, since RNase III processing appears to occur rapidly in cells.
Notes
Radiation safety and guidelines:
Radiation awareness and safe radiation source handling training are required.
Set aside a space in the laboratory for studies with ATP, [γ-32P].
Always limit the amount of time you are exposed to radiation.
To avoid exposure, radioactive samples should be stored in lead-lined containers, and experimental wastes should be disposed of in appropriate containers.
When working with radioactive materials, always use gloves and protective gear, as well as a plexiglass shield to prevent radiation.
Check the radiation level often with a Geiger counter, placing it in front of the protective screen and your gloves, in case of contamination.
Recipes
8% sequencing solution
Reagent Final concentration Amount Acrylamide n/a 38 g Bis-acrylamide n/a 2 g Urea 8 M 250 g 5× TBE 1× 100 mL H2O n/a Up to 500 mL Total 500 mL 8% Urea-PAGE gel
Reagent Final concentration Amount 8% sequencing solution n/a 49.67 mL 10% (w/v) APS solution n/a 300 µL 99% TEMED n/a 30 µL Total 50 mL 2× Stop buffer
Reagent Final concentration Amount 99% Formamide n/a 9 mL EDTA (0.5 M, pH 8.0) 10 mM 200 µL 1% (w/v) Xylene cyanol 0.01 % 100 µL 1% (w/v) Bromophenol blue 0.01 % 100 µL Total 10 mL
Acknowledgments
This work was supported by a grant (2020R1A2C200633611 to H.M.L.) of the Basic Science Research Program of the National Research Foundation of Korea. This protocol was used in ‘sRNA-mediated regulation of gal mRNA in E. coli: Involvement of transcript cleavage by RNase E together with Rho-dependent transcription termination’ (Jeon et al., 2021).
Competing interests
The authors declare no competing interests.
References
- Adhya, S. and Miller, W. (1979). Modulation of the two promoters of the galactose operon of Escherichia coli. Nature 279(5713): 492-494.
- Altman, S. and Robertson, H. D. (1973). RNA precursor molecules and ribonucleases in E. coli. Mol Cell Biochem 1(1): 83-93.
- Burmann, B. M., Schweimer, K., Luo, X., Wahl, M. C., Stitt, B. L., Gottesman, M. E. and Rosch, P. (2010). A NusE:NusG complex links transcription and translation. Science 328(5977): 501-504.
- Chakrabarti, S. L. and Gorini, L. (1975). A link between streptomycin and rifampicin mutation. Proc Natl Acad Sci U S A 72(6): 2084-2087.
- Chen, M. and Fredrick, K. (2018). Measures of single- versus multiple-round translation argue against a mechanism to ensure coupling of transcription and translation. Proc Natl Acad Sci U S A 115(42): 10774-10779.
- Cole, S. D., Miklos, A. E., Chiao, A. C., Sun, Z. Z. and Lux, M. W. (2020). Methodologies for preparation of prokaryotic extracts for cell-free expression systems. Synth Syst Biotechnol 5(4): 252-267.
- Demo, G., Rasouly, A., Vasilyev, N., Svetlov, V., Loveland, A. B., Diaz-Avalos, R., Grigorieff, N., Nudler, E. and Korostelev, A. A. (2017). Structure of RNA polymerase bound to ribosomal 30S subunit. eLife 6: e28560.
- Fan, H., Conn, A. B., Williams, P. B., Diggs, S., Hahm, J., Gamper, H. B., Jr., Hou, Y. M., O'Leary, S. E., Wang, Y. and Blaha, G. M. (2017). Transcription-translation coupling: direct interactions of RNA polymerase with ribosomes and ribosomal subunits. Nucleic Acids Res 45(19): 11043-11055.
- Holden, H. M., Rayment, I. and Thoden, J. B. (2003). Structure and function of enzymes of the Leloir pathway for galactose metabolism. J Biol Chem 278(45): 43885-43888.
- Jeon, H. J., Kang, C., N, M. P. A., Lee, Y., Wang, X., Chattoraj, D. K. and Lim, H. M. (2020). Translation Initiation Control of RNase E-Mediated Decay of Polycistronic gal mRNA. Front Mol Biosci 7: 586413.
- Jeon, H. J., Lee, Y., N, M. P. A., Wang, X., Chattoraj, D. K. and Lim, H. M. (2021). sRNA-mediated regulation of gal mRNA in E. coli: Involvement of transcript cleavage by RNase E together with Rho-dependent transcription termination. PLoS Genet 17(10): e1009878.
- Kim, T. W., Oh, I. S., Keum, J. W., Kwon, Y. C., Byun, J. Y., Lee, K. H., Choi, C. Y. and Kim, D. M. (2007). Prolonged cell-free protein synthesis using dual energy sources: Combined use of creatine phosphate and glucose for the efficient supply of ATP and retarded accumulation of phosphate. Biotechnol Bioeng 97(6): 1510-1515.
- Kohler, R., Mooney, R. A., Mills, D. J., Landick, R. and Cramer, P. (2017). Architecture of a transcribing-translating expressome. Science 356(6334): 194-197.
- Lee, H. J., Jeon, H. J., Ji, S. C., Yun, S. H. and Lim, H. M. (2008). Establishment of an mRNA gradient depends on the promoter: an investigation of polarity in gene expression. J Mol Biol 378(2): 318-327.
- Muratore, G., Goracci, L., Mercorelli, B., Foeglein, A., Digard, P., Cruciani, G., Palu, G. and Loregian, A. (2012). Small molecule inhibitors of influenza A and B viruses that act by disrupting subunit interactions of the viral polymerase. Proc Natl Acad Sci U S A 109(16): 6247-6252.
- Nashimoto, H. and Uchida, H. (1985). DNA sequencing of the Escherichia coli ribonuclease III gene and its mutations. Mol Gen Genet 201(1): 25-29.
- Nudler, E. and Gottesman, M. E. (2002). Transcription termination and anti-termination in E. coli. Genes Cells 7(8): 755-768.
- Proshkin, S., Rahmouni, A. R., Mironov, A. and Nudler, E. (2010). Cooperation between translating ribosomes and RNA polymerase in transcription elongation. Science 328(5977): 504-508.
- Ray-Soni, A., Bellecourt, M. J. and Landick, R. (2016). Mechanisms of Bacterial Transcription Termination: All Good Things Must End. Annu Rev Biochem 85: 319-347.
- Robertson, H. D., Webster, R. E. and Zinder, N. D. (1968). Purification and properties of ribonuclease III from Escherichia coli. J Biol Chem 243(1): 82-91.
- Sambrook, J. and Russell, D. W. (2006). Rapid Amplification of 3' cDNA Ends (3'-RACE). CSH Protoc 2006(1).
- Silverman, A. D., Karim, A. S. and Jewett, M. C. (2020). Cell-free gene expression: an expanded repertoire of applications. Nat Rev Genet 21(3): 151-170.
- Silverman, A. D., Kelley-Loughnane, N., Lucks, J. B. and Jewett, M. C. (2019). Deconstructing Cell-Free Extract Preparation for in Vitro Activation of Transcriptional Genetic Circuitry. ACS Synth Biol 8(2): 403-414.
- Takahashi, M. K., Hayes, C. A., Chappell, J., Sun, Z. Z., Murray, R. M., Noireaux, V. and Lucks, J. B. (2015). Characterizing and prototyping genetic networks with cell-free transcription-translation reactions. Methods 86: 60-72.
- Tando, T., Ishizaka, A., Watanabe, H., Ito, T., Iida, S., Haraguchi, T., Mizutani, T., Izumi, T., Isobe, T., Akiyama, T., Inoue, J., et al. (2010). Requiem protein links RelB/p52 and the Brm-type SWI/SNF complex in a noncanonical NF-kappaB pathway. J Biol Chem 285(29): 21951-21960.
- Tinafar, A., Jaenes, K. and Pardee, K. (2019). Synthetic Biology Goes Cell-Free. BMC Biol 17(1): 64.
- Wang, C., Molodtsov, V., Firlar, E., Kaelber, J. T., Blaha, G., Su, M. and Ebright, R. H. (2020). Structural basis of transcription-translation coupling. Science 369(6509): 1359-1365.
- Wang, X., Jeon, H. J., N, M. P. A., He, J. and Lim, H. M. (2018). Visualization of RNA 3' ends in Escherichia coli Using 3' RACE Combined with Primer Extension. Bio-protocol 8(5): e2752.
- Wang, X., Ji, S. C., Jeon, H. J., Lee, Y. and Lim, H. M. (2015). Two-level inhibition of galK expression by Spot 42: Degradation of mRNA mK2 and enhanced transcription termination before the galK gene. Proc Natl Acad Sci U S A 112(24): 7581-7586.
- Wang, X., Ji, S. C., Yun, S. H., Jeon, H. J., Kim, S. W. and Lim, H. M. (2014). Expression of each cistron in the gal operon can be regulated by transcription termination and generation of a galk-specific mRNA, mK2. J Bacteriol 196(14): 2598-2606.
- Wang, X., N, M. P. A., Jeon, H. J., Lee, Y., He, J., Adhya, S. and Lim, H. M. (2019). Processing generates 3' ends of RNA masking transcription termination events in prokaryotes. Proc Natl Acad Sci U S A 116(10): 4440-4445.
- Webster, M. W., Takacs, M., Zhu, C., Vidmar, V., Eduljee, A., Abdelkareem, M. and Weixlbaumer, A. (2020). Structural basis of transcription-translation coupling and collision in bacteria. Science 369(6509): 1355-1359.
- Wong, R. W. and Blobel, G. (2008). Cohesin subunit SMC1 associates with mitotic microtubules at the spindle pole. Proc Natl Acad Sci U S A 105(40): 15441-15445.
- Zhao, J., Hyman, L. and Moore, C. (1999). Formation of mRNA 3' ends in eukaryotes: mechanism, regulation, and interrelationships with other steps in mRNA synthesis. Microbiol Mol Biol Rev 63(2): 405-445.
- Zhu, M., Mori, M., Hwa, T. and Dai, X. (2019). Disruption of transcription-translation coordination in Escherichia coli leads to premature transcriptional termination. Nat Microbiol 4(12): 2347-2356.
Article Information
Copyright
© 2022 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
N, M. P. A. and Lim, H. M. (2022). An in vitro Assay of mRNA 3’ end Using the E. coli Cell-free Expression System. Bio-protocol 12(4): e4333. DOI: 10.21769/BioProtoc.4333.
Category
Microbiology > Microbial genetics > RNA > Transcription
Biological Engineering > Synthetic biology
Molecular Biology > RNA > miRNA interference
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.