- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Reconstitution of Membrane-associated Components of a G-protein Signaling Pathway on Membrane-coated Nanoparticles (Lipobeads)
Published: Vol 12, Iss 2, Jan 20, 2022 DOI: 10.21769/BioProtoc.4303 Views: 3278
Reviewed by: Khyati Hitesh ShahSaravanan KolandaiveluKathleen Boesze-Battaglia
Original research article
The authors used this protocol in:

Dec 2019
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols


Abstract
G-protein coupled signaling pathways are organized into multi-protein complexes called signalosomes that are located within and on cellular membranes. We describe the use of silica nanoparticles coated with a unilamellar phospholipid bilayer (lipobeads) to reconstitute the activated photoreceptor G-protein α-subunit (Gtα*) with its cognate effector (phosphodiesterase-6; PDE6) for biochemical and structural studies of the activation mechanism regulating this GPCR signaling pathway. Lipobeads are prepared by resuspending dried-down phospholipid mixtures with monodisperse 70 nm silica particles, followed by extrusion through a 100 nm membrane filter. This uniform and supported liposomal preparation is easily sedimented, permitting the separation of soluble from membrane-associated proteins. Upon loading lipobeads with Gtα* and PDE6, we find that activation of PDE6 catalysis by Gtα* occurs much more efficiently than in the absence of membranes. Chemical cross-linking of membrane-confined proteins allows detection of changes in protein-protein interactions, resulting from G-protein activation of PDE6. The advantages of using lipobeads over partially purified membrane preparations or traditional liposomal preparations are generally applicable to the study of other membrane-confined signal transduction pathways.
Background
Supported phospholipid bilayers have been used for a variety of fundamental and applied applications, including membrane biophysics, biosensors, cell-cell interactions, screening assays of membrane proteins, and as drug-delivery vehicles (Ng et al., 2004; Troutier and Ladaviere, 2007; Chemburu et al., 2010; Crites et al., 2015; Schadauer et al., 2015). Microspheres consisting of silica or polymeric materials (e.g., hydrogels) have been used as a solid support for forming unilamellar phospholipid bilayers whose size, density, membrane curvature, and other biophysical properties can be optimized for their intended applications.
We have utilized 70 nm diameter silica particles coated with a unilamellar lipid bilayer (termed “lipobeads”; Figure 1A) to bind the prenylated photoreceptor phosphodiesterase (PDE6) holoenzyme (Figure 1B), as well as the acylated photoreceptor G-protein α-subunit (Gtα), for enzymological and structural studies of the Gtα*-PDE6 activation complex in a membrane-confined environment (Figure 1C) (Irwin et al., 2019). Activation of PDE6 by its cognate G-protein (transducin) occurs on the membrane surface in the light-harvesting outer segment of rod and cone photoreceptors upon photoactivation of rhodopsin (Cote et al., 2021). Studies have shown that the efficiency with which Gtα* can activate PDE6 is greatly enhanced when both proteins are tethered to either the photoreceptor membrane or to liposomal preparations (Melia et al., 2000). Reduction in the dimensionality of diffusional encounters between Gtα* and PDE6, in conjunction with high local protein concentrations, likely account for this phenomenon. The ability to reconstitute the proteins comprising GPCR signalosomes in vitro on well-defined lipobeads enables the integration of biochemical, biophysical, and structural studies to advance our mechanistic understanding of GPCR signaling pathways, in the context of their membrane environment.
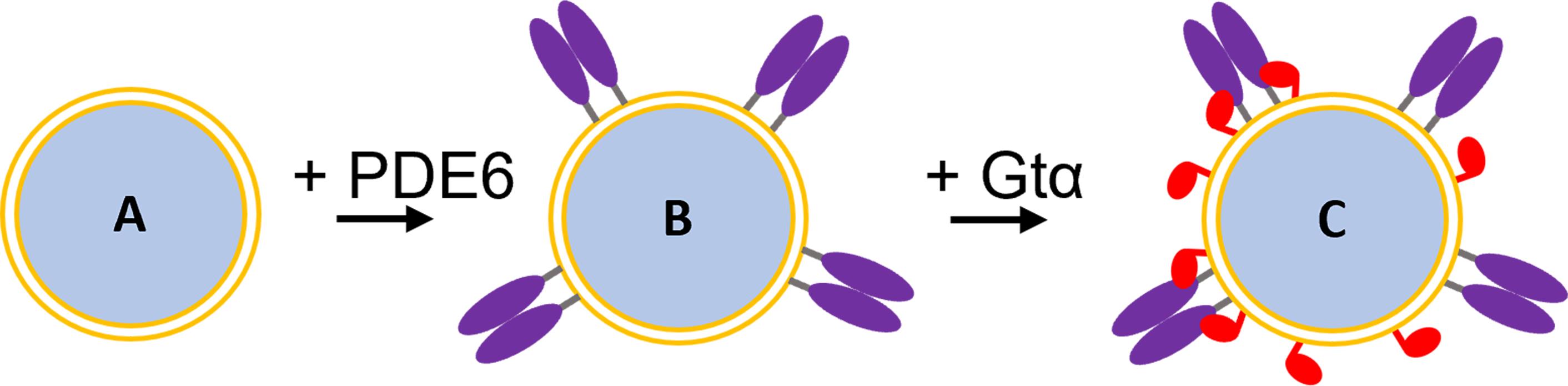
Figure 1. Reconstitution of the Gtα*-PDE6 activation complex on lipobeads. A. Lipobead composed of a silica nanoparticle coated with a unilamellar phospholipid bilayer. B. PDE6 (purple) attached to lipobeads by their prenyl moieties. C. Addition of acylated Gtα* (red) to lipobeads with pre-bound PDE6 results in Gtα* binding and the formation of the Gtα*-PDE6 activation complex.
Materials and Reagents
Amicon Ultra-4 30 kDa or 50 kDa ultrafiltration units (Millipore-Sigma, catalog numbers: UFC803024 and UFC805024)
Phospholipids (Avanti Polar Lipids, Inc.)
DOTAP: 1,2-dioleoyl-3-trimethylammonium-propane (chloride salt; Avanti Polar Lipids, catalog number: 890890C-200 mg) is supplied as 100 mg portions dissolved in chloroform at 25 mg/mL (FW 698.5; 35.8 mM).
DOPC: 1,2-dioleoyl-sn-glycero-3-phosphocholine (Avanti Polar Lipids, catalog number: 850375C-500 mg) is supplied as 100 mg portions dissolved in chloroform at 25 mg/mL (FW 786.1; 31.8 mM).
Monodisperse silica particles [70 nm diameter; 25 mg/mL aqueous suspension (0.11 μM; 7 × 1013 particles/mL); density = 2.0 g/cm3] are obtained from DiagNano (CD Bioparticles, catalog number: DNG-B004).
Purified PDE6 is prepared as described previously (Pentia et al., 2005) and stored at -20°C in PDE6 storage buffer.
Purified Gtα-GDP or purified Gtα*-GTPγS is prepared as described previously (Ting et al., 1993; Irwin et al., 2019) and stored at -20°C in Gtα storage buffer.
NuPAGE 4-12% Bis-Tris gel (Invitrogen, catalog number: NP0321BOX)
Bis(sulfosuccinimidyl) suberate (BS3) (Thermo Scientific, catalog number: 21580)
NaCl (Electron Microscopy Sciences, catalog number: EM-7760)
MgCl2·6H2O (Electron Microscopy Sciences, catalog number: EM-5980)
Dithiothreitol (Electron Microscopy Sciences, catalog number: EM-3860)
Glycerol (IBI Scientific, catalog number: IB15760)
Tris base (J.T. Baker, catalog number: JT4109)
AlCl3·6H2O (Millipore Sigma, catalog number: A0718)
NaF (J.T. Baker, catalog number: JT3689)
HEPES, hemisodium salt (MP Biomedicals, catalog number: IC15245180)
Guanosine-5’-diphosphate, sodium salt (Millipore Sigma, catalog number: G7127)
HNM buffer (see Recipes)
PDE6 storage buffer (see Recipes)
Gtα storage buffer (see Recipes)
745 µM AlCl3 in HNM buffer (see Recipes)
250 mM NaF in HNM buffer (see Recipes)
1 M MgCl2 (see Recipes)
50 mM GDP (see Recipes)
10 mM BS3 (see Recipes)
0.5 mM BS3 (see Recipes)
Equipment
Eppendorf 5415D centrifuge
Vortex mixer (VWR, catalog number: 10153-838)
Avanti Mini Extruder Set (Avanti, catalog number: 610023)
0.1 µm, 19 mm diameter polycarbonate membrane (Avanti, catalog number: 610005 or Whatman catalog number: 800309)
10 mm filter supports (Avanti, catalog number: 610014)
1,000 μL gas-tight syringe (Avanti, catalog number: 610017)
Procedure
Preparation of phospholipids
An 80:20 molar ratio of DOPC:DOTAP (Melia et al., 2000) is prepared by combining 1.26 mL of DOPC (31.5 mg, 40 µmol) and 279 µL of DOTAP (7.0 mg, 10 µmol) with 461 µL of chloroform.
[DOPC] = 20.0 mM, [DOTAP] = 5.0 mM; total [phospholipid] = 25.0 mM
Aliquot 200 µL portions (4.0 μmol DOPC and 1 μmol DOTAP) into glass tubes and evaporate them under a stream of nitrogen gas.
Dried down lipids are stored in darkness at -20°C for up to six months until use.
Preparation of silica nanoparticles
Silica particles are disaggregated by mixing 200 μL (5 mg; 1.4 × 1013 particles) of silica particles with 1 mL of 95% ethanol.
Vortex at maximum speed for 2 min.
Incubate for a minimum of 1 h at room temperature (overnight is preferable).
Centrifuge the silica suspension at 10,000 × g for 10 min in the Eppendorf 5415D centrifuge at 10°C.
Resuspend the pellet with 1.0 mL of ice-cold HNM buffer, vortex for 2 min, and centrifuge again at 10,000 × g for 10 min in the Eppendorf 5415D centrifuge at 10°C, to remove residual ethanol.
Resuspend the pellet with 1.0 mL of ice-cold HNM buffer, vortex for 2 min, and keep on ice until use.
Preparation of lipobeads
The dried-down DOPC/DOTAP lipid mixture is mixed with the silica particle suspension at room temperature without nitrogen gas, and gently vortexed (setting #5-6) for 2 min, then allowed to settle for 1 min. This process is repeated four more times.
Final concentration: 80:20 DOPC:DOTAP = 5 mM (total phospholipid), [lipobead] = 23 nM
Assemble the Mini Extruder (a video created by the manufacturer describing the assembly of the Mini Extruder is found here: https://www.youtube.com/watch?v=8ei62ZmLgSM):
Pre-wet two 10 mm filter supports with HNM buffer and place one inside the O-ring of the internal membrane support.
Place the membrane support into the outer casing.
Place a single polycarbonate 0.1 µm membrane on top of the membrane support.
Place the second filter support on the second membrane support and place the second membrane support into the outer casing (O-rings facing each other).
Place the Teflon bearing into the retainer nut.
Finger-tighten the retainer nut onto the outer casing.
Attach an empty gas-tight syringe to one end of the Mini Extruder.
Pre-wet the assembly by passing HNM buffer through the Mini-Extruder with the second gas-tight syringe.
Extrude the lipid/silica suspension through the Mini Extruder:
Load the lipid/silica mixture into one of the gas-tight syringes.
Firmly depress the syringe barrel to force the lipobeads through the membrane quickly (~10 s duration per cycle)
Repeat a total of 15 times, with the lipobead suspension finally filling the originally empty syringe (this prevents aggregates that never passed through the filter from being recovered).
Separate the unilamellar lipobeads recovered from the syringe from unbound phospholipids by centrifugation at 10,000 × g for 10 min at room temperature in the Eppendorf 5415 D centrifuge.
Resuspend the pellet with 500 µL of HNM buffer for a lipobead concentration of 46 nM.
Lipobead preparations are stable for ~2 days if stored in the dark at room temperature.
Binding PDE6 to lipobeads
Add 10 µL of 46 nM lipobead preparation (0.46 pmol) to purified PDE6 holoenzyme and incubate at room temperature for 30 min.
The amount of PDE6 added to the lipobead preparation can be varied from 10 fmol (1 nM concentration) to at least 10 pmol (1 μM concentration), without exceeding the maximum binding capacity of the lipobeads. The amount of PDE6 added to the lipobeads is determined as described in Step G2b.
The glycerol concentration of the PDE6 stock solution must be less than 10% (v/v) for optimal binding to lipobeads.
To reduce the glycerol concentration, the stock solution of PDE6 can be buffer exchanged using 50 kDa MWCO Amicon Ultra-4 filtration units. The protein is diluted over ten-fold with HNM buffer, followed by centrifugation at 3,000 × g for 10-20 min. Centrifugation time will depend on the initial volume of protein.
Spin the PDE6-lipobead mixture at 10,000 × g for 10 min in the Eppendorf centrifuge in the cold room (~10°C).
Remove supernatant as soon as possible after centrifugation, as the pellet loosens over time.
To determine the extent of PDE6 binding to lipobeads, the supernatant can be recovered and assayed. As seen in Figure 2, over a range from 0.01 to 10 pmol of PDE6 added, 85 ± 6% is bound to lipobeads.
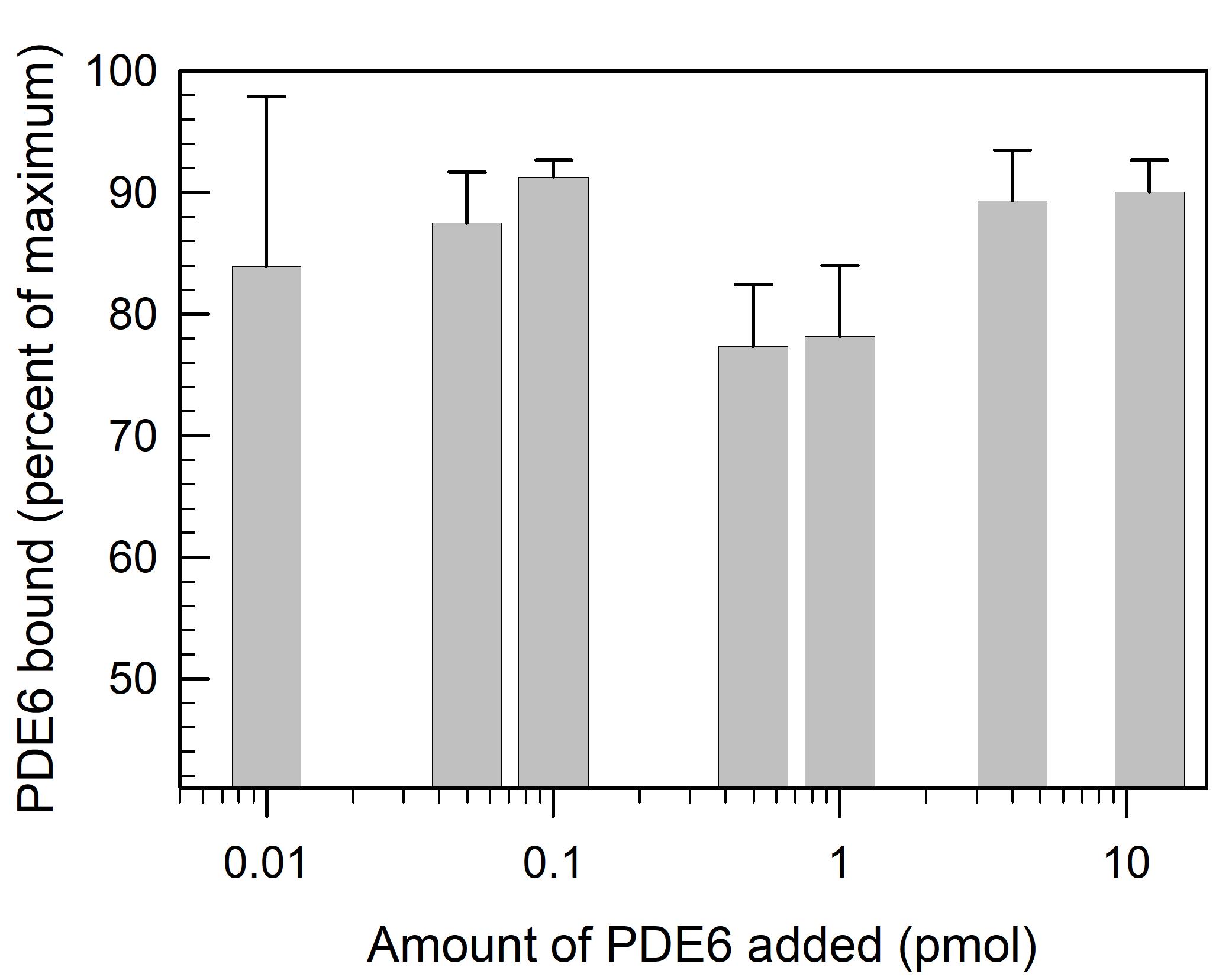
Figure 2. Binding of PDE6 to lipobeads. Lipobeads (0.46 pmol) are mixed with 40 μL of the indicated amount of PDE6 and incubated for 30 min. Following centrifugation, the supernatant fractions are assayed for PDE6 activity (see Step G2b), to calculate the percent of total PDE6 activity bound to the lipobeads. Data represent the mean ± S.D. (n = 3).Use the lipobead pellet containing bound PDE6 for the subsequent incubation with Gtα*.
Preparation of aluminum fluoride-activated Gtα* (Gtα-GDP-AlF4-)
For Gα-GDP preparations stored in 50% (v/v) glycerol, the glycerol concentration is first reduced by buffer exchange using HNM buffer and ultrafiltration.
The glycerol concentration of the Gtα preparation must be less than 10% (v/v) for optimal binding to lipobeads.
Perform buffer exchange by first diluting the Gtα stock solution over ten-fold with HNM buffer, placing the protein sample in the upper chamber of a 30 kDa MWCO Amicon Ultra-4 filtration unit, and centrifuging at 3,000 × g for 10-20 min (depending on the initial volume of the diluted protein).
Purified Gtα-GDP is activated by the addition of aluminum fluoride (Bigay et al., 1987). In a final volume of 50 μL, add the following:
45.6 μL of Gtα-GDP in Gtα storage buffer [containing <10% (v/v) final glycerol concentration].
2.0 µL of 745 µM AlCl3 (final concentration = 30 µM).
2.0 µL of 250 mM NaF (final concentration = 10 mM).
0.4 µL of 1 M MgCl2 (final concentration = 8 mM).
Incubate on ice for 15-30 min.
Formation of Gtα*-PDE6 complex on lipobeads
Add 50 µL of Gtα* (either Gtα-GDP-AlF4- or Gtα-GTPγS) to the PDE6-containing lipobead pellet, mix well, then incubate for 45 min at room temperature.
The concentration of Gtα* added to the PDE6-bound lipobeads can be varied up to a maximum of ~5 μM (this being the limit of solubility of purified Gtα that is added to the lipobead mixture).
For chemical cross-linking experiments (see Figure 3), the cross-linker is added to the lipobead suspension prior to centrifugation. Add BS3 to a final concentration of 0.125 mM, from a 0.5 mM working solution or 10 mM stock solution. Allow crosslinking reactions to proceed for 45 min at room temperature, followed by centrifugation at 10,000 × g for 10 min. Resuspend the pellets in 1× gel loading sample buffer for SDS-PAGE.
For enzymatic assays of cGMP hydrolysis of PDE6 (see Figure 4), the lipobeads containing Gtα*-PDE6 are not centrifuged prior to addition of cGMP substrate to initiate the assay of PDE6 hydrolytic activity.
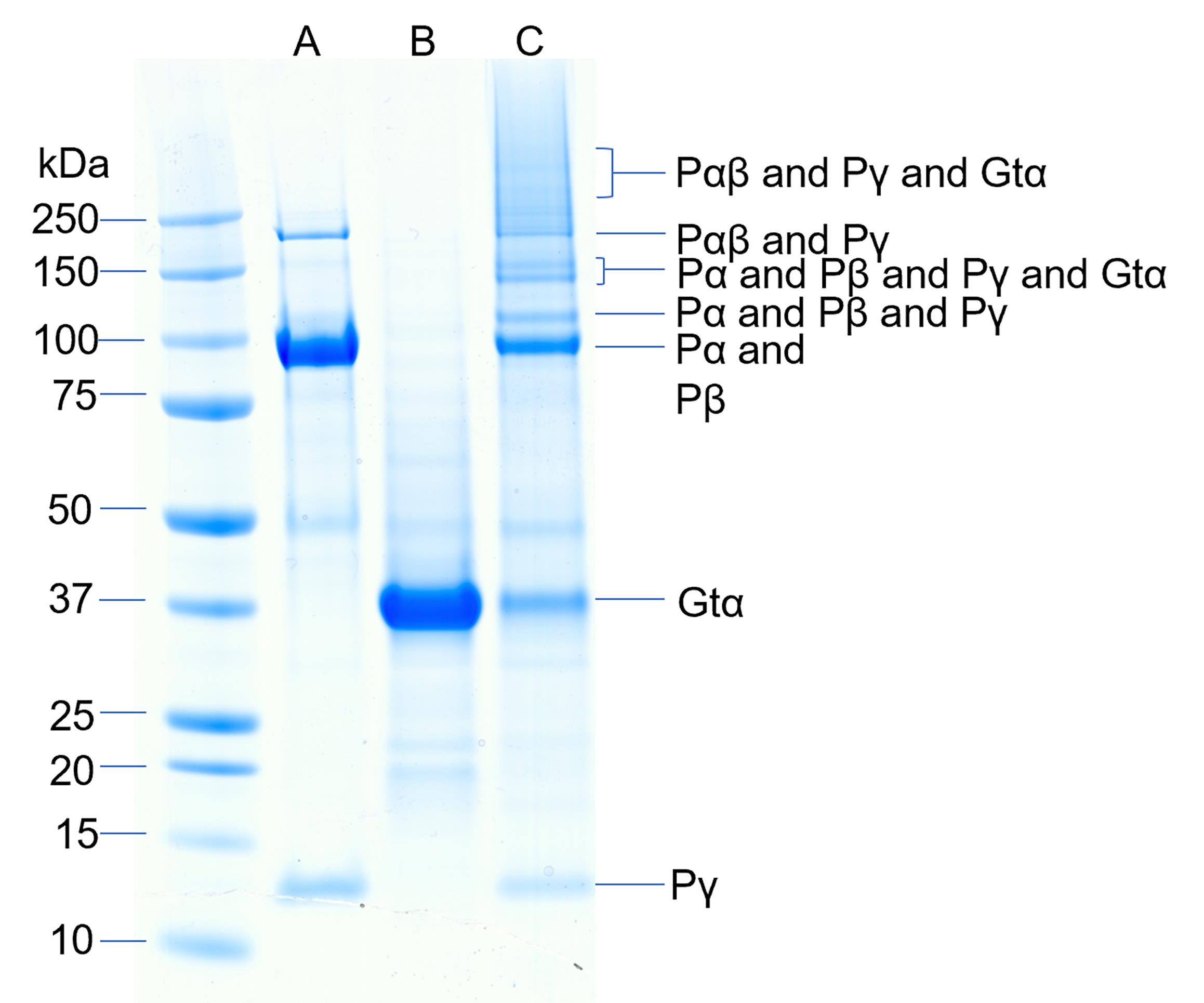
Figure 3. Chemical cross-linking of the activated complex of Gtα and PDE6. Cross-linking of PDE6 and Gtα* was performed by first binding 9 pmol of PDE6 to 0.46 pmol lipobeads for 20 min followed by centrifugation. The lipid pellet was resuspended with 115 pmol Gtα-GDP-ALF and incubated for 45 min followed by centrifugation. The pellet was resuspended in 20 μL of HNM buffer containing 0.125 mM BS3 and crosslinking proceeded for 45 min. Lane A contains purified PDE6, lane B contains purified Gtα*, and lane C contains the crosslinked mixture.Analytical methods
Determination of protein concentration
The concentration of PDE6 or Gtα is determined by the bicinchoninic assay (Thermo Fisher, Inc.) following the manufacturer’s protocol.
For analysis of Gtα samples containing excess unbound nucleotide, buffer exchange using Amicon Ultra-4 30 kDa ultrafiltration units (following the procedure described in Step E1b) with buffer lacking guanine nucleotides is performed, to remove interference with the protein assay.
PDE6 enzymatic activity assay
Quantify the rate of cGMP hydrolysis using a colorimetric assay of inorganic phosphate, in which the breakdown of cGMP to 5’-GMP by PDE6 is followed by addition of 5’-nucleotidase to liberate inorganic phosphate (Cote, 2000).
To measure the total amount of PDE6 present in a sample, incubate the enzyme with trypsin to proteolytically digest the inhibitory γ-subunit that is tightly bound to the holoenzyme (Pentia et al., 2005). Calculate the amount of PDE6 in the sample with knowledge of the catalytic turnover number for trypsin-activated PDE6 [kcat = 5600 cGMP hydrolyzed per s per PDE6 (Mou et al., 1999)].
To calculate the percent of maximal activation induced by addition of Gtα (see Figure 4), the extent of transducin activation of PDE6 catalytic activity on lipobeads is referenced to the maximum trypsin-activated enzyme activity (Step G2b).
Measure the PDE6 basal (i.e., nonactivated) activity on lipobeads in the absence of added Gtα*.
Data analysis
Evaluation of the ability of Gtα* to activate PDE6 catalysis.
There is substantial evidence in the literature (Malinski and Wensel, 1992; Melia et al., 2000) supporting the importance of membrane attachment for efficient activation of PDE6 by Gtα*. Membrane binding of the G-protein and its effector reduces the dimensionality of diffusional encounters, and also creates a high local concentration of these interacting proteins.
Figure 4 demonstrates that Gtα* activation of PDE6 in solution (blue symbols) is much less effective than when the same concentration of PDE6 and Gtα* are pre-incubated with lipobeads prior to assaying PDE6 activation (black symbols). Figure 4 also shows that the efficacy with which Gtα* can activate PDE6 is greatly enhanced as the PDE6 concentration bound to lipobeads increases (compare black and red symbols). Under these conditions, a ten-fold increase in PDE6 bound to lipobeads reduces by five-fold the concentration of Gtα* needed to achieve 50% activation of PDE6.
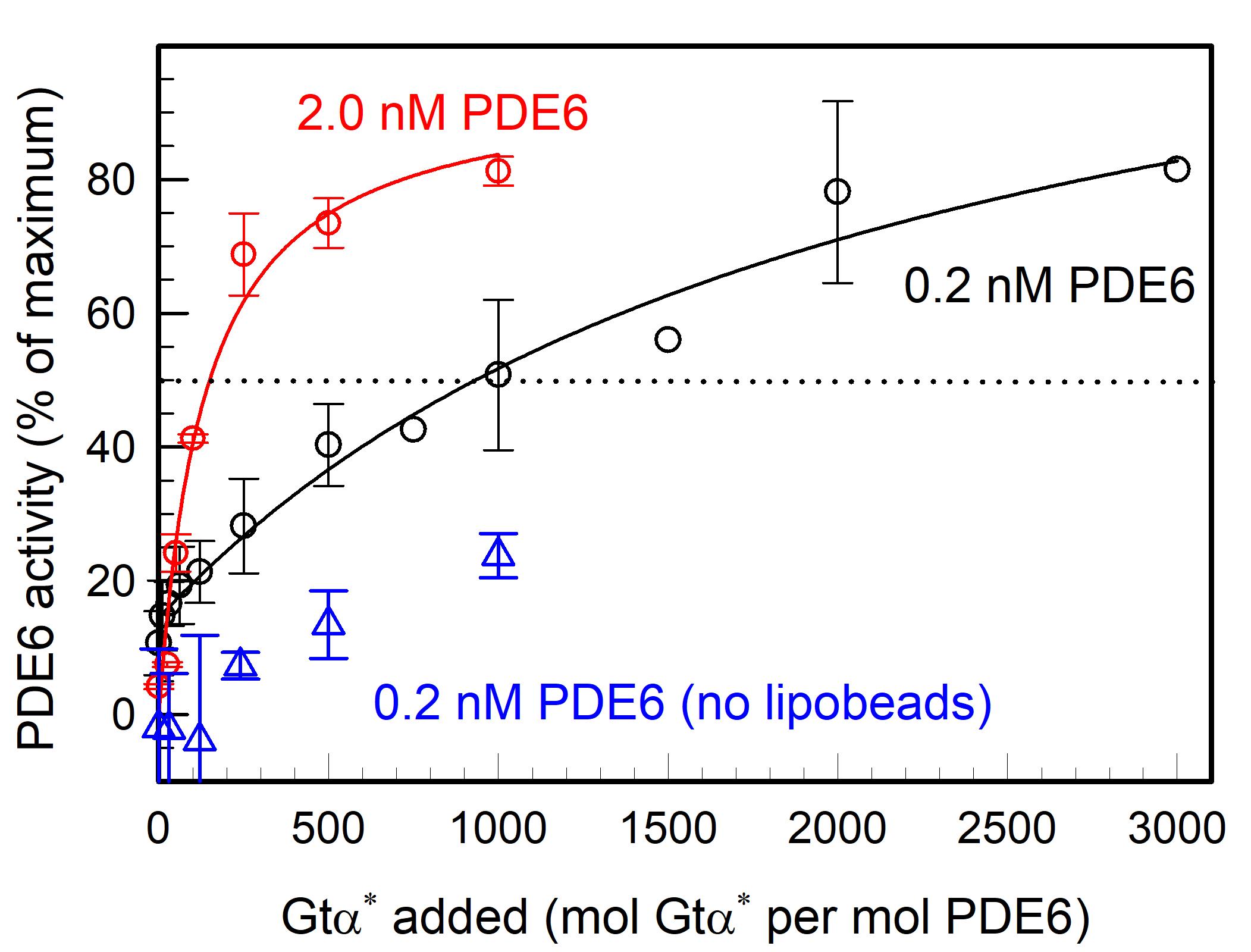
Figure 4. G-protein activation of PDE6 is enhanced on lipobeads in a concentration-dependent manner. PDE6 holoenzyme (either 0.2 nM or 2 nM) was incubated with 1.1 pmol of lipobeads for 30 min at room temperature, prior to the addition of the indicated amounts of Gtα-GTPγS (Gtα*) for 45 min. A separate sample of PDE6 was mixed with Gtα* in the absence of lipobeads. Upon addition of cGMP (2 mM final concentration), the extent to which PDE6 was activated by Gtα* was determined by monitoring the degradation of cGMP. The extent of activation by Gtα* is expressed as the percentage of PDE6 activity measured when the inhibitory Pγ subunits of PDE6 are removed by limited trypsin proteolysis. Data represent the mean ± S.D. for 3 experiments (blue and black symbols; three black symbols without error bars are individual determinations) or the mean and range for 2 experiments (red symbols).
Characterization of protein complex formation using chemical cross-linking.
Protein complex formation is determined by the appearance of higher molecular weight bands on SDS-PAGE following chemical crosslinking (Figure 2).
The identity of proteins present in various cross-linked complexes (Figure 2) is ascertained by excising Coomassie-stained bands from the gel, performing digestion with trypsin, and analysis by mass spectrometry (see Irwin et al., 2019 for details).
Identification of cross-linked peptides enables integrative structural modeling of the Gtα*-PDE6 complex on lipobeads, as described in detail in Irwin et al. (2019).
Notes
Lipobead properties:
A single 100 nm unilamellar liposome encapsulating a silica particle (surface area = 0.031 μm2) is calculated to contain 80,047 phospholipids per particle [using Encapsula NanoSciences calculator (https://encapsula.com/calculator)].
This calculation is based on knowledge of the phospholipid head-group area [for phosphatidylcholine, 0.71 nm2 (Kucerka et al., 2006)] and the thickness of the lipid bilayer [5 nm (Marquardt et al., 2016)].
Distribution of phospholipids in unilamellar membrane is calculated to be 55% in the outer monolayer and 45% in inner monolayer, in agreement with 31P NMR spectroscopy of 100 nm unilamellar liposomes prepared by extrusion (Malinski and Wensel, 1992).
Total phospholipid concentration encapsulating the silica particles: 46 nM particles × 80,047 lipids per particle = 3.7 mM; thus, a 2.7-fold excess of phospholipids are present when initially preparing lipobeads.
Surface-accessible (i.e., outer monolayer) phospholipid concentration = 2.0 mM.
The stability of the lipobead preparations (as judged by the extent of activation of PDE6 by Gtα*, as well as the extent of PDE6 binding to lipobeads) has been found to decline significantly three days after initial preparation and storage of lipobeads at room temperature.
Recipes
HNM buffer (1 L)
20 mM HEPES-0.5Na (4.986 g)
100 mM NaCl (5.844 g)
2 mM MgCl2·6H2O (0.407 g)
pH 7.5
PDE6 storage buffer (1 L)
20 mM HEPES-0.5Na (4.986 g)
100 mM NaCl (5.844 g)
2 mM MgCl2·6H2O (0.407 g)
2 mM DTT (0.308 g)
50% (v/v) glycerol
Gtα storage buffer (1 L)
10 mM Tris base (1.211 g), pH 7.4
2 mM MgCl2·6H2O (0.407 g)
2 mM DTT (0.308 g)
Add 50 μM GDP [1/1,000 dilution of 50 mM GDP and 40% glycerol (v/v)] immediately prior to storage at -20°C.
745 µM AlCl3 in HNM buffer
AlCl3 (9.9 mg) in 100 mL of HNM buffer
250 mM NaF in HNM buffer
NaF (105 mg) in 10 mL of HNM buffer
1 M MgCl2
MgCl2·6H2O (2.033 g) in 10 mL of water
50 mM GDP (1 mL)
GDP-Na (23.3 mg) in 10 mM Tris, pH 7.5
10 mM BS3 (175 μL)
1 mg BS3 in 175 μL of water
0.5 mM BS3 (20 μL)
1 μL of 10 mM BS3 diluted to 20 μL with HNM buffer
Acknowledgments
This work was supported by the National Eye Institute grant R01 EY05798 and the National Institute of General Medical Sciences grants P20 GM113131. An abbreviated version of this protocol first appeared in Irwin et al. (2019; doi: 10.1074/jbc.RA119.011002).
Competing interests
The authors declare no competing financial or non-financial interests.
References
- Bigay, J., Deterre, P., Pfister, C. and Chabre, M. (1987). Fluoride complexes of aluminium or beryllium act on G-proteins as reversibly bound analogues of the gamma phosphate of GTP. EMBO J 6(10): 2907-2913.
- Chemburu, S., Fenton, K., Lopez, G. P. and Zeineldin, R. (2010). Biomimetic silica microspheres in biosensing. Molecules 15(3): 1932-1957.
- Cote, R. H. (2000). Kinetics and regulation of cGMP binding to noncatalytic binding sites on photoreceptor phosphodiesterase. Methods Enzymol 315: 646-72.
- Cote, R. H., Gupta, R., Irwin, M. J. and Wang, X. (2021). Photoreceptor Phosphodiesterase (PDE6): Structure, Regulatory Mechanisms, and Implications for Treatment of Retinal Diseases. Adv Exp Med Biol. doi: 10.1007/5584_2021_649.
- Crites, T. J., Maddox, M., Padhan, K., Muller, J., Eigsti, C. and Varma, R. (2015). Supported Lipid Bilayer Technology for the Study of Cellular Interfaces. Curr Protoc Cell Biol 68: 24.25.21-24.25.31.
- Irwin, M. J., Gupta, R., Gao, X. Z., Cahill, K. B., Chu, F. and Cote, R. H. (2019). The molecular architecture of photoreceptor phosphodiesterase 6 (PDE6) with activated G protein elucidates the mechanism of visual excitation. J Biol Chem 294(51): 19486-19497.
- Kucerka, N., Tristram-Nagle, S. and Nagle, J. F. (2005). Structure of fully hydrated fluid phase lipid bilayers with monounsaturated chains. J Membr Biol 208(3): 193-202.
- Malinski, J. A. and Wensel, T. G. (1992). Membrane stimulation of cGMP phosphodiesterase activation by transducin: comparison of phospholipid bilayers to rod outer segment membranes. Biochemistry 31(39): 9502-9512.
- Marquardt, D., Heberle, F. A., Greathouse, D. V., Koeppe, R. E., Standaert, R. F., Van Oosten, B. J., Harroun, T. A., Kinnun, J. J., Williams, J. A., Wassall, S. R. et al. (2016). Lipid bilayer thickness determines cholesterol's location in model membranes.Soft Matter 12(47): 9417-9428.
- Melia, T. J., Malinski, J. A., He, F. and Wensel, T. G. (2000). Enhancement of phototransduction protein interactions by lipid surfaces. J Biol Chem 275(5): 3535-3542.
- Mou, H., Grazio, H. J., 3rd, Cook, T. A., Beavo, J. A. and Cote, R. H. (1999). cGMP binding to noncatalytic sites on mammalian rod photoreceptor phosphodiesterase is regulated by binding of its gamma and delta subunits. J Biol Chem 274(26): 18813-18820.
- Ng, C. C., Cheng, Y. L. and Pennefather, P. S. (2004). Properties of a self-assembled phospholipid membrane supported on lipobeads. Biophys J 87(1): 323-331.
- Pentia, D. C., Hosier, S., Collupy, R. A., Valeriani, B. A. and Cote, R. H. (2005). Purification of PDE6 isozymes from mammalian retina. Methods Mol Biol 307: 125-140.
- Schadauer, F., Geiss, A. F., Srajer, J., Siebenhofer, B., Frank, P., Reiner-Rozman, C., Ludwig, B., Richter, O. M., Nowak, C. and Naumann, R. L. (2015). Silica nanoparticles for the oriented encapsulation of membrane proteins into artificial bilayer lipid membranes. Langmuir 31(8): 2511-2516.
- Ting, T. D., Goldin, S. B. and Ho, Y. K. (1993). Purification and characterization of bovine transducin and its subunits. Hargrave, P. A. (Ed.). In: Photoreceptor Cells. Academic Press, 180-195.
- Troutier, A. L. and Ladaviere, C. (2007). An overview of lipid membrane supported by colloidal particles. Adv Colloid Interface Sci 133(1): 1-21.
Article Information
Copyright
© 2022 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Irwin, M. J., Wang, X. and Cote, R. H. (2022). Reconstitution of Membrane-associated Components of a G-protein Signaling Pathway on Membrane-coated Nanoparticles (Lipobeads). Bio-protocol 12(2): e4303. DOI: 10.21769/BioProtoc.4303.
- Irwin, M. J., Gupta, R., Gao, X. Z., Cahill, K. B., Chu, F. and Cote, R. H. (2019). The molecular architecture of photoreceptor phosphodiesterase 6 (PDE6) with activated G protein elucidates the mechanism of visual excitation. J Biol Chem 294(51): 19486-19497.
Category
Neuroscience > Cellular mechanisms > Protein isolation
Neuroscience > Cellular mechanisms > Intracellular signalling
Biochemistry > Lipid > Lipid-protein interaction
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.