- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

ATAC Sequencing Protocol For Cryopreserved Mammalian Cells
Published: Vol 12, Iss 2, Jan 20, 2022 DOI: 10.21769/BioProtoc.4294 Views: 5459
Reviewed by: Giusy TornilloYogita JethmalaniFernando A Gonzales-Zubiate

Original research article
The authors used this protocol in:

Dec 2020
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
ATAC-seq (assay for transposase-accessible chromatin with high-throughput sequencing) is a powerful method to evaluate chromatin accessibility and nucleosome positioning at a genome-wide scale. This assay uses a hyperactive Tn5 transposase, to simultaneously cut open chromatin and insert adapter sequences. After sequencing, the reads generated through this technique are generally indicative of transcriptional regulatory elements that are located in accessible chromatin. This method was originally developed by Buenrostro et al. (2013), and since then it has been improved by the same authors several times, until their last update called OMNI ATAC-seq (Corces et al., 2017). Here, we describe an ATAC-seq protocol based on the OMNI-ATAC method, with a special focus on the initial steps of thawing cryopreserved cells, and the final steps of library purification using magnetic beads. This protocol can be of interest for laboratories working in a fast-paced environment.
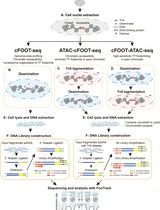
Graphic abstract:
Flowchart of the protocol
Background
In eukaryotic organisms, the packaging of long DNA molecules into the nucleus is achieved by wrapping the DNA around histone proteins to form nucleosomes, and further compacting this structure into chromatin (Kornberg, 1974). The control of chromatin condensation is a major player in the regulation of gene expression. Since high DNA compaction restricts access for the transcriptional machinery, closed chromatin is usually associated with repressed gene expression. By contrast, open chromatin is associated with active gene expression, and the presence of regulatory elements like enhancers and promoters (Wegel and Shaw, 2005). The potential to reveal areas of high and low transcriptional activity has led to genome-wide mapping of chromatin accessibility becoming an area of broad interest.
Until now, several technologies for assessing the genomic profile of chromatin compaction have been developed. Some examples are micrococcal nuclease sensitivity (MNase-seq) (Zaret, 2005), formaldehyde-assisted isolation of regulatory elements (FAIRE-Seq) (Giresi et al., 2007), and deoxyribonuclease I hypersensitivity (DNase-Seq) (Boyle et al., 2008). In 2013, Greenleaf and colleagues developed a new method to probe chromatin accessibility, called assay for transposase-accessible chromatin with high-throughput sequencing (ATAC-seq). ATAC-seq is a technique that assesses open chromatin using a mutated hyperactive Tn5 transposase, which simultaneously cuts DNA and tags the resulting fragments with sequencing adaptors, preferentially in decondensed chromatin areas (Buenrostro et al., 2013). DNA sequencing libraries are then generated by PCR amplification and analyzed by high-throughput sequencing. The main advantages of ATAC-seq over the methods mentioned above are the low number of cells required as input and the length of the protocol (see Sun et al., 2019 for a full comparison between several chromatin accessibility assays).
Since development of the ATAC-seq method, the original authors have advanced (Buenrostro et al., 2015) and improved (OMNI ATAC-seq, Corces et al., 2017) the protocol, to overcome some of the initial limitations of this technique. For example, to remove mitochondrial DNA from the transposition reaction, the OMNI ATAC-seq protocol includes a washing step with non-ionic detergent after the cell lysis. Another improvement is the addition of PBS in the transposition mix, to reduce the background noise. Other researchers from different laboratories have also helped to improve and optimize ATAC-seq protocols (Fujiwara et al., 2019), or have adapted them for specific organisms and cell lines (Doganli et al., 2017, Shashikant and Ettensohn, 2019).
Being a core sequencing facility, our group handles a large number of ATAC-seq projects on a regular basis. We have adopted the OMNI ATAC-seq protocol for most of the ATAC-seq experiments, with two main modifications: (i) to handle a large number of projects, cell samples from researchers are sent cryopreserved, to be stored and processed at a convenient time. In this protocol, we include a section focused on the process of thawing cryopreserved cells, which is very important to preserve cell viability, and therefore to the success of an ATAC-seq experiment. (ii) After the purification of the final PCR reaction with the different silica column DNA purification kits available in the market, it is very common to find primer dimers and unwanted DNA fragments, due to over/under transposase digestion or an excess of PCR cycles. Thus, instead of using silica column-based methods, we purify the ATAC-seq libraries with magnetic beads. In addition to the removal of impurities, this allows us to size select the DNA fragments as needed for different samples. The use of magnetic beads for this step also enables the integration of automation platforms for processing a large number of libraries (Hess et al., 2020).
In summary, we present here an ATAC-seq protocol adapted from Corces et al. (2017) that can be used by sequencing core facilities and other laboratories that routinely handle large quantities of samples.
Materials and Reagents
Solid Phase Reversible Immobilization (SPRI) select beads (Beckman Coulter, catalog number: B23318). Store at room temperature.
High Sensitivity DNA Chips and reagents (Agilent Technologies, catalog number: 5067-4626). Store reagents at 4°C and DNA Chips at room temperature.
Hanks Balanced Salt Solution (Sigma, catalog number: H6648-500ML). Store at room temperature.
Phosphate Buffered Saline (PBS) pH 7.4 (Gibco, Thermofisher, catalog number: 10010-023). Store at 4°C.
Iscove’s Modified Dulbecco’s Medium (IMDM) (Gibco, Thermofisher, catalog number: 12440-053). Store at 4°C.
Fetal Bovine Serum (FBS) (Gibco,Thermofisher, catalog number: 10082147). Store at -20°C.
1 M Tris-HCl pH 7.4 (Teknova, catalog number: T5074). Store at room temperature.
5 M NaCl (Teknova, catalog number: S5845). Store at room temperature.
1 M MgCl2 (Teknova, catalog number: M0308). Store at room temperature.
Digitonin (Promega, catalog number: G9441). Dilute the original 2% digitonin solution 1:1 in water. Make aliquots and store at -20°C.
Tween-20 (Sigma/Roche, catalog number: 11332465001). Store at 4°C.
NP40 (Sigma/Roche, catalog number: 11332473001). Store at 4°C.
Tagment DNA Enzyme (transposase) and 2× TD buffer (Illumina, catalog number: 20034197). Store at -20°C.
NEBNext 2× MasterMix (New England Biolabs, catalog number: M0541S). Store at -20°C.
Zymo DNA Clean & ConcentratorTM-5 Kit (Zymo Research, catalog number: D4014). Store at room temperature.
DNase (Worthington, catalog number: LS002007)
20,000 Units/mL DNase stock solution (see Recipes)
ATAC-Resuspension buffer (ATAC-RSB) (see Recipes)
ATAC-RSB with detergents buffer (see Recipes)
Wash out lysis buffer (see Recipes)
Transposition mix (see Recipes)
Equipment
Pipettes (P1000, P200, P20, P10)
Cell counter (Nexcelom, model: Cellometer Auto 2000 Cell Viability Counter)
ThermoMixer (Eppendorf, model: C with heated lid)
Thermal Cycler (Bio-Rad, model: T100)
Bioanalyzer (Agilent Technologies, model: 2100)
Software
Agilent Technologies 2100 Bioanalyzer 2100 Expert (Agilent Technologies, https://www.agilent.com/en/product/automated-electrophoresis/bioanalyzer-systems/bioanalyzer-software/2100-expert-software-228259)
Procedure
Notes:
Prepare buffers, mixes, and stock solutions as described in the Recipes section, before starting the protocol.
We advise starting with at least 2 biological replicates. If this is not available, 2 technical replicates should be prepared from the same sample.
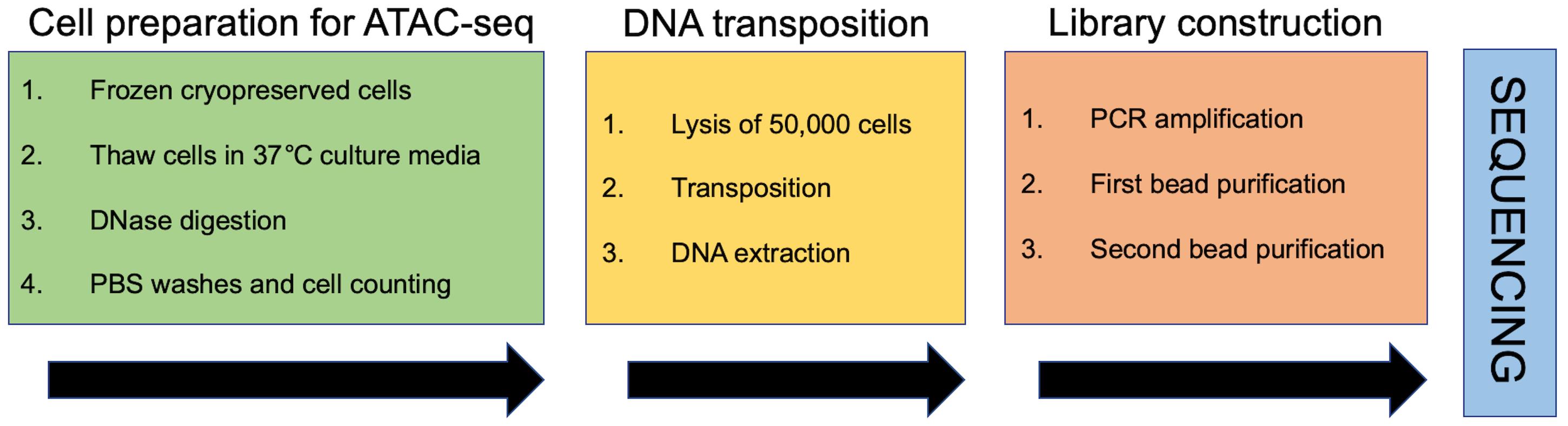
Preparation of cryopreserved cells for ATAC-seq (day 1)
Notes:
Cell viability is one of the most important parameters for a successful ATAC-seq experiment. To avoid cell death and lysis, the cells from cryopreserved vials should be carefully thawed and rapidly transferred to a conducive environment.
The addition of a DNase digestion step in this section will digest the DNA from lysed cells that could otherwise interfere with the final data and interpretations.
Warm the cell culture media (IMDM + 10% FBS) to 37°C.
Add 4 mL of warm media to a 15 mL tube (one tube per sample).
Remove cryopreserved cell vials from the -80°C freezer and place them in a bucket with ice.
Place the cryopreserved cell vials at 37°C (no more than 3 vials at a time).
When cells are half thawed (typically 1-2 min), add 1:1 volume of warm media to the vial.
Pipet up and down several times, until the cells are completely thawed.
Immediately transfer the cells into the 15 mL tube with warm media.
Spin cells down at 500 × g for 5 min at room temperature.
Discard the supernatant and resuspend the cell pellet in 1 mL of warm media.
Transfer the resuspended cells to a microfuge tube.
Add 10 μL of the aliquoted DNase stock solution (see Recipe 1) to the resuspended cells and incubate for 30 min at 37°C (final DNase concentration in each sample is 200 Units/mL).
Spin cells down at 500 × g for 5 min at 4°C.
Discard the supernatant and resuspend the cell pellet with 1 mL of ice-cooled PBS (first PBS wash).
Spin cells down at 500 × g for 5 min at 4°C.
Repeat Steps A13-A14.
Resuspend the final pellet with 1 mL of ice-cooled PBS, count cells, and estimate viability using a cell counter. Using a dual-fluorescence (e.g., acridine orange and propidium iodide) imaging-based automated cell counter gives rapid and fairly accurate cell counts and viability estimates.
Cell lysis, transposition, and DNA extraction (day 1)
Notes:
To perform a successful ATAC-seq experiment, it is crucial to use the right cell-to-transposase ratio. Using too few cells will over-digest the chromatin, causing enrichment of smaller DNA fragments that may get preferentially sequenced, increasing the background noise in the data. On the other hand, an excess of cells will result in under-digestion, causing production of large DNA molecules that may be difficult to sequence.
The authors of the OMNI-ATAC manuscripts (Buenrostro et al., 2015; Corces et al., 2017) have optimized the number of cells for this assay to 50,000. Thus, we have used this number for our routine experiments. If more or less cells need to be used, the cell-to-reagents ratio should be maintained, by adjusting the volumes of all reagents accordingly.
Spin 50,000 viable cells down at 500 × g for 5 min at 4°C in an Eppendorf centrifuge.
Remove the supernatant very carefully, without disturbing the pelleted cells. This process can be difficult since sometimes the pellets are almost invisible. To avoid accidentally aspirating the cells, we recommend first using a P1000 pipette until the last 150-200 μL, then a P200 pipette until the last 15-20 μL is left in the tube, and finally removing the remaining volume with a P20 pipette. If during this process some of the cells are accidentally disturbed, a second centrifugation at 500 × g, for 5 min at 4°C will be required.
To lyse the cells, add 50 μL of the cold ATAC-RSB with detergents buffer previously prepared (see Recipes 2 and 3).
Resuspend the cells with a P200 pipette up and down three times, and immediately incubate on ice for 3 min. If preparing more than one sample, ensure that all the samples have the same incubation time.
Add 1 mL of the wash-out lysis buffer (see Recipe 4) and invert the tube three times to mix.
Spin down the resulting nuclei solution at 500 × g for 10 min at 4°C in an Eppendorf centrifuge.
Aspirate the supernatant very carefully, as described in Step B2.
Resuspend the nuclei pellet in 50 μL of the transposition mix (see Recipe 5), and mix it by pipetting up and down six times.
Using a thermomixer, incubate the samples at 37°C with agitation (1,000 RPM) for 30 min. In this step, the transposase will fragment and tag the accessible DNA with adaptors.
Extract the DNA from the transposed nuclei using a Zymo DNA Clean and Concentrator-5 kit, according to the manufacturer’s instructions. Elute DNA in 21 μL of elution buffer. At this step, DNA can be stored at -20°C, until ready to prepare the library by PCR amplification.
Library preparation (day 2)
Notes:
During this part of the protocol, the DNA that was fragmented and tagged with adaptors in section B is PCR amplified using barcoded primers.
To pool up to 24 libraries together, a list of 24 barcoded oligos (Ad2.1-Ad2.24) is in Table 1.
In our experience, when starting with approximately 50,000 cells, the number of PCR cycles to have a successful ATAC-seq experiment without library over-amplification is between 7 and 9. In case of high cell viability (over 80%), we run the PCR for 7 cycles. In cases of low viability (less than 50%), we do 9. However, to avoid the risk of size and GC content PCR bias, the appropriate number of cycles can be determined by qPCR. Buenrostro et al. (2015) has a full explanation on how to calculate the precise number of PCR cycles.
Prepare the following PCR reaction mix in a PCR tube.
25 μL of 2× NEBNext Master Mix
2.5 μL of 25 µM primer Ad1
2.5 μL of 25 µM barcoded primer Ad2.1
20 μL of transposed DNA sample
When preparing different samples in the same pool for sequencing, each individual sample PCR reaction mix will have a different barcoded primer Ad2.
Perform PCR using the following cycling conditions:
1 = 72°C for 5 min
2 = 98°C for 30 s
3 = 98°C for 10 s
4 = 63°C for 30 s
5 = 72°C for 1 min
6 = Go to step 3, six times
7 = Hold at 4°C
Table 1. List of barcoded primers.
Ad1_noMX: AATGATACGGCGACCACCGAGATCTACACTCGTCGGCAGCGTCAGATGTG Ad2.1_TAAGGCGA CAAGCAGAAGACGGCATACGAGATTCGCCTTAGTCTCGTGGGCTCGGAGATGT Ad2.2_CGTACTAG CAAGCAGAAGACGGCATACGAGATCTAGTACGGTCTCGTGGGCTCGGAGATGT Ad2.3_AGGCAGAA CAAGCAGAAGACGGCATACGAGATTTCTGCCTGTCTCGTGGGCTCGGAGATGT Ad2.4_TCCTGAGC CAAGCAGAAGACGGCATACGAGATGCTCAGGAGTCTCGTGGGCTCGGAGATGT Ad2.5_GGACTCCT CAAGCAGAAGACGGCATACGAGATAGGAGTCCGTCTCGTGGGCTCGGAGATGT Ad2.6_TAGGCATG CAAGCAGAAGACGGCATACGAGATCATGCCTAGTCTCGTGGGCTCGGAGATGT Ad2.7_CTCTCTAC CAAGCAGAAGACGGCATACGAGATGTAGAGAGGTCTCGTGGGCTCGGAGATGT Ad2.8_CAGAGAGG CAAGCAGAAGACGGCATACGAGATCCTCTCTGGTCTCGTGGGCTCGGAGATGT Ad2.9_GCTACGCT CAAGCAGAAGACGGCATACGAGATAGCGTAGCGTCTCGTGGGCTCGGAGATGT Ad2.10_CGAGGCTG CAAGCAGAAGACGGCATACGAGATCAGCCTCGGTCTCGTGGGCTCGGAGATGT Ad2.11_AAGAGGCA CAAGCAGAAGACGGCATACGAGATTGCCTCTTGTCTCGTGGGCTCGGAGATGT Ad2.12_GTAGAGGA CAAGCAGAAGACGGCATACGAGATTCCTCTACGTCTCGTGGGCTCGGAGATGT Ad2.13_GTCGTGAT CAAGCAGAAGACGGCATACGAGATATCACGACGTCTCGTGGGCTCGGAGATGT Ad2.14_ACCACTGT CAAGCAGAAGACGGCATACGAGATACAGTGGTGTCTCGTGGGCTCGGAGATGT Ad2.15_TGGATCTG CAAGCAGAAGACGGCATACGAGATCAGATCCAGTCTCGTGGGCTCGGAGATGT Ad2.16_CCGTTTGT CAAGCAGAAGACGGCATACGAGATACAAACGGGTCTCGTGGGCTCGGAGATGT Ad2.17_TGCTGGGT CAAGCAGAAGACGGCATACGAGATACCCAGCAGTCTCGTGGGCTCGGAGATGT Ad2.18_GAGGGGTT CAAGCAGAAGACGGCATACGAGATAACCCCTCGTCTCGTGGGCTCGGAGATGT Ad2.19_AGGTTGGG CAAGCAGAAGACGGCATACGAGATCCCAACCTGTCTCGTGGGCTCGGAGATGT Ad2.20_GTGTGGTG CAAGCAGAAGACGGCATACGAGATCACCACACGTCTCGTGGGCTCGGAGATGT Ad2.21_TGGGTTTC CAAGCAGAAGACGGCATACGAGATGAAACCCAGTCTCGTGGGCTCGGAGATGT Ad2.22_TGGTCACA CAAGCAGAAGACGGCATACGAGATTGTGACCAGTCTCGTGGGCTCGGAGATGT Ad2.23_TTGACCCT CAAGCAGAAGACGGCATACGAGATAGGGTCAAGTCTCGTGGGCTCGGAGATGT Ad2.24_CCACTCCT CAAGCAGAAGACGGCATACGAGATAGGAGTGGGTCTCGTGGGCTCGGAGATGT
Library purification (day 2)
Note: After the PCR is completed, double-sided bead purification is necessary, to remove primer dimers and fragments over 1,000 bp.
Perform a double-sided SPRI bead purification.
Add 0.5× volume (25 μL) of SPRI beads to the PCR samples (50 μL), and pipet up and down 10 times to mix thoroughly.
Quickly spin the tubes down for 3 s and incubate for 10 min at room temperature.
Place PCR tubes in a magnetic rack for 5 min, or until the supernatant is clear.
Carefully transfer the supernatant to a new PCR tube.
Add 1.3× original volume (65 μL) of SPRI beads, and pipet up and down 10 times to mix thoroughly.
Quickly spin the tubes down for 3 s and incubate for 10 min at room temperature.
Place PCR tubes in a magnetic rack for 5 min or until the supernatant is clear.
Carefully remove the supernatant and discard it.
Wash beads twice with 200 µL of 80% freshly made ethanol.
Eliminate ethanol traces by spinning the tubes down for 3 s, placing them back in a magnetic rack, and aspirating the ethanol with a P10 pipette tip.
Resuspend beads in 20 μL of nuclease-free water.
Quickly spin the tubes down for 3 s and incubate for 1 min at room temperature.
Place PCR tubes in a magnetic rack for 5 min or until the supernatant is clear.
Transfer 19 μL of supernatant to a new tube.
Check fragment size distribution profiles of the samples by running 0.5 μL on high sensitivity chip on a Bioanalyzer (Figure 1).
Samples can be stored at -20°C until the next step.
Note: Figure 1A shows a typical ATAC-seq bioanalyzer profile, after the first bead clean-up. Depending on the initial cell viability, cell type, experimental treatment, number of transposed nuclei,and number of PCR cycles, the nucleosomal banding pattern may have a different outcome.
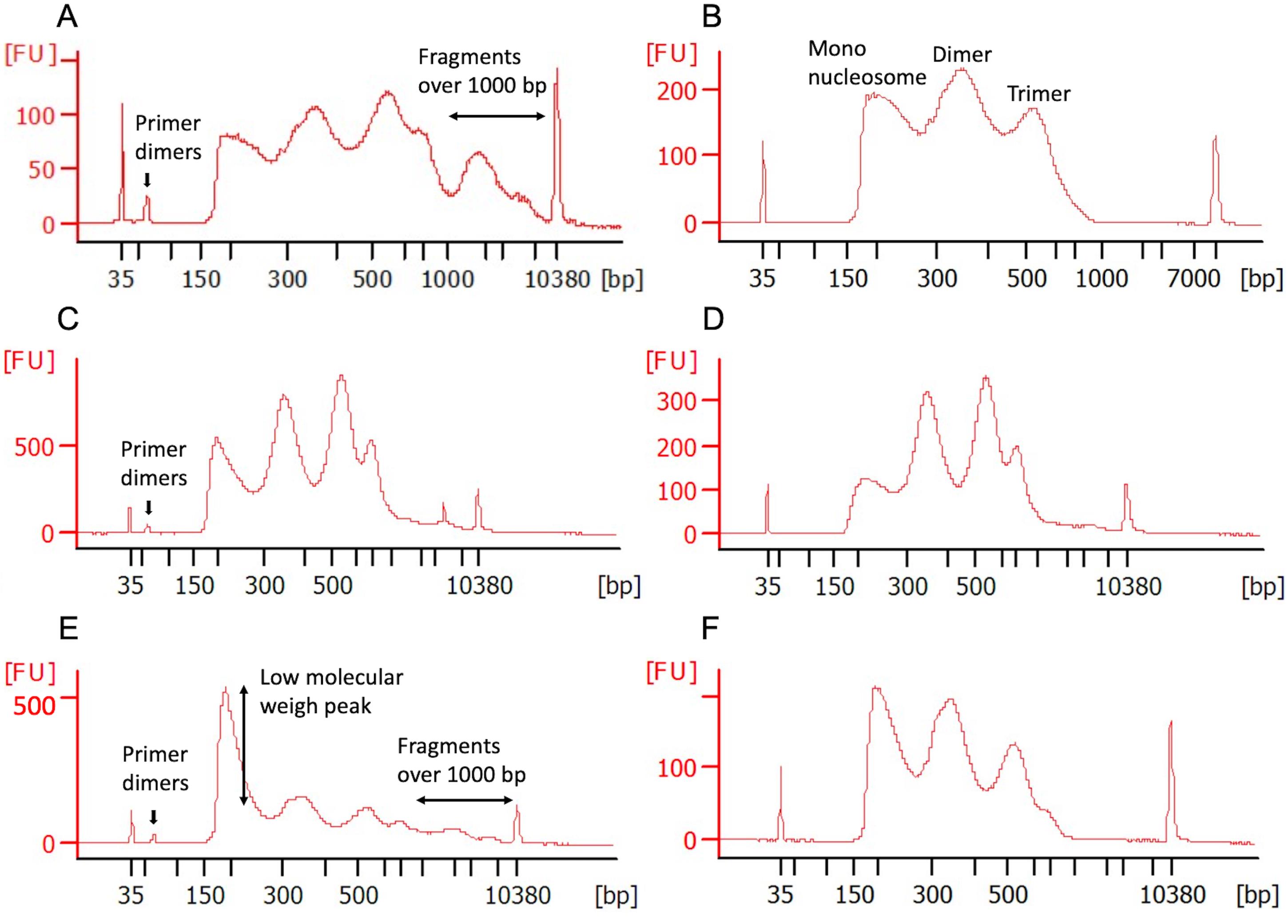
Figure 1. High Sensitivity Bioanalyzer profile of several ATAC-seq libraries after a variety of magnetic bead purifications. A. ATAC-seq profile after the first double size selection clean-up. Primer dimers and fragments over 1,000 bp are still present in this library. B. To remove all the unwanted DNA, a second double size selection purification was performed. The peaks in the final library indicate the mono-, di-, and tri-nucleosome-associated fragments. C. ATAC-seq profile after the first double size selection clean-up, showing the presence of primer dimers. D. To remove all the primer dimers, a second clean-up as described in Step D2 was conducted. E. ATAC-seq profile after the first double size selection clean-up, showing the presence of primer dimers, fragments over 1000 bp, and a big first peak. F. To reduce the size of the low molecular weight peak, a 0.95× instead of a 1× beads ratio was applied during the second purification (see notes in section D for more details).
Notes:
In our experience, cells with very low viability (less than 30%), usually do not show good amplification after 9 cycles of PCR. Though these libraries can be further amplified and sometimes a pattern resembling the nucleosomal banding pattern may appear, the risk of PCR bias is very high for such samples, and we recommend not to proceed with the sequencing of such samples.
If after 9 PCR cycles, samples with good starting cell viability do not show amplification, we recommend a few extra amplification cycles (no more than 4).
After the first library purification, most of the experiments will still have primer dimers and/or fragments over 1,000 bp (Figure 1A and 1C). Another issue could be the presence of a big low molecular weight peak (3 to 4 times taller than the other 2 peaks, Figure 1E). The presence of this peak could be due to over-digestion with the transposase or over-PCR amplification. This big peak can be problematic during the sequencing process, due to the fact that the DNA in this peak will be preferentially sequenced, because of its lower molecular weight compared to the other two peaks. In all three cases, the library should be submitted to a second bead purification: (i) Libraries with primer dimers and fragments over 1,000 bp (Figure 1A) will need the double-sided bead clean-up described in Step D1. On this occasion, the beads ratio in Step D1e should be 1×, and the incubation times in Steps D1b and D1f should only be 5 min (Figure 1B). (ii) If the library only has primer dimers (Figure 1C), we recommend doing a left-side clean-up with a 1× beads ratio (Step D2) (Figure 1D). 3) If in both situations the big low molecular size peak is also present (Figure 1E), the beads ratio in Steps D2b and D1e should be 0.95× (Figure 1F).
If needed, perform a left-sided SPRI bead purification.
Add nuclease-free water to the sample from Step E1n up to 50 μL.
Add 1× volume (50 μL) SPRI beads to the PCR samples, and pipet up and down 10 times to mix thoroughly.
Quickly spin the tubes down for 3 s and incubate for 5 min at room temperature.
Place PCR tubes in a magnetic rack for 5 min or until the supernatant is clear.
Carefully remove supernatant and discard it.
Wash beads twice with 200 μL of 80% freshly made ethanol.
Eliminate ethanol traces by spinning the tubes down for 3 s, placing them back in a magnetic rack, and aspirating the ethanol with a P10 pipet tip.
Resuspend beads in 15 μL of nuclease-free water.
Quickly spin the tubes down for 3 s and incubate for 1 min at room temperature.
Place PCR tubes in a magnetic rack for 5 min or until the supernatant is clear.
Transfer 14 μL of supernatant to a new tube.
Run 1 μL of the sample with high sensitivity chip Bioanalyzer (Figure 1D).
Samples can be stored at -20°C.
Data analysis
The minimum library quantity required for pooling and sequencing for any Illumina platform is 5 nM in 10 μL.
Add libraries in the same pool by equimolarity. We advise qPCR for accurate estimation of library concentrations.
We recommend paired-end sequencing on an Illumina platform NextSeq1000/2000 or NovaSeq6000, with 100 million minimum reads per sample with 2×100 bps read length, or NextSeq500/550, with 2×75 bps read length.
For downstream data analysis it is necessary to have at least two replicates per sample.
Recipes
20,000 Units/mL DNase stock solution
Make a 20,000 Units/mL DNase stock solution. Resuspend the lyophilized DNase powder (100 mg) in 10 mL of Hanks balanced salt solution (HBSS). This corresponds to 10 mg/mL. As 1 mg of DNase equals to 2,000 Units, this means that 10 mg/mL are 20,000 Units/mL. Aliquot the stock solution in microfuge tubes and keep them at 4°C for up to 3 weeks. Otherwise, keep the aliquots at -20°C.
ATAC-RSB buffer (Table 2)
Prepare buffer ATAC-RSB as described in Table 2. This buffer can be stored at room temperature for months.
Table 2. Recipe for ATAC-RSB buffer.
Reagent Initial concentration Final concentration Volume for 100 mL Tris-HCl pH 7.4 1 M 10 mM 1,000 μL NaCl 5 M 10 mM 200 μL MgCl2 1 M 3 mM 300 μL Nuclease-free water - - 98.5 μL ATAC-RSB with detergents buffer (Table 3)
Prepare ATAC-RSB with detergents buffer on the day of the experiment and store it at 4°C.
Table 3. Recipe for ATAC-RSB with detergents buffer (50 μL per sample).
Reagent Initial concentration Final concentration Volume for 1,000 μL NP40 10% 0.1% 10 μL Tween-20 10% 0.1% 10 μL Digitonin 1% 0.01% 10 μL ATAC-RSB buffer - - 970 μL Wash-out buffer (Table 4)
Prepare wash-out buffer on the day of the experiment and store it at 4°C.
Table 4. Recipe for wash out lysis buffer (1 mL per sample).
Reagent Initial concentration Final concentration Volume for 10 mL NP40 10% 0.1% 100 μL ATAC-RSB buffer - - 9,900 μL Transposition mix (Table 5)
Prepare transposition mix during step 6 in the ATAC-seq protocol section according to Table 5, and keep it on ice. If preparing more than one sample, add an additional excess of each reagent.
Table 5. Recipe for transposition mix (50 μL per sample).
Reagent Initial concentration Final concentration Volume for 50 μL TD buffer 2× 1× 25 μL Transposase
PBS
Digitonin
Tween-20
Nuclease-free water
20,000 nM
-
1%
10%
-
100 nM
-
0.01%
0.1%
-
2.5 μL
16.5 μL
0.5 μL
0.5 μL
5 μL
Acknowledgments
We are thankful to the authors of OMNI ATAC-seq manuscript Dr. Chang and Dr. Greenleaf for their consent for publication of this protocol. We thank all members of the CCR Sequencing Facility at the Frederick National Laboratory for Cancer Research for their help during sample preparation and sequencing, especially Yuliya Kriga and Yunlong He for assistance in library preparation, Oksana German for library QC, and Tatyana Smirnova for running the sequencers. We would also like to thank all members of the Sequencing Facility Bioinformatics Group for data analysis. This protocol was used in the research manuscript available at Krishna et al. (2020, DOI: 10.1126/science.abb9847).
Competing interests
No competing interests to declare.
References
- Boyle, A. P., Guinney, J., Crawford, G. E. and Furey, T. S. (2008). F-Seq: a feature density estimator for high-throughput sequence tags. Bioinformatics 24(21): 2537-2538.
- Buenrostro, J. D., Giresi, P. G., Zaba, L. C., Chang, H. Y. and Greenleaf, W. J. (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10(12): 1213-1218.
- Buenrostro, J. D., Wu, B., Chang, H. Y. and Greenleaf, W. J. (2015). ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr Protoc Mol Biol 109: 21 29 21-21 29 29.
- Corces, M. R., Trevino, A. E., Hamilton, E. G., Greenside, P. G., Sinnott-Armstrong, N. A., Vesuna, S., Satpathy, A. T., Rubin, A. J., Montine, K. S., Wu, B., et al. (2017). An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat Methods 14(10): 959-962.
- Doganli, C., Sandoval, M., Thomas, S. and Hart, D. (2017). Assay for Transposase-Accessible Chromatin with High-Throughput Sequencing (ATAC-Seq) Protocol for Zebrafish Embryos. Methods Mol Biol 1507: 59-66.
- Fujiwara, S., Baek, S., Varticovski, L., Kim, S. and Hager, G. L. (2019). High quality ATAC-seq data recovered from cryopreserved breast cell lines and tissue. Sci Rep 9(1): 516.
- Giresi, P. G., Kim, J., McDaniell, R. M., Iyer, V. R. and Lieb, J. D. (2007). FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome Res 17(6): 877-885.
- Hess, J. F., Kohl, T. A., Kotrova, M., Ronsch, K., Paprotka, T., Mohr, V., Hutzenlaub, T., Bruggemann, M., Zengerle, R., Niemann, S. and Paust, N. (2020). Library preparation for next generation sequencing: A review of automation strategies. Biotechnol Adv 41: 107537.
- Kornberg, R. D. (1974). Chromatin structure: a repeating unit of histones and DNA. Science 184(4139): 868-871.
- Krishna, S., Lowery, F. J., Copeland, A. R., Bahadiroglu, E., Mukherjee, R., Jia, L., Anibal, J. T., Sachs, A., Adebola, S. O., Gurusamy, D., et al. (2020). Stem-like CD8 T cells mediate response of adoptive cell immunotherapy against human cancer. Science 370(6522): 1328-1334.
- Shashikant, T. and Ettensohn, C. A. (2019). Genome-wide analysis of chromatin accessibility using ATAC-seq. Methods Cell Biol 151: 219-235.
- Sun, Y., Miao, N. and Sun, T. (2019). Detect accessible chromatin using ATAC-sequencing, from principle to applications. Hereditas 156: 29.
- Wegel, E. and Shaw, P. (2005). Gene activation and deactivation related changes in the three-dimensional structure of chromatin. Chromosoma 114(5): 331-337.
- Zaret, K. (2005). Micrococcal nuclease analysis of chromatin structure. Curr Protoc Mol Biol Chapter 21: Unit 21.1.
Article Information
Copyright
© 2022 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Caravaca, J. M., Mehta, M., Gowda, S. and Tran, B. (2022). ATAC Sequencing Protocol For Cryopreserved Mammalian Cells . Bio-protocol 12(2): e4294. DOI: 10.21769/BioProtoc.4294.
- Krishna, S., Lowery, F. J., Copeland, A. R., Bahadiroglu, E., Mukherjee, R., Jia, L., Anibal, J. T., Sachs, A., Adebola, S. O., Gurusamy, D., et al. (2020). Stem-like CD8 T cells mediate response of adoptive cell immunotherapy against human cancer. Science 370(6522): 1328-1334.
Category
Molecular Biology > DNA > Chromatin accessibility
Cancer Biology > Cancer biochemistry > Cancer metabolism
Biochemistry > DNA
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.