- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Heterologous Expression and High Degree Purification of the Restriction Endonuclease SauUSI
Published: Vol 12, Iss 1, Jan 5, 2022 DOI: 10.21769/BioProtoc.4275 Views: 3935
Reviewed by: Alba BlesaNingfei AnAnonymous reviewer(s)
Original research article
The authors used this protocol in:
Feb 2021
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Mechanisms that target and destroy foreign nucleic acids are major barriers to horizontal gene transfer (HGT) in prokaryotes. Amongst them, restriction-modification (R-M) systems are found in ≥75% of the sequenced genomes in Bacteria and Archaea. Due to their high target sequence specificity and potent nucleolytic activity, R-M systems are used as a paradigm to elucidate the mechanisms of DNA binding and cleavage. Since these enzymes modulate HGT, they are one of the machineries implicated in the ability of a bacterium to gain antibiotic resistance. This protocol provides a detailed purification strategy for the Type IV restriction endonuclease SauUSI from Staphylococcus aureus. This protocol eventually leads to ≥95% purity of protein which can then be used for crystallographic and biochemical purposes.
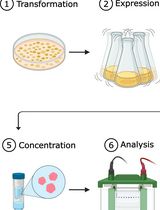
Graphic abstract:
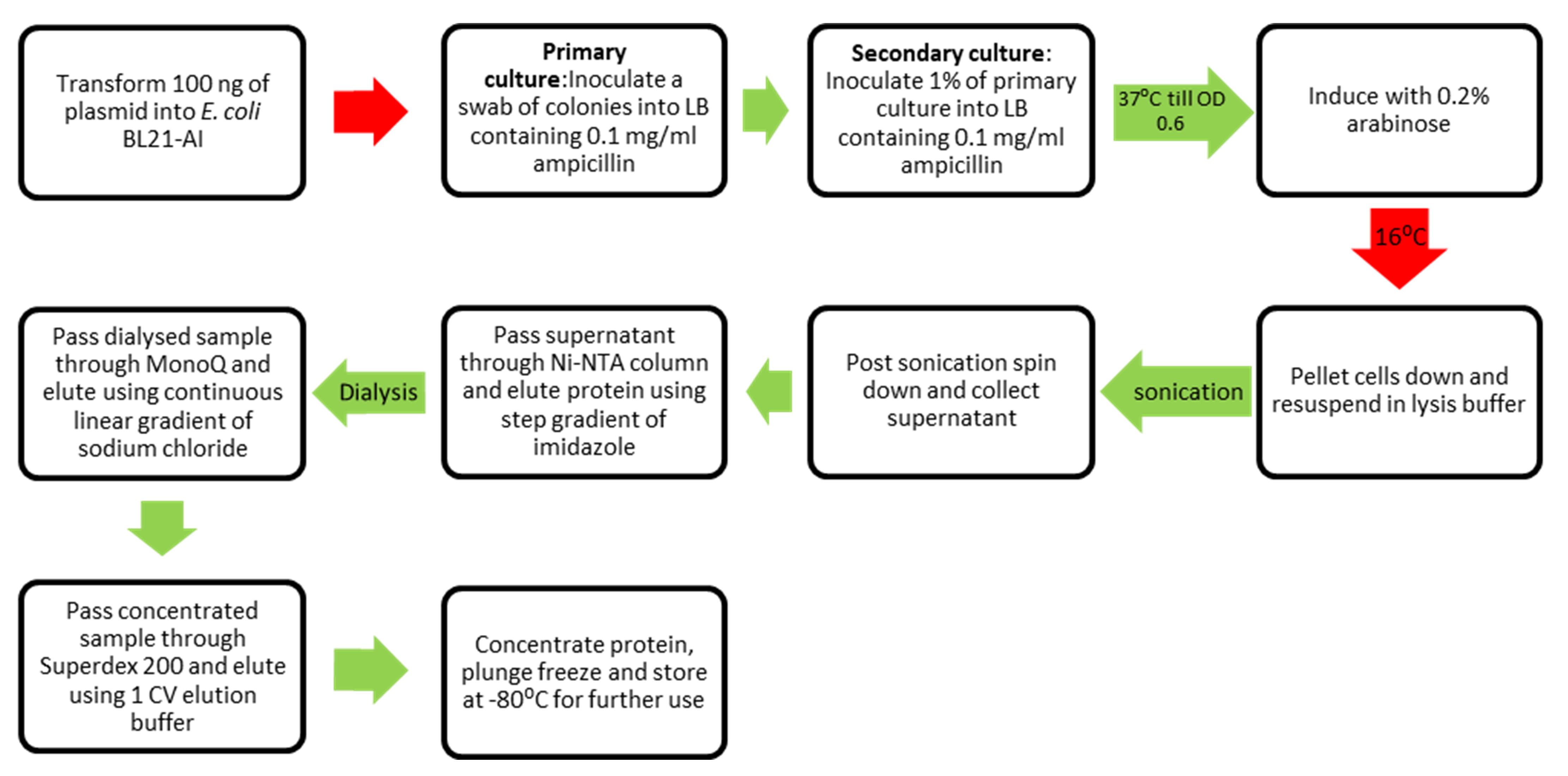
Workflow for purification of SauUSI.
Background
Restriction-modification (R-M) systems function as the innate immune system of bacteria, thus preventing not only infection by bacteriophages but also other potentially harmful mobile genetic elements. There are a variety of bacterial R-M systems that differ in their target site epigenetic status, oligomeric structure, cofactor requirements, and mode of DNA cleavage (Loenen et al., 2014). SauUSI is a Type IV restriction endonuclease present in Staphylococcus aureus that binds specifically to the methylated DNA target sequence 5’-S5mCNGS-3’ (S=C/G and N=A/T/G/C) (Xu et al., 2011). SauUSI has the ability to cleave DNA by two distinct mechanisms; they are: a) a highly efficient convergence-based cleavage mechanism when there are two or more target sequences, and b) a lesser efficient switch-based mechanism on DNA with only one target sequence (Tumuluri et al., 2021). Studies on the cleavage activity of SauUSI highlight its role in preventing the acquisition of exogenous genetic elements that have the ability to transform methicillin-resistant Staphylococcus aureus (MRSA) into vancomycin-resistant Staphylococcus aureus (VRSA) (Corvaglia et al., 2010; Monk et al., 2012; Tumuluri et al., 2021).
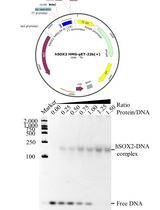
For the in-vitro biochemical characterization and crystallographic studies, SauUSI was purified post expression in the heterologous Escherichia coli BL21-AI cells. For this purpose, the sauUSIR gene was cloned into a pHis17 (ampicillin resistant) vector (Kunzelmann and Webb, 2009), between the NdeI (upstream) and BamHI (downstream) restriction sites (Figure 1), using a restriction-free (RF) cloning strategy (van den Ent and Löwe, 2006; Tumuluri et al., 2021). The RF cloning primarily consisted of two sequential polymerase chain reactions (PCRs): the first reaction was to amplify the sauUSIR gene with flanking regions complementary to the pHis17 vector, and the second reaction was an extension PCR to incorporate the amplified sauUSIR gene into the vector. The reaction was treated with DpnI to cleave the parent template, and subsequentially transformed into Escherichia coli NEB Turbo® cells. The presence of the sauUSIR gene was confirmed by DNA sequencing.
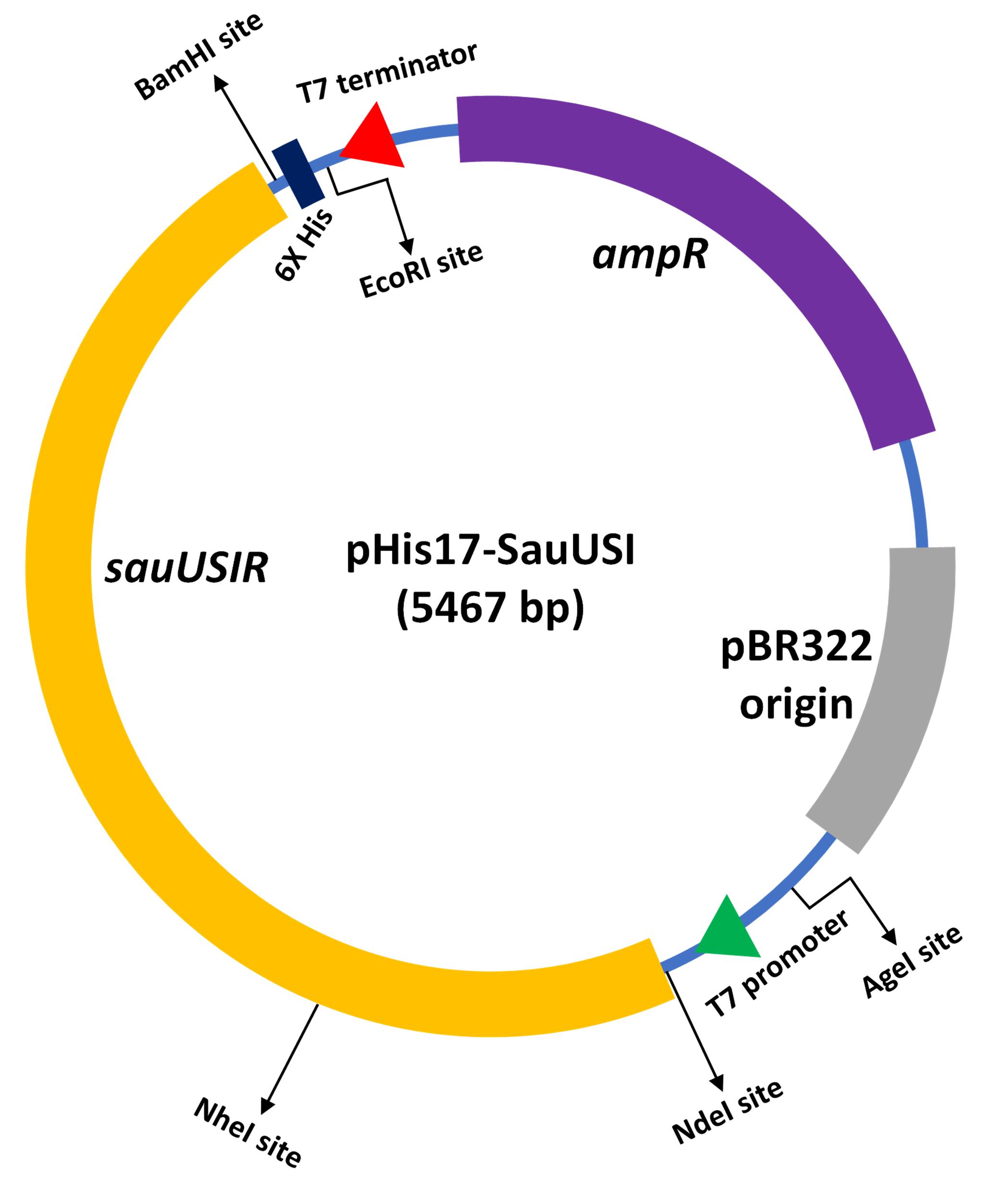
Figure 1. Cartoon representation of the expression vector integrated with sauUSIR gene. The sauUSIR gene is 2859 bp and codes of a 953 amino acid single polypeptide protein. This gene is integrated between NdeI and BamHI sites in a pHis 17 vector thus providing a C-terminal 6× Histidine tag for ease of purification.
Materials and Reagents
100 mm Petri dishes (HiMedia, catalog number: PW1305-1x100NO)
50 mL beaker (Borosil)
200 mL conical flask (Borosil)
2000 mL culture flask (Borosil)
50 mL centrifuge tubes (Eppendorf)
15 mL centrifuge tubes (Eppendorf)
1.5 mL fraction tubes (Eppendorf)
0.5 mL fraction tubes (Eppendorf)
SnakeSkinTM dialysis tubing 10000 MWCO (ThermoFisher, catalog number: 68100)
0.2 µm syringe filter (WhatmanTM, catalog number: 9914-2502)
0.2 µm filter membranes (Millipore, catalog number: GVWP04700)
Whatman filter paper (No. 1) (Filtration laboratory, catalog number: T-06648-13)
Lysogeny broth (LB) (HiMedia, catalog number: M1245-1KG)
Luria-Bertani (LB) Agar (HiMedia, catalog number: M1151-1KG)
Ampicillin (Sigma-Aldrich, catalog number: A9518-25G)
Arabinose (SRL, catalog number: 52392)
Sodium Chloride (NaCl) (Sigma-Aldrich, catalog number: S9888-1KG)
Tris(hydroxymethyl)aminomethane (Tris-Base) (Sigma-Aldrich, catalog number: 252859-500G)
Hydrochloric acid (HCl) for molecular biology (Qualigens, catalog number: Q29505)
Imidazole (Sigma-Aldrich, catalog number: I2399-500G)
Magnesium Chloride (MgCl2) (Sigma-Aldrich, catalog number: M8266-100G)
3-[(3-cholamidopropyl) dimethylammonio]-1-propanesulfonate (CHAPS) (Sigma-Aldrich, catalog number: 226947-5G)
Glycerol for molecular biology (Sigma-Aldrich, catalog number: 356352-1L-M)
Ethylenediaminetetraacetic acid (EDTA) (Sigma-Aldrich, catalog number: ED-500G)
1,4-Dithiothreitol (DTT) (Sigma-Aldrich, catalog number: 10708984001)
Glycine (Sigma-Aldrich, catalog number: G8898-1KG)
Sodium dodecyl sulfate (SDS) (Sigma-Aldrich, catalog number: L3771-100G)
Ammonium persulfate (APS) (Sigma-Aldrich, catalog number: A3678-25G)
Tetramethylethylenediamine (TEMED) (Sigma-Aldrich, catalog number: T9281-25ML)
Acrylamide (Sigma-Aldrich, catalog number: A8887-500G)
Bis-acrylamide (Sigma-Aldrich, catalog number: 146072-500G)
Bio-Rad Precision Plus ProteinTM Standards (Bio-Rad, catalog number: 1610374)
2× Laemmli sample buffer (Bio-Rad, catalog number: 1610737EDU)
Coomassie Brilliant Blue (R-250) (Sigma-Aldrich, catalog number: 1125530025)
Ethanol for molecular biology
Glacial Acetic acid for molecular biology (Qualigens, catalog number: Q21055)
Liquid nitrogen
Stock solutions (see Recipes)
20% (w/v) Arabinose
5 M NaCl
2 M Tris-HCl (pH = 8)
2 M Imidazole
4% (w/v) CHAPS
2 M MgCl2
0.5 M EDTA (pH = 8)
1 M DTT
Buffers for purification (see Recipes)
Lysis buffer
Buffers for affinity (Ni-NTA) chromatography (Buffer A and Buffer B)
Buffer for Dialysis (Buffer B0)
Buffers for anion exchange chromatography (Buffer B50 and Buffer B1000)
Buffer for size exclusion chromatography (Buffer B100)
SDS-PAGE (see Recipes)
10× stock of TGS (Tris base, glycine, and SDS) running buffer
Coomassie staining solution
Coomassie destaining solution
Solutions required for Gel mix
10% resolving gel
5% stacking gel
Equipment
Thermoblock (Eppendorf ThermoMixer C, catalog number: 5382000015)
Shaker incubator (ThermoScientific, model: MXQ 6000)
Avanti J26S-XP centrifuge (Beckman Coulter, model: Avanti J26S-XP, Rotor: JLA-9.1000)
Optima-L-100K ultracentrifuge (Beckman Coulter, model: Optima L-100K, Rotor: Type 45 Ti)
GE AKTAprime plus FPLC system (GMI, catalog number: 8149-30-0004)
Bio-Rad NGCTM chromatography system (Bio-Rad, catalog number: 788001)
Vacuum filter assembly (Millipore, model: XX1014720)
Sonicator (Sonics-Vibra cell) (Sonics, model: VCX 130)
HisTrapTM HP 5 mL Ni-NTA column (GE, catalog number: 17524802)
MonoQTM 10/100 GL column (GE, catalog number: 17516701)
SuperloopTM (50 mL) (GE, catalog number: 18111382)
Superdex 200TM 16/600 pg column (GE, catalog number: GE28-9893-35)
SDS-PAGE apparatus (Bio-Rad, catalog number: 1658001FC)
VivaspinR 10000 MWCO centrifugal concentrator (Sartorius, catalog number: Z614602-12EA)
Software
Microsoft Excel
GraphPad Prism
ImageJ
Procedure
Bacterial cell transformation
Thaw BL21 (AI) competent cells on ice.
Under sterile conditions, add 100 ng of plasmid to 50 µL of competent cells, and place on ice for 12-15 min.
Transfer the cells to a thermoblock (heat shock) at 37°C for 3 min, or at 42°C for 1.5 min.
Transfer the cells back to ice for 2 min.
Add 100 µL of 1× LB to the cells, place at 37°C for 10 min, and plate all the cells on a LB Agar plate containing 0.1 mg/mL Ampicillin.
Incubate the plate at 37°C overnight.
Culture growth
Primary culture
Inoculate a swab (~15-20 colonies) of the transformed cells into 40 mL of 1× LB containing 0.1 mg/mL Ampicillin, and grow at 37°C in a shaker incubator (180 RPM) for 5-6 h.
Secondary culture
Inoculate 1% of primary culture in 1 L of 1× LB containing 0.1 mg/mL Ampicillin, grow at 37°C in a shaker incubator (180 RPM), and monitor the optical density (OD) of the secondary culture.
Once the OD reaches 0.4-0.5, change the temperature of the shaker incubator (180 RPM) to 16°C.
Once the OD at 600 nm reaches 0.6, induce the culture with 0.2% (w/v) Arabinose, and grow overnight (12-14 h) at 16°C.
Cell pelleting, lysis, and lysate clarification
Cell pelleting
Pellet the cells down using a Avanti J26X-SP centrifuge at an RCF of 5180 (~4,000-5,000 RPM) for 20 min, maintaining the temperature at 4°C, and then discard the supernatant.
Either store the pellet at -80°C after plunge freezing in liquid nitrogen, or process it immediately for lysis.
Bacterial cell lysis
Weigh the cell pellet and resuspend it in lysis buffer (volume of the lysis buffer to be added is 5 times the weight of the cell pellet, i.e., 5 mL of lysis buffer for every 1 g of cell pellet).
Lyse the cells on ice using a sonicator set at 60% amplitude, with a pulse of 1 s and a recovery of 3 s, for 3 min. Post this, allow the lysate to sit on ice for 5 min. Then, continue the sonication protocol for another 2 min.
Lysate clarification
Centrifuge the lysate using an Optima-L-100K ultracentrifuge for 50 min at 100,000 × g, maintaining the temperature at 4°C.
Transfer the lysate to a pre-chilled tube or conical flask, for loading onto the Ni-NTA column.
Ni-NTA column chromatography (perform at 4°C, using a GE AKTAprime plus FPLC system)
Equilibrate a GE HisTrapTM HP 5 mL Ni-NTA column with 3 column volumes (CV) of Buffer A.
Load the supernatant onto the columns at a slow flow rate ~1 mL/min.
After the supernatant is loaded, wash the column with 20 CV Buffer A at a flow rate of 2 mL/min.
Elute the sample using a step gradient (5%, 10%, 20%, 30%, 50%, and 100%) of Buffer B. Gradients of Buffer B were made by mixing of Buffer A and Buffer B. Elute two fractions of 5 mL each at each step.
Load alternate fractions on a 10% SDS-PAGE gel with an appropriate protein ladder (Bio-Rad Precision Plus ProteinTM Standards) (Figure 2). The protein usually elutes in the step gradients of 5% to 30% of Buffer B. Subsequently, based on the SDS-PAGE gel, pool the fractions with most protein, which are usually fractions corresponding to 10%, 20%, and 30% of Buffer B (Figure 2).
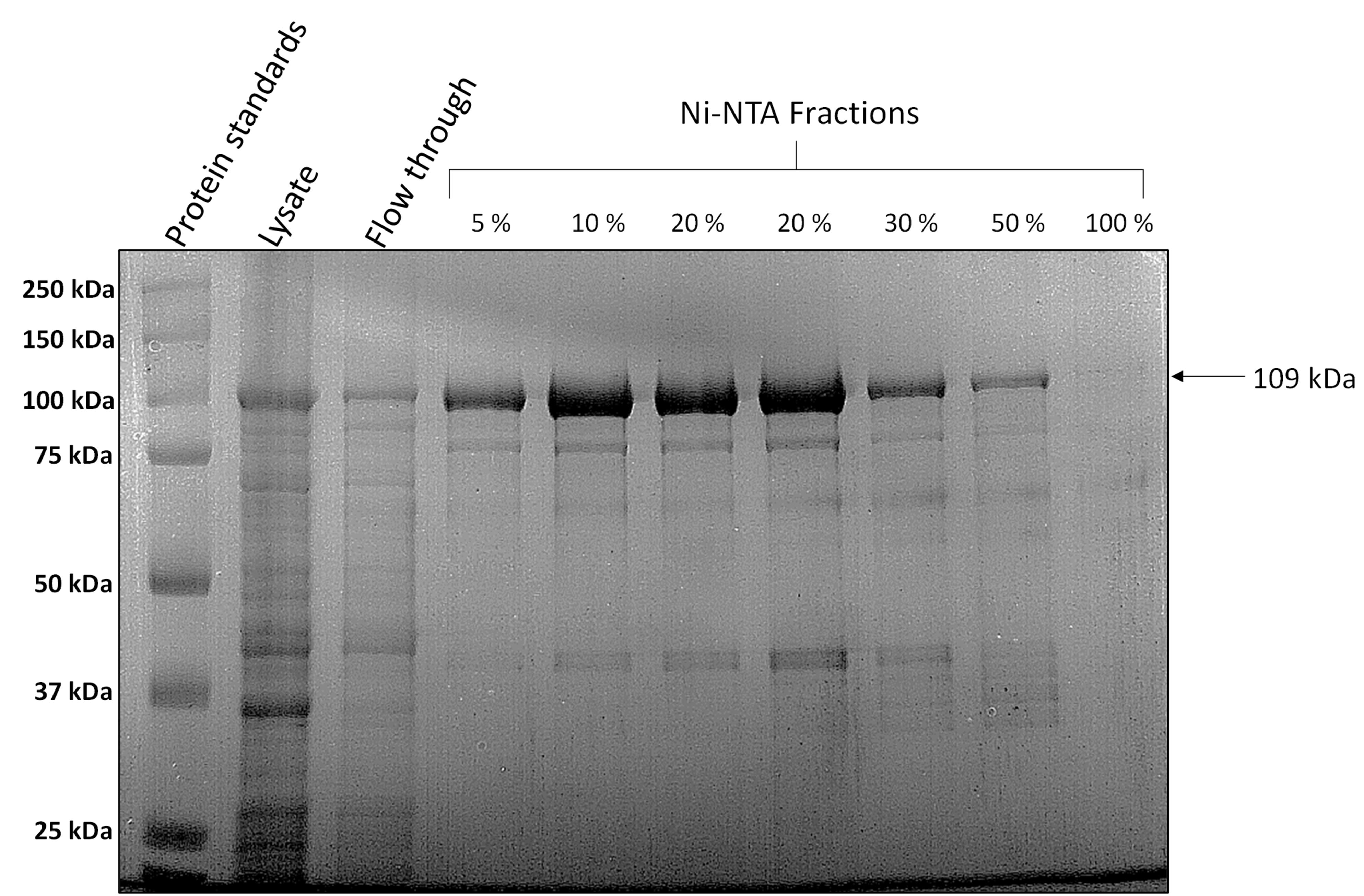
Figure 2. Representative 10% SDS-PAGE after Ni-NTA chromatography. Load 10 µL of sample (5 µL of the protein fraction mixed with 5 µL of 2× Laemmli sample buffer) on a 10% SDS-PAGE gel. The fractions with the most protein are pooled and dialysed. The SDS-PAGE gel is stained with Coomassie R-250.
Dialysis (perform at 4°C, using SnakeSkinTM dialysis tubing 10000 MWCO)
Dialyse the pooled fractions to remove excess salt and imidazole, using a 10,000 MWCO dialysis tubing against 2 L of Buffer B0 for 3 h.
Anion exchange chromatography (perform at 4°C, using a Bio-Rad NGCTM chromatography system)
Equilibrate a GE MonoQTM 10/100 GL column with 3 CV of Buffer B50.
Using a 50 mL GE SuperloopTM, load the dialysed sample onto the column, and wash the column with 10 CV of Buffer B50.
Elute the sample in 1 mL fractions, using a linear gradient (0-50%) of Buffer B1000 across 20 CV. Monitor the absorbance at 280 nM (UV280) and conductivity measurements. The protein begins to elute when the conductivity reaches ~38 mS/cm (Figure 3A).
Based on the chromatograph, load alternate fractions of the protein peak on a 10% SDS-PAGE gel with an appropriate protein ladder (Bio-Rad Precision Plus ProteinTM Standards) (Figure 3B). Once the presence of the protein is confirmed, pool the fractions.
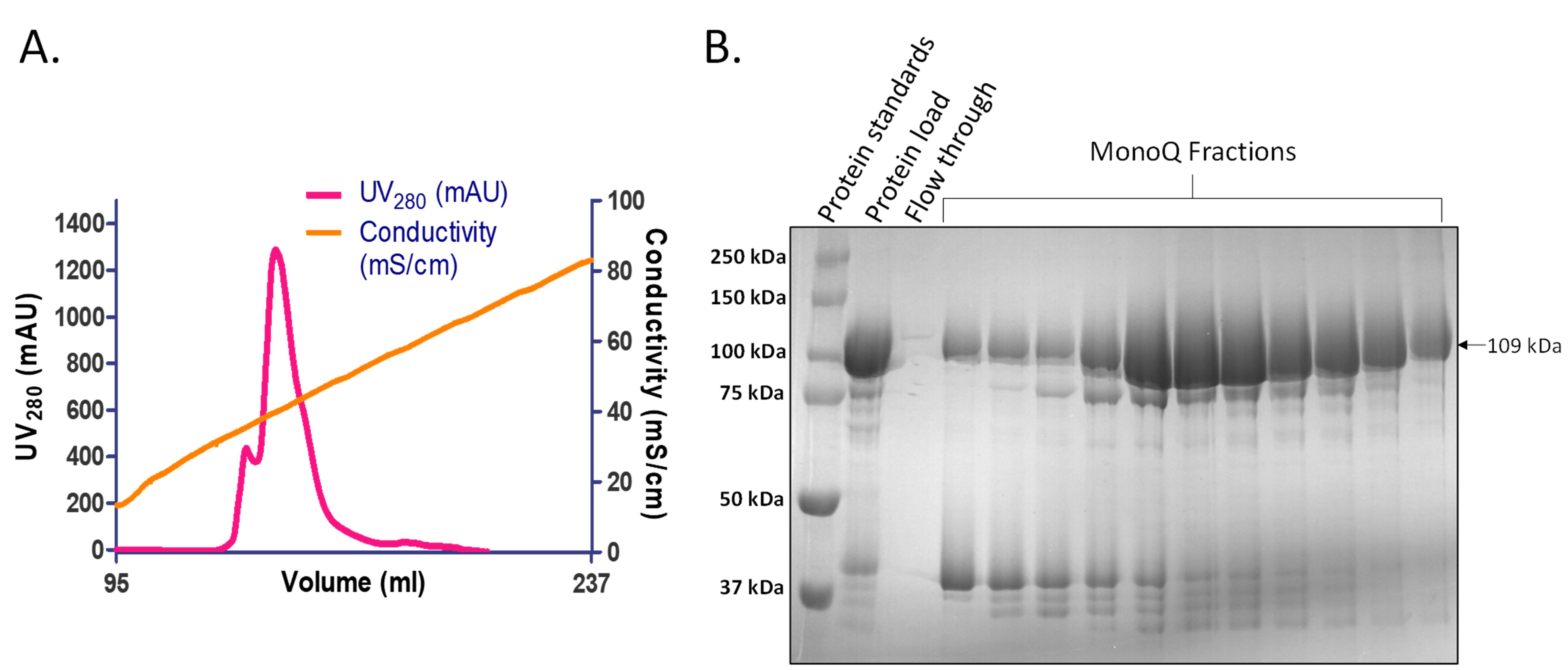
Figure 3. Representative chromatogram and 10% SDS-PAGE after anion exchange chromatography. A. Elution profile of SauUSI after anion exchange chromatography using MonoQTM 10/100 GL column. The protein begins to elute as the conductivity approaches ~38 mS/cm. B. Representative 10% SDS-PAGE (stained with Coomassie R-250) image after anion exchange chromatography. 10 µL of sample (5 µL of the protein fraction mixed with 5 µL of 2× Laemmli sample buffer) is loaded on the gel. The fractions with the most protein are pooled and concentrated, using Vivaspin Turbo 10,000 MWCO centrifugal concentrator.
Concentration of pooled fractions (perform at 4°C, using a Vivaspin Turbo 10,000 MWCO centrifugal concentrator)
Equilibrate the centrifugal concentrator using Buffer B100.
Add the pooled fractions into the concentrator, and spin at a RCF of 3180 (~4,000-5,000 RPM) until the sample is concentrated to ~600 µL, which takes approximately 1 h.
Size exclusion chromatography (perform at 4°C, using a Bio-Rad NGCTM chromatography system)
Equilibrate a GE Superdex 200TM 16/600 pg column with 1.5 CV of Buffer B100.
Load the concentrated protein using a 1 mL loop onto the column.
Elute the sample in 1 mL fractions across 1 CV of Buffer B100. This elution position represents a molecular weight of ~200 kDa, which is in accordance with the dimeric state of SauUSI (~219 kDa) (Figure 4A).
Load the relevant fractions on a 10% SDS-PAGE gel with an appropriate protein ladder (Bio-Rad Precision Plus ProteinTM Standards) (Figure 4B). Once the presence of the protein is confirmed, pool the fractions (since the sample is denatured prior to loading onto a SDS-PAGE, the protein would run at the monomeric molecular weight of ~109 kDa).
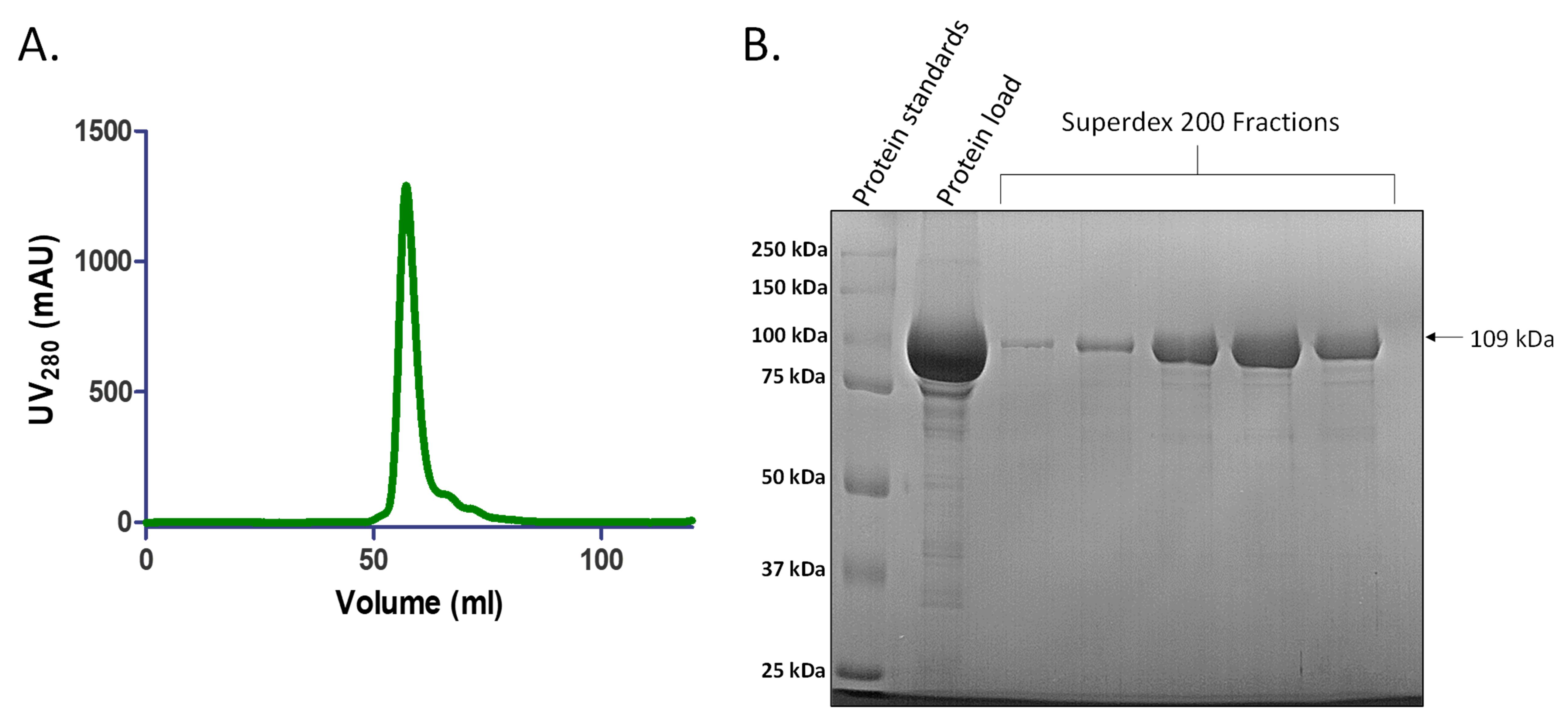
Figure 4. Representative chromatogram and 10% SDS-PAGE after size exclusion chromatography. A. Elution profile of SauUSI after anion exchange chromatography using Superdex 200TM 16/600 GL column. The protein begins to elute at ~0.45 CV. B. Representative 10% SDS-PAGE (stained with Coomassie R-250) image after size exclusion chromatography. 10 µL of sample (5 µL of the protein fraction mixed with 5 µL of 2× Laemmli sample buffer) is loaded on the gel. Size exclusion chromatography helps to separate the protein of interest from other contaminating proteins based on molecular weight, eventually leading to ≥95% purity of protein, and also to elute the protein in an appropriate buffer for further experiments. The fractions with protein are pooled and concentrated using a Vivaspin Turbo 10,000 MWCO centrifugal concentrator.
Concentration of pooled fractions (perform at 4°C, using a Vivaspin Turbo 10,000 MWCO centrifugal concentrator)
Equilibrate the centrifugal concentrator using Buffer B100, add the pooled fractions into the concentrator, and spin at 3,180 RCF (~4,000-5,000 RPM).
Divide the concentrated protein (~10 mg/mL) into suitable aliquots (~5 µL), plunge freeze in liquid nitrogen, and store at -80°C.
Running of 10% SDS-PAGE (Using Bio-Rad SDS-PAGE apparatus)
Add 5 µL of sample to 5 µL of 2× Laemmli sample buffer, and heat at 99°C for 10 min.
Load the samples onto a 10% SDS-PAGE immersed in a running tank containing 1× TGS running buffer. Allow the gel to run at a constant voltage of 200 V for 45 min.
After the run, allow the gel to stain in Coomassie (R-250) staining solution with gentle heating, using a commercial microwave until the solution boils.
Destain the gel in Coomassie destaining solution with gentle heating, using a commercial microwave until the solution boils.
Data analysis
The original research paper for the above purification protocol was published in Tumuluri et al. (2021), https://doi.org/10.1093/nar/gkab042.
Recipes
Materials that need to be Autoclaved
50 mL of 1× Lysogeny broth
Dissolve 1.25 g of LB broth using reverse osmosis (RO) water made up to 50 mL in a 200 mL conical flask. Close the mouth with a cotton plug.
1,000 mL of 1× Lysogeny broth
Dissolve 25 g of LB broth using RO water made up to 1,000 mL in a 2,000 mL conical flask. Close the mouth with a cotton plug. The composition of HiMedia LB (per litre) is 10 g NaCl, 10 g peptone, and 5 g yeast extract.
Stock solutions
20% (w/v) Arabinose
Dissolve 5 g of Arabinose in RO water made up to 25 mL. After dissolution, filter the solution into a sterile 50 mL centrifuge tube using a sterile 0.2 µm syringe filter.
5 M NaCl
Dissolve 146.1 g of NaCl in RO water made up to 500 mL.
2 M Tris-HCl (pH = 8)
Dissolve 121.14 g of Tris base in 300 mL of RO water. Adjust the pH using concentrated HCl. Make the volume up to 500 mL after pH adjustment.
2 M Imidazole
Dissolve 68.07 g of Imidazole in RO water and make up to 500 mL.
4% (w/v) CHAPS
Dissolve 1 g CHAPS in RO water and make up to 25 mL.
2 M MgCl2
Dissolve 1.90 g of MgCl2 in RO water and make up to 10 mL.
0.5 M EDTA (pH = 8)
Dissolve 3.362 g of disodium EDTA salt in RO water (adjust the pH using NaOH pellets), make up to 20 mL, and sterilize by autoclaving.
1 M DTT
Dissolve 3.09 g of DTT in 0.01 M sodium acetate (pH = 5.2) and make up to 20 mL. Filter the solution using a 0.2 µm syringe filter.
Buffers for purification
Note: Prepare the buffers as per the compositions given below, and prechill at 4°C before use.
Lysis buffer (500 mL)
50 mM Tris-HCl (pH = 8) (12.5 mL, 2 M)
500 mM NaCl (50 mL, 5 M)
25 mM Imidazole (6.25 mL, 2 M)
5 mM MgCl2 (1.25 mL, 2 M)
10% glycerol (50 mL, 100%)
0.2% CHAPS (25 mL, 4%)
Make the volume up to 500 mL in RO water and filter, using a vacuum filter assembly attached to a 0.2 µm filter.
Buffers for affinity (Ni-NTA) chromatography (Buffer A and Buffer B) (500 mL of each)
50 mM Tris-HCl (pH = 8) (Buffer A 12.5 mL, Buffer B 12.5 mL, 2 M)
500 mM NaCl (Buffer A 50 mL, Buffer B 50 mL, 5 M)
25 mM Imidazole (Buffer A 6.25 mL, 2 M)
500 mM Imidazole (Buffer B 125 mL, 2 M)
Make the volume up to 500 mL in RO water and filter, using a vacuum filter assembly attached to a 0.2 µm filter. Allow the buffers to degas for 20 min using the same assembly.
Buffer for dialysis (Buffer B0) (2 L)
50 mM Tris-HCl (pH = 8) (50 mL, 2 M)
1 mM EDTA (4 mL, 0.5 M)
1 mM DTT (2 mL, 1 M)
Buffers for anion exchange chromatography (Buffer B50 and Buffer B1000) (500 mL of each)
50 mM Tris-HCl (pH = 8) (Buffer B50 12.5 mL, Buffer B1000 12.5 mL, 2 M)
50 mM NaCl (Buffer B50 5 mL, 5 M)
1000 mM NaCl (Buffer B1000 100 mL, 5 M)
1 mM EDTA (Buffer B50 1 mL, Buffer B1000 1 mL, 0.5 M)
1 mM DTT (Buffer B50 0.5 mL, Buffer B1000 0.5 mL, 1 M)
Make the volume up to 500 mL in RO water and filter, using a vacuum filter assembly attached to a 0.2 µm filter. Allow the buffers to degas for 20 min using the same assembly.
Buffer for size exclusion chromatography (Buffer B100) (500 mL)
50 mM Tris-HCl (pH = 8) (12.5 mL, 2 M)
100 mM NaCl (10 mL, 5 M)
1 mM DTT (0.5 mL, 1 M)
Make the volume up to 500 mL in RO water and filter, using a vacuum filter assembly attached to a 0.2 µm filter. Allow the buffers to degas for 20 min using the same assembly.
SDS-PAGE
10× stock of TGS (Tris base, glycine, and SDS) running buffer
Dissolve 30 g of Tris base, 144 g of glycine, and 10 g of SDS in RO water made up to 1,000 mL. Working concentration: 1×.
Coomassie staining solution
Dissolve 2.5 g of Coomassie Brilliant Blue (R-250) in a solution containing 500 mL of ethanol, 100 mL of glacial acetic acid, and 100 mL of RO water. After complete solvation, filter the solution through a Whatman filter paper (No. 1).
Coomassie destaining solution
Add 200 mL of ethanol and 100 mL of glacial acetic acid to 700 mL of RO water.
Solutions required for the Gel mix
30% Acrylamide/Bis-Acrylamide (AB) mix
Dissolve 58 g of Acrylamide and 2 g of Bis-Acrylamide in RO water made up to 200 mL.
1.5 M Tris-HCl (pH = 8.8)
Dissolve 90.85 g of Tris base in 300 mL of RO water. Adjust the pH using concentrated HCl. Make the volume up to 500 mL after pH adjustment.
1.5 M Tris-HCl (pH = 6.8)
Dissolve 90.85 g of Tris base in 300 mL of RO water. Adjust the pH using concentrated HCl. Make the volume up to 500 mL after pH adjustment.
10% SDS
Dissolve 1 g of SDS in RO water made up to 10 mL.
10% APS
Dissolve 1 g of APS in RO water made up to 10 mL.
10% resolving gel
Water (RO) (4 mL)
AB-mix (3.3 mL, 30%)
Tris-HCl (pH = 8.8) (2.5 mL, 1.5 M)
SDS (0.1 mL, 10%)
APS (0.1 mL, 10%)
TEMED (0.004 mL)
5% stacking gel
Water (RO) (1.4 mL)
AB-mix (0.33 mL, 30%)
Tris-HCl (pH = 6.8) (0.25 mL, 1.5 M)
SDS (0.02 mL, 10%)
APS (0.02 mL, 10%)
TEMED (0.002 mL)
Acknowledgments
We thank Vrunda Rajgor, Shuang Yong Xu and Om Prakash Chouhan for their contribution to the study of SauUSI. Finally, we thank IISER Pune for the infrastructure to perform experiments. Funding: V.S.T. acknowledges IISER Pune for the Graduate Fellowship; K.S. acknowledges the Department of Biotechnology (DBT) for the S. Ramachandran National Bioscience Award grant. The biochemical and structural characterization of SauUSI is published in Tumuluri et al. (2021), https://doi.org/10.1093/nar/gkab042
Competing interests
There are no conflicts of interest or competing interests.
References
- Corvaglia, A. R., François, P., Hernandez, D., Perron, K., Linder, P. and Schrenzel, J. (2010). A type III-like restriction endonuclease functions as a major barrier to horizontal gene transfer in clinical Staphylococcus aureus strains. Proc Natl Acad Sci U S A 107(26): 11954-11958.
- Kunzelmann, S. and Webb, M. R. (2009). A biosensor for fluorescent determination of ADP with high time resolution. J Biol Chem 284(48): 33130-33138.
- Loenen, W. A., Dryden, D. T., Raleigh, E. A., Wilson, G. G. and Murray, N. E. (2014). Highlights of the DNA cutters: a short history of the restriction enzymes. Nucleic Acids Res 42(1): 3-19.
- Monk, I. R., Shah, I. M., Xu, M., Tan, M. W. and Foster, T. J. (2012). Transforming the untransformable: application of direct transformation to manipulate genetically Staphylococcus aureus and Staphylococcus epidermidis. mBio 3(2): e00277-11.
- Tumuluri, V. S., Rajgor, V., Xu, S. Y., Chouhan, O. P. and Saikrishnan, K. (2021). Mechanism of DNA cleavage by the endonuclease SauUSI: a major barrier to horizontal gene transfer and antibiotic resistance in Staphylococcus aureus. Nucleic Acids Res 49(4): 2161-2178.
- van den Ent, F. and Löwe, J. (2006). RF cloning: a restriction-free method for inserting target genes into plasmids. J Biochem Biophys Methods 67(1): 67-74.
- Xu, S. Y., Corvaglia, A. R., Chan, S. H., Zheng, Y. and Linder, P. (2011). A type IV modification-dependent restriction enzyme SauUSI from Staphylococcus aureus subsp. aureus USA300. Nucleic Acids Res 39(13): 5597-5610.
Article Information
Copyright
© 2022 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Tumuluri, V. S. and Saikrishnan, K. (2022). Heterologous Expression and High Degree Purification of the Restriction Endonuclease SauUSI. Bio-protocol 12(1): e4275. DOI: 10.21769/BioProtoc.4275.
Category
Biochemistry > Protein > Expression
Molecular Biology > Protein > Expression
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.