- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Construction of a Highly Diverse mRNA Library for in vitro Selection of Monobodies
Published: Vol 11, Iss 16, Aug 20, 2021 DOI: 10.21769/BioProtoc.4125 Views: 4040
Reviewed by: Luis Alberto Sánchez VargasZHILEI CHENAnonymous reviewer(s)

Original research article
The authors used this protocol in:

Oct 2020

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Recently, we developed transcription/translation coupled with the association of puromycin linker (TRAP) display as a quick in vitro selection method to obtain antibody-like proteins. For the in vitro selection, it is important to prepare mRNA libraries among which the diversity is high. Here, we describe a method for the preparation of monobody mRNA libraries with greater than 1013 theoretical diversity. First, we synthesized two long single-stranded DNAs that corresponded to fragments of monobody DNA, with random codons in the BC and FG loops. These oligonucleotides were ligated by T4 DNA ligase with the support of guide oligonucleotides containing 3′ ends that were protected by a modification. After amplifying the product DNAs by PCR, one end of each DNA fragment was digested with the type II restriction enzyme BsaI, and the resulting DNA fragments were ligated using T4 DNA ligase. After amplification of the DNA product, mRNAs were synthesized by T7 RNA polymerase. This method is simple and could be used for the preparation of mRNA libraries for various antibody-like proteins.
Graphic abstract:
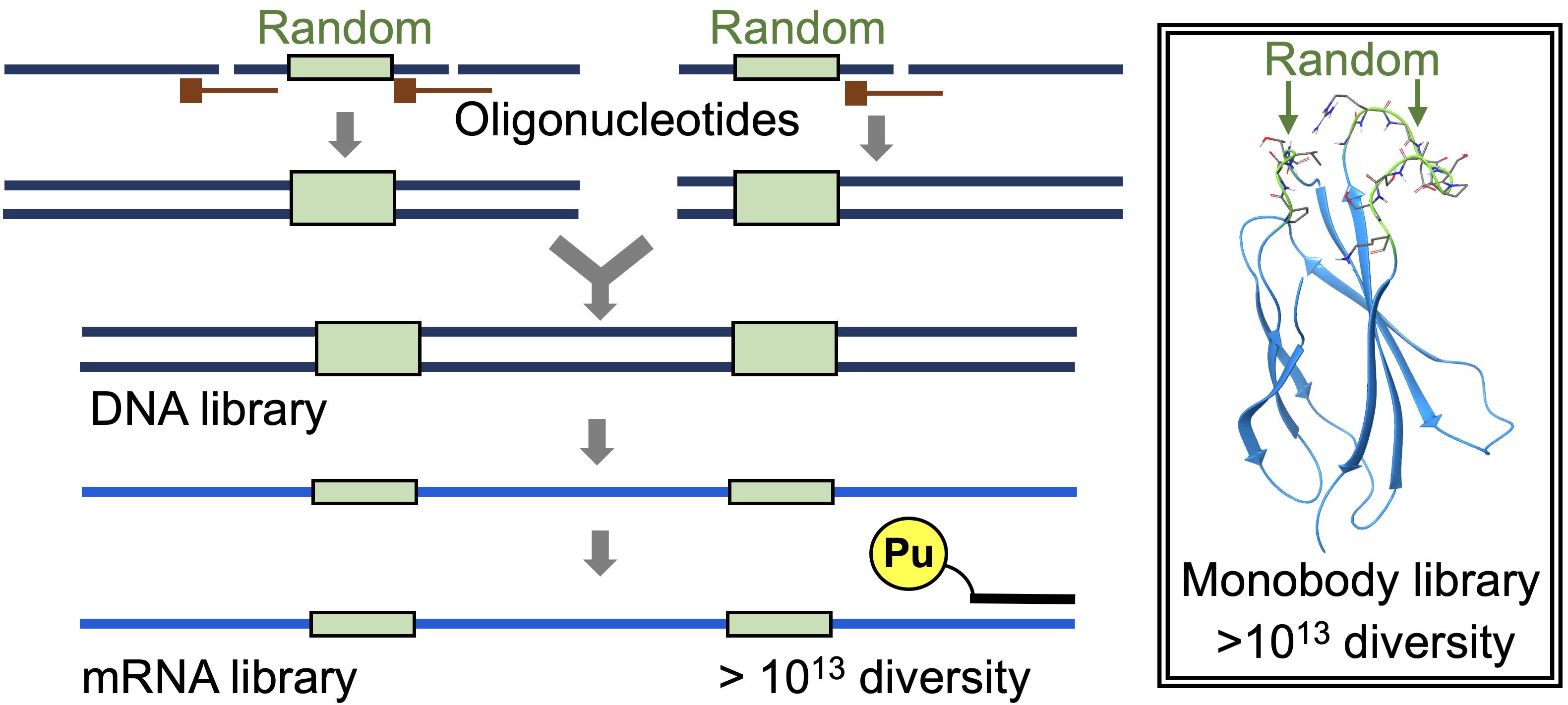
Construction of a highly diverse mRNA library.
Background
Various antibody-like proteins (ALPs) have been obtained using an in vitro selection approach and employed in the biological and medical fields (Schumacher et al., 2018; Simeon and Chen 2018). During this in vitro selection, initial library construction is an important step in obtaining high-affinity binders. For example, to develop a nanobody, the VHH domains of camel antibodies obtained after immunization were used for the construction of an initial library (Arbabi Ghahroudi et al., 1997). Alternatively, the VHH domains from naïve genes or a semi-synthetic or synthetic library could be used as the initial library (Goldman et al., 2006; Monegal et al., 2009; Yan et al., 2014).
For other non-immunoglobulin scaffolds, synthetic libraries were used exclusively as the initial library (Nord et al., 1997; Koide et al., 1998; Beste et al., 1999; Binz et al., 2003; Grabulovski et al., 2007; Olson et al., 2008). Oligonucleotides containing random codons were assembled by extension PCR and ligated to other DNA fragments for synthesis of the DNA library. As randomized codons, NNK codons (with N representing A, C, G, or T; and K representing T or G) were generally used because they cover all 20 amino acids and can reduce the redundancy of codons and the frequency of stop codons. Alternatively, direct codon synthesis using trinucleotide phosphoramidites (Virnekäs et al., 1994) was used to mimic the amino acid frequency in the CDR3 sequences of antibodies (Zemlin et al., 2003; Wojcik et al., 2010; Koide et al., 2012).
The construction of medium-sized libraries (1010 diversity) is relatively simple, whereas that of large-sized libraries (1013 to 1014 diversity) requires special care to maintain the diversity (Olson and Roberts 2007). For example, when a 50 nM DNA product is obtained after 10 cycles of PCR, the concentration of the original template can be calculated as 50 pM. If we use only 100 µl reaction mixture, the maximum diversity that can be achieved using the conditions described above is limited to 3 × 109.
Recently, by modifying the traditional mRNA display (Nemoto et al., 1997; Roberts and Szostak 1997) and the original version of the TRAP display (Ishizawa et al., 2013; Kawakami et al., 2013), we developed an improved TRAP display for the quick in vitro selection of ALPs from a large library (>1013 theoretical diversity), from which we selected high-affinity monobodies (sub-nM KD) against the SARS-CoV-2 spike protein (Kondo et al., 2020). In the present article, we describe the preparation of a monobody mRNA library with >1013 diversity (Figures 1 and 2). We first divided the monobody gene into two fragments, one containing 8 or 10 random codons in the BC loop sequence (A-fragment) and the other containing 10 or 12 random codons in the FG loop sequence (B-fragment). As a random codon, we used a codon mix with the following ratios: 20% Tyr, 10% Ser, 15% Gly, 10% Trp, and 3% each of all remaining amino acids, with the exception of Cys, which was similar to the original cocktail (30% Tyr, 15% Ser, 10% Gly, 5% Phe, 5% Trp, and 2.5% each of all remaining amino acids, with the exception of Cys) (Wojcik et al., 2010; Koide et al., 2012). To prepare a template DNA for each of the two fragments, we ligated 2 to 3 oligonucleotides assembled by guide DNAs using the T4 DNA ligase. We added a modification at the 3′ end of the guide DNAs to prevent them from becoming a primer in the following amplification step. After amplifying the product DNAs by large-scale PCR with minimum amplification cycles (15 ml, 7 cycles), these DNA fragments were digested with BsaI and ligated using T4 DNA ligase. After amplification of the DNA product by large-scale PCR (60 ml, 4 cycles), mRNAs were synthesized by T7 RNA polymerase. This procedure would be useful for the preparation of high-diversity mRNA libraries for various ALPs in the future.

Figure 1. Monobody library sequences. A. DNA sequence of the monobody library [T7 promoter, green; codon mix (20% Tyr, 10% Ser, 15% Gly, 10% Trp, and 3% each of all remaining amino acids, with the exception of Cys), yellow; an21 sequence (the 21-mer sequence for annealing to HEX-PuL), cyan]. B. Protein sequence of the monobody library (randomized residues X, yellow).
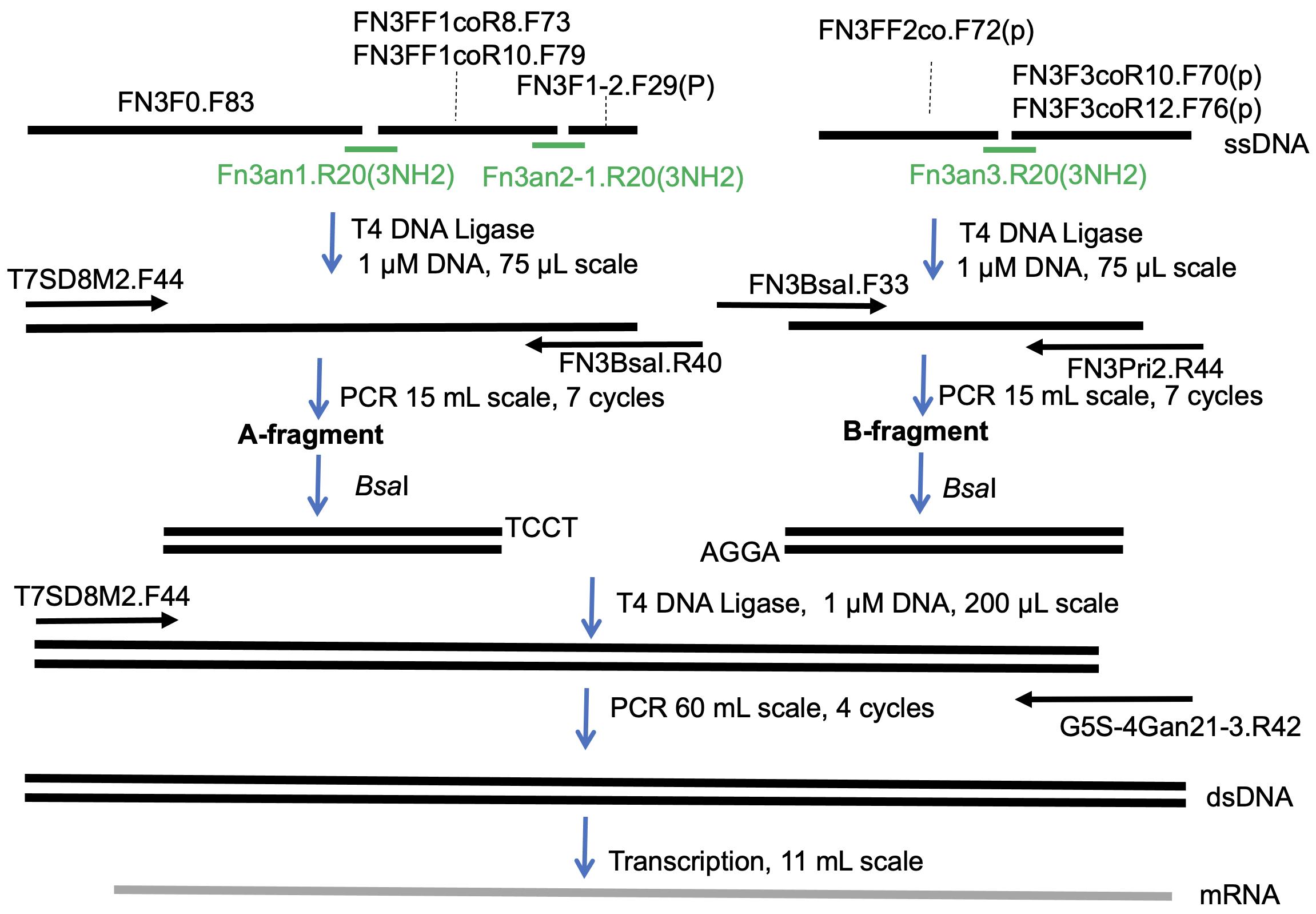
Figure 2. Scheme used for preparation of the monobody mRNA library.
Materials and Reagents
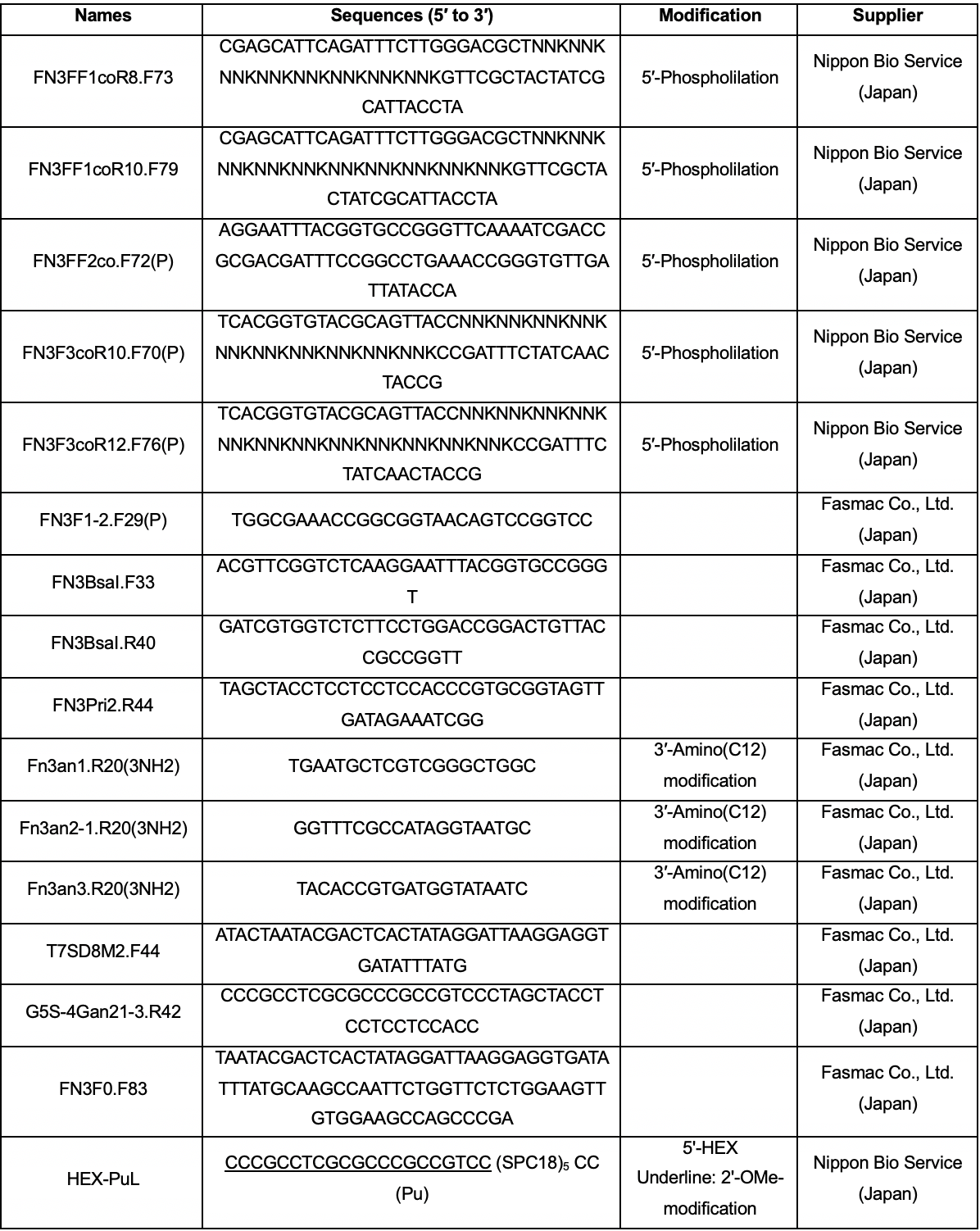
Oligonucleotides (sequences and suppliers are listed in Table 1)
T4 DNA ligase (New England Biolabs, catalog number: M0202L)
Bsa I-HF® (New England Biolabs, catalog number: R3535L)
Note: This has been replaced with a new version (Bsa I-HFv2®, catalog number: R3733L).
Pfu-S DNA polymerase prepared in-house (Wang et al., 2004; Kondo et al., 2020)
T7 RNA polymerase prepared in-house (Abramochkin and Shrader, 1995; Kondo et al., 2020)
ExcelBand 100-bp DNA Ladder (Smobio Technology, catalog number: DM2100)
0.5 mol/L-EDTA Solution (pH 8.0) (Nacalai Tesque, catalog number: 14347-21)
Formamide (Nacalai Tesque, catalog number: 16229-95)
Glycerol (Nacalai Tesque, catalog number: 17018-25)
40 (w/v) %-Acrylamide/Bis Mixed Solution (37.5:1) (Nacalai Tesque, catalog number: 06121-95)
Trimethylolaminomethane (Tris) (Nacalai Tesque, catalog number: 35406-75)
Polyethylene Glycol 6000 (PEG 6000) (FUJIFILM Wako Pure Chemicals, catalog number: 169-22945)
1,4‐Dithiothreitol (DTT) (Nacalai Tesque, catalog number: 14128-62)
Magnesium Chloride Hexahydrate (MgCl2) (Nacalai Tesque, catalog number: 20908-65)
Potassium Chloride (KCl) (Nacalai Tesque, catalog number: 28514-75
Sodium Chloride (NaCl) (Nacalai Tesque, catalog number: 31320-05)
Polyethylene Glycol Mono-p-isooctylphenyl Ether (Triton X-100) (Nacalai Tesque, catalog number: 12967-45)
Dimethyl Sulfoxide (DMSO) (Nacalai Tesque, catalog number: 13407-03)
Magnesium Sulfate Heptahydrate (Nacalai Tesque, catalog number: 06296-25)
Spermidine Trihydrochloride (Nacalai Tesque, catalog number: 32110-54)
100 mM dNTP Set (NIPPON GENE CO., LTD., Custom order)
CTP, GTP, UTP (JENA BIOSCIENCE, Custom order)
ATP (FUJIFILM Wako Pure Chemicals, catalog number: 019-09672)
Phenol:Chloroform:Isoamyl Alcohol 25:24:1 Mixed, pH 6.7 (Nacalai Tesque, catalog number: 25967-74)
Chloroform (Nacalai Tesque, catalog number: 08402-55)
2-Propanol (Nacalai Tesque, catalog number: 29113-95)
Ethanol (EtOH) (Nacalai Tesque, catalog number: 14713-95)
2× Ligation solution (see Recipes)
1× PCR solution (see Recipes)
1× RNA Pol solution (see Recipes)
DNA loading buffer (see Recipes)
Table 1. Oligonucleotide sequences and suppliers. NNK codons were prepared via trinucleotide phosphoramidite synthesis (20% Tyr, 10% Ser, 15% Gly, 10% Trp, and 3% each of all the remaining amino acids, with the exception of Cys). Abbreviations: SPC18, spacer 18; Pu, puromycin; HEX, hexachloro-fluorescein.
Equipment
T100 thermal cycler (Bio-Rad, catalog number: 1861096J1)
Gel Doc EZ (Bio-Rad, catalog number: 1708270)
Mini-PROTEAN System (Bio-Rad, catalog number: 1658000FC)
Cooling slab electrophoresis device (BIO CRAFT, catalog number: BE130G)
Procedure
Preparation of A-fragment DNA of the monobody library
Prepare the following solution (35 μl), heat at 95°C for 2 min, and place at room temperature for 5 min:
FN3F0.F83 (2 μM final concentration), FN3F1-2.F29(P) (2 μM final concentration), FN3FF1coR8.F73 (1 μM final concentration), FN3FF1coR10.F79 (1 μM final concentration), Fn3an1.R20(3NH2) (4 μM final concentration), and Fn3an2-1.R20(3NH2) (4 μM final concentration) in 10 mM Tris-HCl pH 7.5.
Add 2× ligation solution (35 μl) to the solution described in (1) and incubate at 25°C for 1 h (see Figure 3A for analysis of the ligation reaction).
Add the mixture to 1× PCR solution (15 ml) and dispense 77 µl resulting solution into each well of two 96-well PCR plates. Amplify the DNA using T7SD8M2.F44 and FN3BsaI.R40 under the following conditions: initial activation step at 94°C for 1 min, followed by seven cycles of heat denaturation at 94°C for 20 s, primer annealing at 55°C for 20 s, and primer extension at 72°C for 45 s (see Figure 3B for optimization of the number of thermal cycles).
Add 3 M NaCl (1.5 ml).
Mix 3 ml phenol:chloroform:isoamyl alcohol (25:24:1) with the resulting solution and centrifuge at room temperature for 5 min at 16,000 × g.
Mix 6 ml chloroform with the water layer and centrifuge at room temperature for 5 min at 16,000 × g.
Mix 16 ml 2-propanol with the water layer and centrifuge at room temperature for 15 min at 16,000 × g.
After removing the supernatant, add 1 ml 70% EtOH and centrifuge at room temperature for 5 min at 16,000 × g.
After removing the supernatant, dry the pellet and dissolve in 0.5 ml 10 mM Tris-acetic acid pH 7.8.
Determine the DNA concentration by 12% polyacrylamide gel electrophoresis using the ExcelBand 100-bp DNA Ladder.
Preparation of B-fragment DNA of the monobody library
Prepare the following solution (35 μl), heat at 95°C for 2 min, and place at room temperature for 5 min:
FN3FF2co.F72(p) (2 μM final concentration), FN3F3coR10.F70(p) (1 μM final concentration), FN3F3coR12.F76(p) (1 μM final concentration), and Fn3an3.R20(3NH2) (4 μM final concentration) in 10 mM Tris-HCl pH 7.5.
Add 2× ligation solution (35 μl) to the solution described in (1) and incubate at 25°C for 1 h (see Figure 3A for analysis of the ligation reaction).
Add the mixture to 1× PCR solution (15 ml) and dispense 77 µl resulting solution into each well of two 96-well PCR plates. Amplify the DNA using FN3BsaI.F33 and FN3Pri2.R44 under the following conditions: initial activation step at 94°C for 1 min, followed by seven cycles of heat denaturation at 94°C for 20 s, primer annealing at 55°C for 20 s, and primer extension at 72°C for 45 s (see Figure 3B for optimization of the number of thermal cycles).
Add 3 M NaCl (1.5 ml).
Mix 3 ml phenol:chloroform:isoamyl alcohol (25:24:1) with the resulting solution and centrifuge at room temperature for 5 min at 16,000 × g.
Mix 6 ml chloroform with the water layer and centrifuge at room temperature for 5 min at 16,000 × g.
Mix 16 ml 2-propanol with the water layer and centrifuge at room temperature for 15 min at 16,000 × g.
After removing the supernatant, add 1 ml 70% EtOH and centrifuge at room temperature for 5 min at 16,000 × g.
After removing the supernatant, dry the pellet and dissolve in 0.5 ml 10 mM Tris-acetic acid pH 7.8.
Determine the DNA concentration by 12% polyacrylamide gel electrophoresis using the ExcelBand 100-bp DNA Ladder.
Digestion of the A-fragment and B-fragment DNA with Bsa I
Incubate the reaction mixture (1× CutSmart® buffer, 2 μM A-fragment or B-fragment, 200 units/ml Bsa I-HF®; in a total of 400 μl) at 37°C for 4 d.
Add 3 M NaCl (40 µl).
Mix 100 µl phenol:chloroform:isoamyl alcohol (25:24:1) with the resulting solution and centrifuge at room temperature for 5 min at 16,000 × g.
Mix 200 µl chloroform with the water layer and centrifuge at room temperature for 5 min at 16,000 × g.
Mix 400 µl 2-propanol with the water layer and centrifuge at room temperature for 15 min at 16,000 × g.
After removing the supernatant, add 100 µl 70% EtOH and centrifuge at room temperature for 5 min at 16,000 × g.
After removing the supernatant, dry the pellet and dissolve in 10 µl 10 mM Tris-acetic acid pH 7.8.
Mix 10 µl resulting solution with 10 µl DNA loading buffer.
Subject the solution to 12% polyacrylamide gel electrophoresis (10 cm × 7.3 cm × 0.75 mm, 300 V, 20 min).
After cutting the band out of the gel, elute the DNA using 0.3 M NaCl (1.2 ml).
Mix 1.2 ml 2-propanol with the solution and centrifuge at room temperature for 15 minutes at 16,000 × g.
After removing the supernatant, add 100 µl 70% EtOH and centrifuge at room temperature for 5 minutes at 16,000 × g.
After removing the supernatant, dry the pellet and dissolve in 100 µl 10 mM Tris-acetic acid pH 7.8.
Determine the DNA concentration by 12% polyacrylamide gel electrophoresis using the ExcelBand 100 bp DNA Ladder.
Ligation of the digested A-fragment and B-fragment DNA
Mix the solution containing 2 μM each A-fragment and B-fragment DNA (100 μl) with the 2× ligation solution (100 μl).
Incubate the resulting mixture at 25°C for 1 h (see Figure 3C for analysis of the ligation reaction).
PCR amplification of the DNA fragment
Add the mixture to 1× PCR solution (60 ml) and dispense 77 µl resulting solution into each well of eight 96-well PCR plates.
Amplify the DNA fragment using T7SD8M2.F44 and G5S-4Gan21-3.R42 under the following conditions: initial activation step at 94°C for 1 min, followed by four cycles of heat denaturation at 94°C for 20 s, primer annealing at 55°C for 20 s, and primer extension at 72°C for 45 s (see Figure 3D for optimization of the number of thermal cycles).
Add 3 M NaCl (6 ml).
Mix 8 ml phenol:chloroform:isoamyl alcohol (25:24:1) with the resulting solution and centrifuge at room temperature for 5 min at 16,000 × g.
Mix 16 ml chloroform with the water layer and centrifuge at room temperature for 5 min at 16,000 × g.
Mix 66 ml 2-propanol with the water layer and centrifuge at room temperature for 15 min at 16,000 × g.
After removing the supernatant, add 1 ml 70% EtOH and centrifuge at room temperature for 5 min at 16,000 × g.
After removing the supernatant, dry the pellet and dissolve in 6 ml 10 mM Tris-acetic acid pH 7.8.
Determine the DNA concentration by 12% polyacrylamide gel electrophoresis using the ExcelBand 100-bp DNA Ladder.
Preparation of the mRNA library
Incubate a mixture of 1× RNA Pol and 50 nM amplified DNA (in a total of 11 ml) at 37°C for 6 h.
Add 0.55 ml 0.5 M EDTA pH 8.0 and 1.2 ml 3 M NaCl.
Mix 2 ml phenol:chloroform:isoamyl alcohol (25:24:1) with the resulting solution and centrifuge at room temperature for 5 min at 16,000 × g.
Mix 4 ml chloroform with the water layer and centrifuge at room temperature for 5 min at 16,000 × g.
Mix 9 ml 2-propanol with the water layer and centrifuge at room temperature for 15 min at 16,000 × g.
After removing the supernatant, add 1 ml 70% EtOH and centrifuge at room temperature for 5 min at 16,000 × g.
After removing the supernatant, dry the pellet and dissolve in 0.5 ml ultrapure water.
Mix 20 µl resulting solution with 150 µl formamide.
Subject the solution to 6 M urea 5% polyacrylamide gel electrophoresis (13.8 cm × 12 cm × 2 mm, 40 W, 40 min).
After cutting the band out of the gel, elute the DNA using 0.3 M NaCl 50% formamide solution (18 ml).
Mix 4 ml phenol:chloroform:isoamyl alcohol (25:24:1) with the resulting solution and centrifuge at room temperature for 5 min at 16,000 × g.
Mix 8 ml chloroform with the water layer and centrifuge at room temperature for 5 min at 16,000 × g.
Mix 18 ml 2-propanol with the water layer and centrifuge at room temperature for 15 min at 16,000 × g
After removing the supernatant, add 1 ml 70% EtOH and centrifuge at room temperature for 5 min at 16,000 × g.
After removing the supernatant, dry the pellet and dissolve in 20 µl ultrapure water.
Determine the mRNA concentration by measuring the OD at 260 nm. Store at -80°C.
Preparation of the mRNA/HEX-PuL complex
Before the first round of selection, heat the solution (25 mM HEPES-K pH 7.8, 200 mM potassium acetate, 4-8 μM RNA, and 4 μM HEX-PuL, in a total of 80 μl) at 95°C for 2 min, then cool to 25°C. Use the resulting complex for the first round of selection.
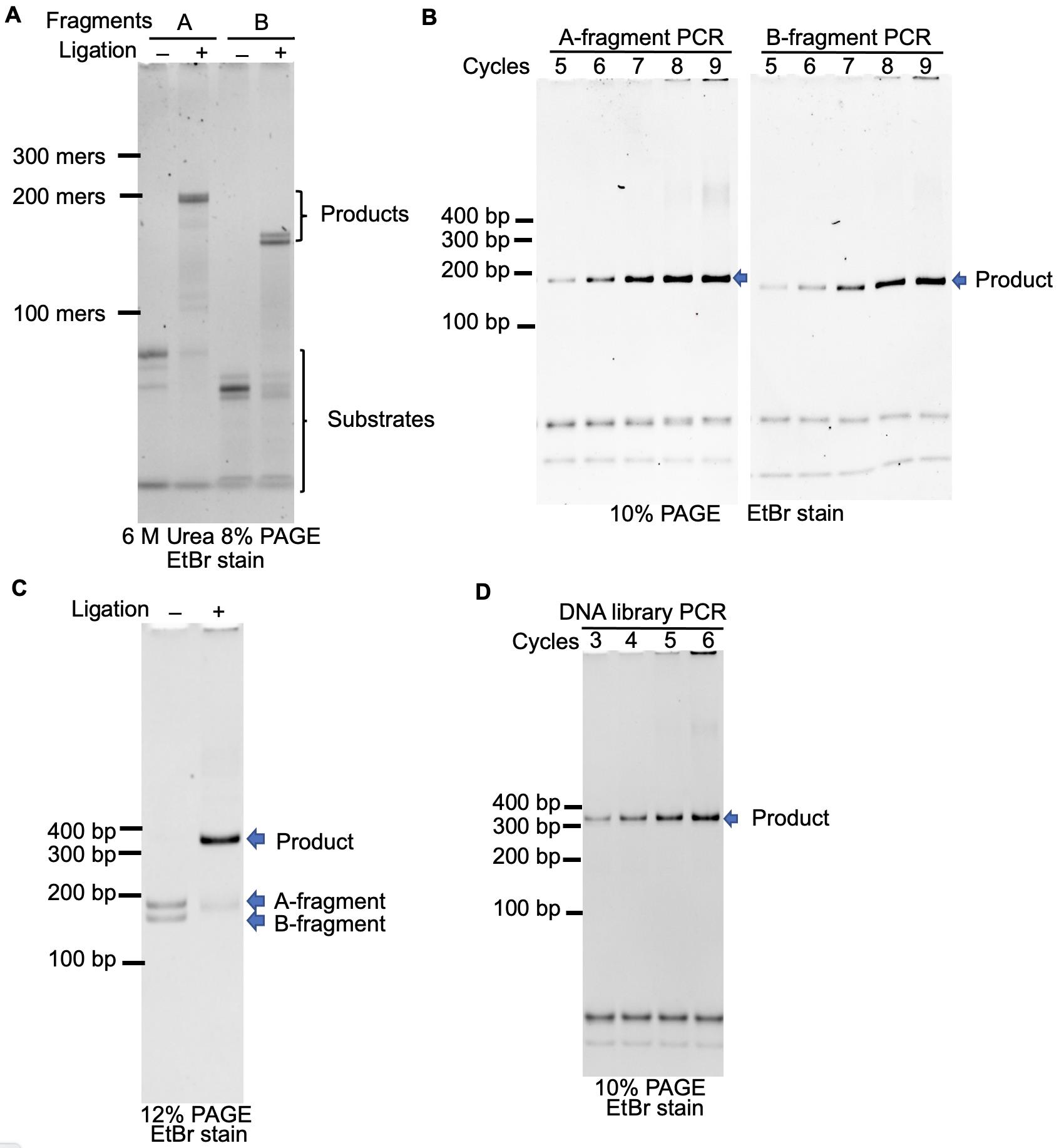
Figure 3. PAGE analysis of the reaction in each procedure. A. Ligation of oligonucleotides during Procedures A and B. B. Optimization of the number of thermal cycles in Procedures A and B. The reaction was performed in 10 µl solution. C. Ligation of the BsaI-treated A- and B-DNA fragments in Procedure D. D. Optimization of the number of thermal cycles in Procedure E. The reaction was performed in 10 µl solution.
Data analysis
The 44 clones in the DNA library were cloned, and the sequences were analyzed by Sanger sequencing. Fourteen single-base deletions and 19 single-base insertions were observed in the backbone sequence; they were likely by-products of the oligonucleotides used in the construction of the DNA template. As a result, the reading frame was maintained in 19 out of 44 clones. The number of codons in the random sequences is summarized in Table 2. The observed frequency showed good agreement with the theoretical frequency.
Table 2. Analysis of the codons in the BC and FG loop sequences of 44 clones.Amino acids Number of observed codons Observed frequency (%) Theoretical frequency (%) Ala 25 3.3 3 Cys 0 0.0 0 Asp 26 3.4 3 Glu 31 4.1 3 Phe 22 2.9 3 Gly 98 12.8 15 His 39 5.1 3 Ile 22 2.9 3 Lys 17 2.2 3 Leu 16 2.1 3 Met 28 3.7 3 Asn 17 2.2 3 Pro 18 2.4 3 Gln 26 3.4 3 Arg 18 2.4 3 Ser 52 6.8 10 Thr 30 3.9 3 Val 52 6.8 3 Trp 65 8.5 10 Tyr 162 21.2 20 Stop 0 0.0 0
Notes
The theoretical diversity of the ssDNA, dsDNA, and mRNA libraries was calculated as follows:
# A-fragment in Procedure A: 1 µM oligonucleotides (75 µl scale); 75 pmol, 4.5 × 1013 molecules
# B-fragment in Procedure B: 1 µM oligonucleotides (75 µl scale); 75 pmol, 4.5 × 1013 molecules
# A-fragment in Procedure A: 43 nM dsDNA product after 7 cycles (15 ml scale); 0.64 nmol, 3.9 × 1014 molecules, 0.6 × 1013 diversity with 64 copies
# B-fragment in Procedure B: 38 nM dsDNA product after 7 cycles (15 ml scale); 0.57 nmol, 3.4 × 1014 molecules, 0.5 × 1013 diversity with 64 copies (only the complementary DNA was synthesized in the 1st cycle)
# dsDNA product in Procedure D: 1 µM dsDNA (200 µl scale); 200 pmol, 1.2 × 1014 molecules
# dsDNA product in Procedure E: 33 nM dsDNA product after 4 cycles (60 ml scale), 2.0 nmol, 1.2 × 1015 molecules, 7.5 × 1013 diversity with 16 copies
# mRNA product in Procedure F: 50 nM DNA template (11 ml scale); 0.55 nmol, 3.3 × 1014 molecules, the diversity was limited by Procedure E.
Recipes
- 2× Ligation solution
100 mM Tris-HCl pH 7.5
12 mM MgCl2
1 mM ATP
16% PEG 6000
20 mM DTT
4,000 U/ml T4 DNA ligase - 1× PCR solution
10 mM Tris-HCl pH 8.4
100 mM KCl
0.1% (v/v) Triton X-100
2% (v/v) DMSO
2 mM MgSO4
0.2 mM each dNTP
0.375 μM each primer
2 nM Pfu-S DNA polymerase - 1× RNA Pol solution
40 mM Tris-HCl pH 8.0
1 mM spermidine
0.01% (v/v) Triton X-100
10 mM DTT
25 mM MgCl2
5 mM each NTP
1 μM T7 RNA polymerase - DNA loading buffer
80 mM Tris-acetate pH 8.0
2 mM EDTA
20% (v/v) glycerol
Acknowledgments
This work was supported by Grant-in-Aid for Scientific Research (A) (General) [Grant Number 15H02006 to H.M.], Grant-in-Aid for Scientific Research on Innovative Areas [Grant Number 20H04704 to G.H.], and Grant-in-Aid for Early-Career Scientists [Grant Number 18K14332 to T.F.] from the Japan Society for the Promotion of Science; and a donation from Dr. Hiroshi Murakami. This protocol was derived from our previous research paper (Kondo et al., 2020).
Competing interests
The authors declare no competing interests.
References
- Abramochkin, G. and Shrader, T. E. (1995). The leucyl/phenylalanyl-tRNA-protein transferase. Overexpression and characterization of substrate recognition, domain structure, and secondary structure. J Biol Chem 270(35): 20621-20628.
- Arbabi Ghahroudi, M., Desmyter, A., Wyns, L., Hamers, R. and Muyldermans, S. (1997). Selection and identification of single domain antibody fragments from camel heavy-chain antibodies. FEBS Lett 414(3): 521-526.
- Beste, G., Schmidt, F. S., Stibora, T. and Skerra, A. (1999). Small antibody-like proteins with prescribed ligand specificities derived from the lipocalin fold. Proc Natl Acad Sci U S A 96(5): 1898-1903.
- Binz, H. K., Stumpp, M. T., Forrer, P., Amstutz, P. and Plückthun, A. (2003). Designing repeat proteins: well-expressed, soluble and stable proteins from combinatorial libraries of consensus ankyrin repeat proteins. J Mol Biol 332(2): 489-503.
- Goldman, E. R., Anderson, G. P., Liu, J. L., Delehanty, J. B., Sherwood, L. J., Osborn, L. E., Cummins, L. B. and Hayhurst, A. (2006). Facile generation of heat-stable antiviral and antitoxin single domain antibodies from a semisynthetic llama library. Anal Chem 78(24): 8245-8255.
- Grabulovski, D., Kaspar, M. and Neri, D. (2007). A novel, non-immunogenic Fyn SH3-derived binding protein with tumor vascular targeting properties. J Biol Chem 282(5): 3196-3204.
- Ishizawa, T., Kawakami, T., Reid, P. C. and Murakami, H. (2013). TRAP display: a high-speed selection method for the generation of functional polypeptides. J Am Chem Soc 135(14): 5433-5440.
- Kawakami, T., Ishizawa, T., Fujino, T., Reid, P. C., Suga, H. and Murakami, H. (2013). In vitro selection of multiple libraries created by genetic code reprogramming to discover macrocyclic peptides that antagonize VEGFR2 activity in living cells. ACS Chem. Biol. 8(6): 1205-1214.
- Koide, A., Bailey, C. W., Huang, X. and Koide, S. (1998). The fibronectin type III domain as a scaffold for novel binding proteins1. J Mol Biol 284(4): 1141-1151.
- Koide, A., Wojcik, J., Gilbreth, R. N., Hoey, R. J. and Koide, S. (2012). Teaching an old scaffold new tricks: monobodies constructed using alternative surfaces of the FN3 scaffold. J Mol Biol 415(2): 393-405.
- Kondo, T., Iwatani, Y., Matsuoka, K., Fujino, T., Umemoto, S., Yokomaku, Y., Ishizaki, K., Kito, S., Sezaki, T., Hayashi, G. and Murakami, H. (2020). Antibody-like proteins that capture and neutralize SARS-CoV-2. Sci Adv 6(42).
- Monegal, A., Ami, D., Martinelli, C., Huang, H., Aliprandi, M., Capasso, P., Francavilla, C., Ossolengo, G. and de Marco, A. (2009). Immunological applications of single-domain llama recombinant antibodies isolated from a naïve library. Protein Eng Des Sel 22(4): 273-280.
- Nemoto, N., Miyamoto-Sato, E., Husimi, Y. and Yanagawa, H. (1997). In vitro virus: bonding of mRNA bearing puromycin at the 3′-terminal end to the C-terminal end of its encoded protein on the ribosome in vitro. FEBS Lett 414(2): 405-408.
- Nord, K., Gunneriusson, E., Ringdahl, J., Stahl, S., Uhlen, M. and Nygren, P. A. (1997). Binding proteins selected from combinatorial libraries of an alpha-helical bacterial receptor domain. Nat Biotechnol 15(8): 772-777.
- Olson, C. A., Liao, H. I., Sun, R. and Roberts, R. W. (2008). mRNA display selection of a high-affinity, modification-specific phospho-IkappaBalpha-binding fibronectin. ACS Chem Biol 3(8): 480-485.
- Olson, C. A. and Roberts, R. W. (2007). Design, expression, and stability of a diverse protein library based on the human fibronectin type III domain. Protein Sci 16(3): 476-484.
- Roberts, R. W. and Szostak, J. W. (1997). RNA-peptide fusions for the in vitro selection of peptides and proteins. Proc Natl Acad Sci U S A 94(23): 12297-12302.
- Schumacher, D., Helma, J., Schneider, A. F. L., Leonhardt, H. and Hackenberger, C. P. R. (2018). Nanobodies: chemical functionalization strategies and intracellular applications. Angew Chem Int Ed 57(9): 2314-2333.
- Simeon, R. and Chen, Z. L. (2018). In vitro-engineered non-antibody protein therapeutics. Protein Cell 9(1): 3-14.
- Virnekäs, B., Ge, L., Plückthun, A., Schneider, K. C., Wellnhofer, G. and Moroney, S. E. (1994). Trinucleotide phosphoramidites: ideal reagents for the synthesis of mixed oligonucleotides for random mutagenesis. Nucleic Acids Res 22(25): 5600-5607.
- Wang, Y., Prosen, D. E., Mei, L., Sullivan, J. C., Finney, M. and Vander Horn, P. B. (2004). A novel strategy to engineer DNA polymerases for enhanced processivity and improved performance in vitro. Nucleic Acids Res 32(3): 1197-1207.
- Wojcik, J., Hantschel, O., Grebien, F., Kaupe, I., Bennett, K. L., Barkinge, J., Jones, R. B., Koide, A., Superti-Furga, G. and Koide, S. (2010). A potent and highly specific FN3 monobody inhibitor of the Abl SH2 domain. Nat Struct Mol Biol 17(4): 519-527.
- Yan, J., Li, G., Hu, Y., Ou, W. and Wan, Y. (2014). Construction of a synthetic phage-displayed Nanobody library with CDR3 regions randomized by trinucleotide cassettes for diagnostic applications. J Transl Med 12: 343.
- Zemlin, M., Klinger, M., Link, J., Zemlin, C., Bauer, K., Engler, J. A., Schroeder, H. W. and Kirkham, P. M. (2003). Expressed murine and human CDR-H3 intervals of equal length exhibit distinct repertoires that differ in their amino acid composition and predicted range of structures. J Mol Biol 334(4): 733-749.
Article Information
Copyright
© 2021 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Kondo, T., Eguchi, M., Tsuzuki, N., Murata, N., Fujino, T., Hayashi, G. and Murakami, H. (2021). Construction of a Highly Diverse mRNA Library for in vitro Selection of Monobodies. Bio-protocol 11(16): e4125. DOI: 10.21769/BioProtoc.4125.
- Kondo, T., Iwatani, Y., Matsuoka, K., Fujino, T., Umemoto, S., Yokomaku, Y., Ishizaki, K., Kito, S., Sezaki, T., Hayashi, G. and Murakami, H. (2020). Antibody-like proteins that capture and neutralize SARS-CoV-2.Sci Adv 6(42).
Category
Immunology > Antibody analysis
Microbiology > Microbial biochemistry > Protein > Immunodetection
Biochemistry > Protein
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.