- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Past Issue in 2025
Volume: 15, Issue: 16
Biochemistry
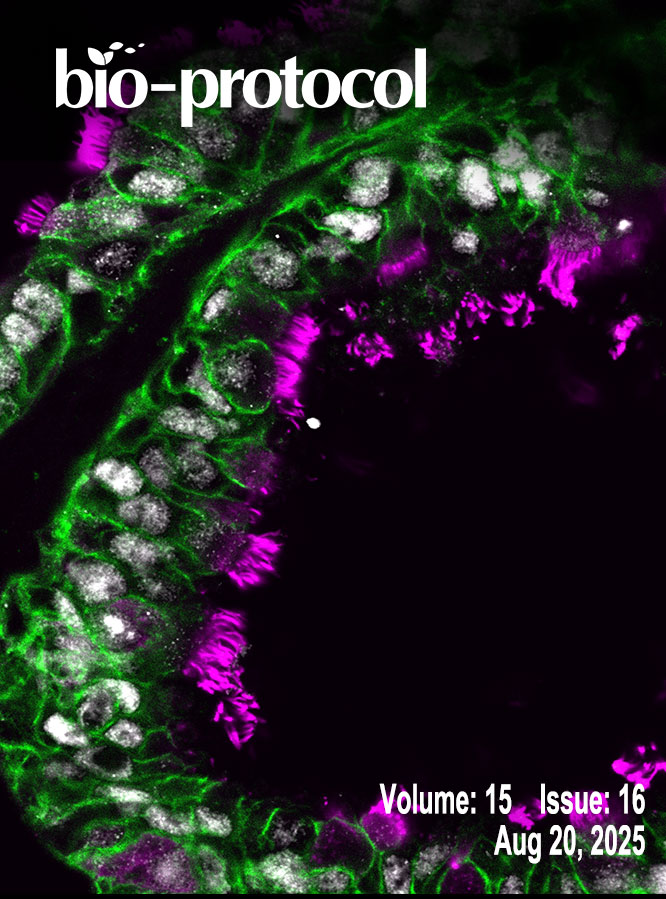
An Optimized Enzyme-Coupled Spectrophotometric Method for Measuring Pyruvate Kinase Kinetics
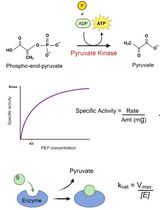
Prokaryotic Expression and Purification of the hSox2-HMG Domain
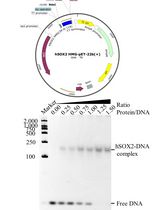
Production of Homogeneous, Functional Zinc-Finger Arrays in High Yield With Two Chromatographic Steps
Cancer Biology
Isolation and Ex Vivo Testing of CD8+ T-Cell Division and Activation Using Mouse Splenocytes
Protocol for Quantifying γH2AX Foci in Irradiated Cells Using Immunofluorescence and Fiji Software
Cell Biology
Establishing and Maintaining 3D Organoid Cultures From Human Fallopian Tube Epithelium
Transplantation of Cultured Myoblasts Into Intact Skeletal Muscle and Analysis of Muscle Contraction Force in Mice Model
Immunology
Rapid Isolation and Flow Cytometry Analysis of Murine Intestinal Immune Cells After Chemically Induced Colitis

Assessing Human Treg Suppression at Single-Cell Resolution Using Mass Cytometry
Microbiology
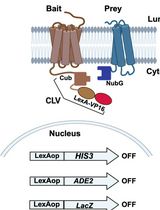
Integrated Membrane Yeast Two-Hybrid System for the Analysis of Membrane Protein Complexes
Assembly and Mutagenesis of Human Coronavirus OC43 Genomes in Yeast via Transformation-Associated Recombination
Molecular Biology
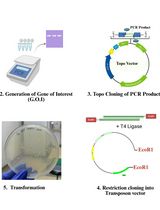
A Simple and Adaptable Method for Cloning Genes Into Transposon Vectors Using Topo and Restriction Systems for Chicken Embryo Transgenesis
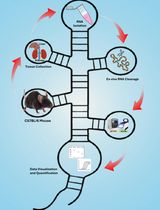
An Ex Vivo Protocol to Assess IRE1α-Dependent RNA Cleavage Using Total RNA Isolated from Mouse Tissues
Neuroscience
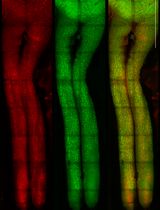
Intravitreal NHS-Biotin Injection and Immunohistochemistry to Label and Image Protein Transport in the Mouse Optic Nerve
Plant Science

ClearDepth Method for Evaluations of Root Depth in Soil-Filled Pots