- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Past Issue in 2025
Volume: 15, Issue: 9

Bioinformatics and Computational Biology

A Guide to Basic RNA Sequencing Data Processing and Transcriptomic Analysis
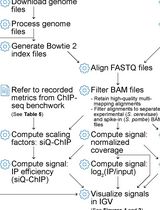
ChIP-seq Data Processing and Relative and Quantitative Signal Normalization for Saccharomyces cerevisiae
Analysis of qRT-PCR Data to Identify the Most Stable Reference Gene Using gQuant

Applying LFQRatio Normalization in Quantitative Proteomic Analysis of Microbial Co-culture Systems
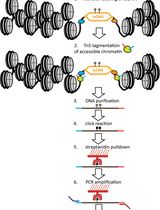
Mapping the Simultaneously Accessible and ssDNA-Containing Genome With KAS-ATAC Sequencing

Reconstruction of Single-Neuron Projectomes in Mice
Cell Biology
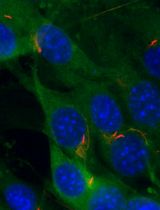
TurboID Labeling and Analysis of Proteins in the Primary Cilium
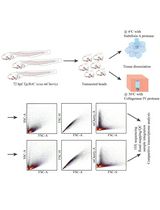
A Cold-Active Protease Tissue Dissociation Protocol for the Preservation of the Tendon Fibroblast Transcriptome
snPATHO-seq: A Detailed Protocol for Single Nucleus RNA Sequencing From FFPE

Developmental Biology
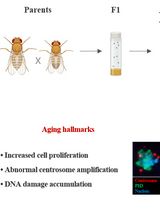
Measuring Anti-aging Effects in Drosophila
Environmental science
From Colonization to High Production and Plasmodium vivax Infection of Anopheles darlingi and Anopheles deaneorum: a Platform for Malaria Research
Immunology

Standardized Flow Cytometry Method for Absolute Counting of Intraepithelial Lymphocytes in the Intestinal Mucosa Using TruCountTM Beads
Plant Science

Optimized Protocol for DNA Extraction in Three Theobroma Species

Assessing Metabolite Interactions With Chloroplastic Proteins via the PISA Assay

Image-Based Lignin Detection in Nematode-Induced Feeding Sites in Arabidopsis Roots

Stem Cell
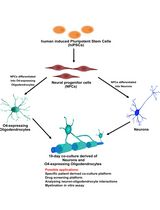
Human iPSC-Derived Neuron and Oligodendrocyte Co-culture as a Small-Molecule Screening Assay for Myelination
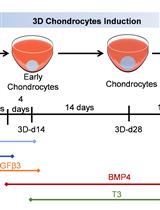
A Cartilaginous Organoid System Derived From Human Expanded Pluripotent Stem Cells (hEPSCs)

Systems Biology
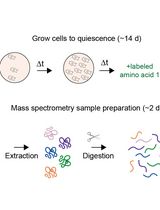
Proteome Birthdating: A Single-Sample Approach for Measuring Global Turnover Dynamics and “Protein Age”