- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Cell Cycle Analysis in the Vertebrate Brain Using Immunolabeled Fresh Cell Nuclei


Published: Vol 3, Iss 22, Nov 20, 2013 DOI: 10.21769/BioProtoc.973 Views: 17841
Reviewed by: Xuecai Ge
Original research article
The authors used this protocol in:
Apr 2013
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Flow cytometry, a standard technique used for quantitative analysis of isolated cells, is routinely employed by immunologists and oncologists to study DNA content, protein expression, and other functional parameters in blood and tumor cells. Unfortunately, the use of this technique by neurobiologists has been hampered by the complexity of the nervous system, whose constituting cells can hardly be dissociated to obtain samples of sufficient quality. We have developed a simplified and quick method to purify and immunolabel cell nuclei with high sensitivity and low background. Our protocol allows the discrimination of single nuclei from doublets and larger aggregates, obtaining low coefficients of variation for cell cycle analysis with propidium iodide. In addition, due to the reduced sample handling this method has high recovery and good reproducibility. As an example, in this protocol we describe the isolation of cell nuclei from adult cerebral cortex, which are subsequently immunostained with antibodies against NeuN (a general neuronal marker) and EGR1 (an early response gene expressed by functionally active neurons), and subjected to flow cytometric gating and analysis. Nevertheless, the protocol can also be applied to other neural tissues from adult and embryonic brain.
Keywords: Flow cytometryMaterials and Reagents
- Frozen tissue samples (adult mouse hemicortex)
- Phosphate-buffered saline (PBS)
- Triton X-100 (Sigma-Aldrich, catalog number: T8787 )
- Protease inhibitors cocktail tablets (cOmplete, Mini, EDTA-free) (Roche Diagnostics, catalog number: 11 836 170 001 )
- Bovine serum albumin (BSA) (stock 30 mg/ml in PBS) (Sigma-Aldrich, catalog number: A4503 )
- Calf serum (Life Technologies, catalog number: 16170-086 )
- Primary antibodies:
Mouse Anti-NeuN antibody (clone A60) (EMD Millipore, catalog number: mab377 )
Rabbit anti-EGR1 antibody (clone T.126.1) (Thermo Fisher Scientific, catalog number: MA5-15009 ) - Secondary antibodies:
Alexa Fluor 647 Goat Anti-Mouse IgG (H + L) Antibody (Life Technologies, catalog number: A21236 )
Alexa Fluor 488 Donkey Anti-Rabbit IgG (H + L) Antibody (Life Technologies, catalog number: A21206 ) - Propidium iodide (stock 1 mg/ml, prepared in autoclaved water) (Sigma-Aldrich, catalog number: P4170 )
- RNase A (stock 1 mg/ml, inactivated by boiling as indicated by the manufacturer) (Sigma-Aldrich, catalog number: R5000 )
- DAPI (stock 100 μg/ml, prepared in autoclaved water) (Sigma-Aldrich, catalog number: D9542 )
Equipment
- 15 ml tube
- 1.5 ml Eppendorf tube
- 7 ml Dounce homogenizer (WHEATON, model: 357542 )
- Microscope
- Refrigerated centrifuge
- Autoclaved 40 μm nylon filters
- Emission filters: BP 530/30, BP 616/23, BP 660/20, APC, FITC
- FACSAria cytometer (BD Biosciences) equipped with a double argon (488 nm) and helium-neon (633 nm) laser
Software
- FACSDiva (BD Biosciences)
- Modfit LT software (Verity Software)
Procedure
- Cell nuclei isolation
- Prepare homogenization buffer containing PBS, 0.1% Triton X-100, and one tablet of protease inhibitors cocktail per 10 ml of buffer.
Note: Keep buffers and sample on ice during processing. - Transfer one mouse hemicortex (or an equivalent mass of adult nervous tissue) to a Dounce homogenizer containing 3 ml of homogenization buffer. The tissue is then homogenized on ice with the “loose” and “tight” pestles, as indicated by the manufacturer.
Note: Volumes can be scaled according to sample size and complexity (for instance, 1 ml homogenization buffer for one telencephalic vesicle from an E17 embryo or one adult murine hippocampus). Several tissues can be processed in parallel, this will allow to get aliquots for control immunostainings (see step B2). - Collect the sample and add 1.5 ml of PBS-0.1%Triton X-100 (final volume 4.5 ml).
- Centrifuge at 200 x g for 1.5 min at 4 °C to remove undissociated tissue (mostly blood vessels).
Optional: Here and in subsequent centrifugation steps the sample can be aliquoted in 1.5 ml Eppendorf tubes to improve the visualization of the pellets. - Discard pellet and transfer the supernatant to a 15 ml tube.
Note: At this step you can remove a small aliquot of the sample, which should be labeled for at least 1 min with either DAPI (100 ng/ml) or propidium iodide (25 μg/ml) to check it under the microscope (Figure 1, Homog.). - Add ice-cold PBS to a final volume of 12 ml.
Note: Volumes can be scaled according to sample size and tissue structure. For instance, for embryonic tissues the volume can be reduced due to their low complexity (one telencephalic vesicle from an E17 embryo can be washed in 1.5 ml PBS). For one adult hippocampus 5 ml PBS are required. - Centrifuge at 400 x g for 4 min at 4 °C.
- Discard supernatant and carefully add PBS to the pellet (total volume of 1-1.2 ml).
Notes:- CRITICAL STEP: Do not disturb the pellet nor try to resuspend it at this point. It is crucial not to disturb the cell nuclei to maintain their integrity!
- At this step you can remove a small aliquot of the supernatant and, after adding either DAPI (100 ng/ml) or propidium iodide (25 μg/ml), check under the microscope that cell nuclei are absent (Figure 1, Supernat.).
- CRITICAL STEP: Do not disturb the pellet nor try to resuspend it at this point. It is crucial not to disturb the cell nuclei to maintain their integrity!
- Maintain the cell nuclei pellet on ice for 20-30 min.
- Resuspend the pellet by gently swirling of the vial (until no visible lumps can be observed), and then by pipetting up and down.
Notes:- At this step remove a small aliquot of the sample and, after adding either DAPI (100 ng/ml) or propidium iodide (25 μg/ml) check the quality of cell nuclei under the microscope (Figure 1, Sample).
- Samples can be stored at 4 °C for a few hours until immunostaining and for 24 h for cell cycle analysis.
- At this step remove a small aliquot of the sample and, after adding either DAPI (100 ng/ml) or propidium iodide (25 μg/ml) check the quality of cell nuclei under the microscope (Figure 1, Sample).
- If immunostaining is not required proceed to step B6.
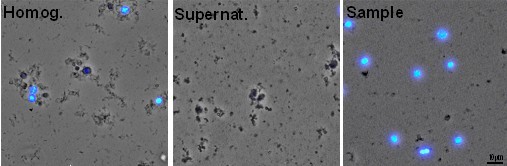
Figure 1. Microscopic images of the aliquots obtained during the cell nuclei isolation procedure. Aliquots of the samples obtained in steps A5 (Homog.), A8 (Supernat.), and A10 (Sample) stained with DAPI (blue).
- Prepare homogenization buffer containing PBS, 0.1% Triton X-100, and one tablet of protease inhibitors cocktail per 10 ml of buffer.
- Fresh/unfixed nuclei immunostaining
- To block nonspecific immunostaining, add 4-5 μl calf serum and 6.4-8 μl BSA (30 mg/ml) to 400-500 μl of the cell nuclei suspension obtained in step A10. Mix by inversion and proceed without further delay.
- Prepare three control samples (400-500 μl each) by pooling small aliquots of the cell nuclei suspensions obtained in step A10, and block nonspecific immunostaining as indicated above:
- Secondary antibody control (CONT).
- Positive control for NeuN (POSITIVE NEUN+).
- Positive control for EGR1 (POSITIVE EGR1+).
Note: This allows to keep enough volume from the cell nuclei suspensions obtained in step A10 to perform at least two different immunostainings (even for small tissues such as the hippocampus).
- Secondary antibody control (CONT).
- Add primary antibody/antibodies against nuclear proteins and mix by inversion:
- Secondary antibody control: No primary antibody.
- Positive control for NeuN: 0.6 μl of mouse anti-NeuN in 500 μl of sample.
- Positive control for EGR1: 1 μl of rabbit anti-EGR1 in 500 μl of sample.
- Sample: 0.6 μl of mouse anti-NeuN and 1 μl of rabbit anti-EGR1 in 500 μl of sample.
Note: Dilutions for other primary antibodies should be empirically optimized (usually the same or double concentration as used for immunohistochemistry should be a good choice). If possible, select antibodies known to recognize unfixed/native epitopes (i.e. validated for immunoprecipitation).
- Secondary antibody control: No primary antibody.
- Without washing, add 1/500 dilution of secondary antibody/antibodies to all the samples (including the controls) and mix by inversion.
Note: We combine primary and secondary antibodies to avoid repeated washing steps that could result in loss of material. Be sure that the fluorophores you use are both excitable and detectable by the cytometer and that they are compatible with the excitation/emission spectra of each other, as well as with that of propidium iodide. - Incubate over night at 4 °C in the dark (without shaking).
Note: Alternatively, samples can be incubated for 2 h at room temperature. - Carefully resuspend the sedimented nuclei and, without washing, filter the sample through a 40 μm autoclaved (i.e. DNase-free) nylon filter.
Note: At this step remove a small aliquot of the sample and, after adding either DAPI (100 ng/ml) or propidium iodide (25 μg/ml), check under the microscope the immunstaining signals (Figure 2).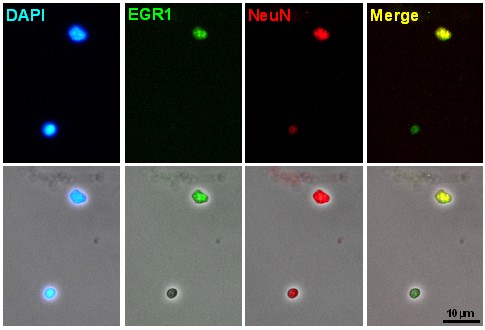
Figure 2. Microscopic images of the aliquots of cell nuclei obtained after immunostaining - Add propidium iodide to the samples to a final concentration of 25-50 μg/ml and DNase-free RNase A to a final concentration of 25 μg/ml.
- To block nonspecific immunostaining, add 4-5 μl calf serum and 6.4-8 μl BSA (30 mg/ml) to 400-500 μl of the cell nuclei suspension obtained in step A10. Mix by inversion and proceed without further delay.
- Flow cytometric gating and analysis
- Analyze the samples using a flow cytometer.
Note: In our case, we use a FACSAria cytometer equipped with a double argon (488 nm) and helium-neon (633 nm) laser. Emission filters: BP 530/30 (for Alexa 488), BP 616/23 (for propidium iodide), and BP 660/20 (for Alexa 647). Data are collected using a linear digital signal process. For data analysis, we use FACSDiva and Modfit LT software. - Localize the nuclei population in the (forward scatter) FSC vs. (side scatter) SSC plot, and create a first gate: P1 (Figure 3A).
- Simultaneously eliminate the debris (i.e. the population unstained for IP) by creating a P2 gate in the Propidium Iodide-A vs. Propidium Iodide-H plot (Figure 3B). This should greatly improve the identification of the nuclei in the FCS-A vs. SSC-A plot (Figure 3C), as well as the fine-tuning of P1 (Figure 3A).
- Select the singlet population and exclude doublets and clumps by gating (P3) on the Propidium Iodide-A vs. Propidium Iodide-H plot (Figure 3D) (see Figure 4 for further explanation).
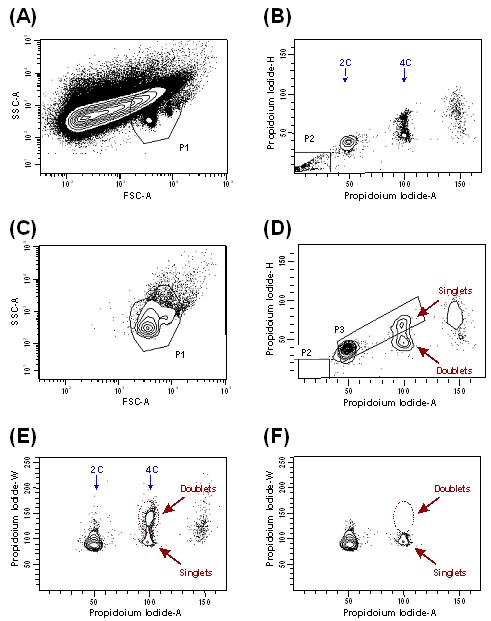
Figure 3. Contour plots and gated scheme used to detect nuclei from cerebral cortex of adult mice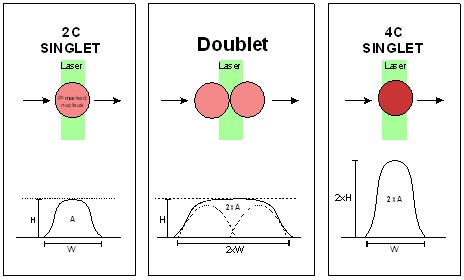
Figure 4. Scheme explaining the procedure used for doublet discrimination. As propidium iodide-labeled nuclei transit through the laser beam, the fluorescence signal is converted into a voltage pulse defined by its height (H), width (W), and area (A). The integrated area of the signal (Propidium iodide-A) is proportional to the DNA content. The Propidium Iodide-W variable directly depends on the time spent by the particle crossing the laser beam and the Propidium Iodide-H reflects the maximal intensity of fluorescence signal. A 4C singlet (tetraploid nucleus) can be discriminated from a doublet (two diploid nuclei adhered to each other) due to the higher H/A ratio and lower W/A ratio of its fluorescence signal (adapted from López-Sánchez and Frade, 2013). - Confirm the correct exclusion of doublets in the selected population by checking the Propidium Iodide-A vs. Propidium Iodide-W plot and adjust P3 if needed. The population with 4C DNA content and high Propidium iodide-W should disappear (compare Figure 3E with Figure 3F). The resulted population is constituted by the singlets to be analyzed henceforth.
- In the control sample (CONT), determine the background threshold of fluorescence signals using emission filter APC (for NeuN) and emission filter FITC (for EGR1). The threshold can be determined representing individual signals in a histogram (Figure 5A-B) or simultaneously representing both signals on the same dot plot (Figure 6A).
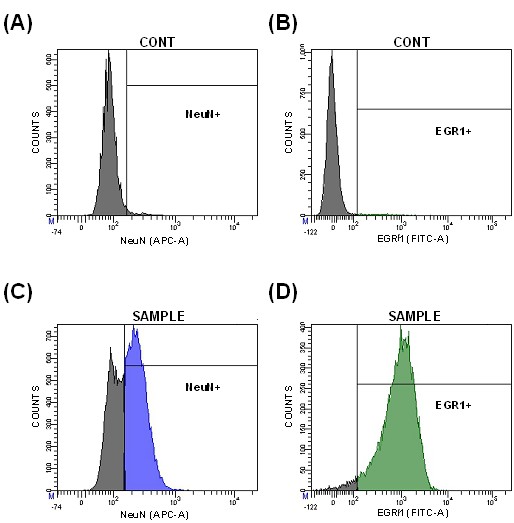
Figure 5. Histograms representing the fluorescence signals for NeuN and EGR1 in secondary antibody controls (CONT) for NeuN (A) and EGR1 (B), as well as sample populations (SAMPLE) immunostained for NeuN (C) and EGR1 (D)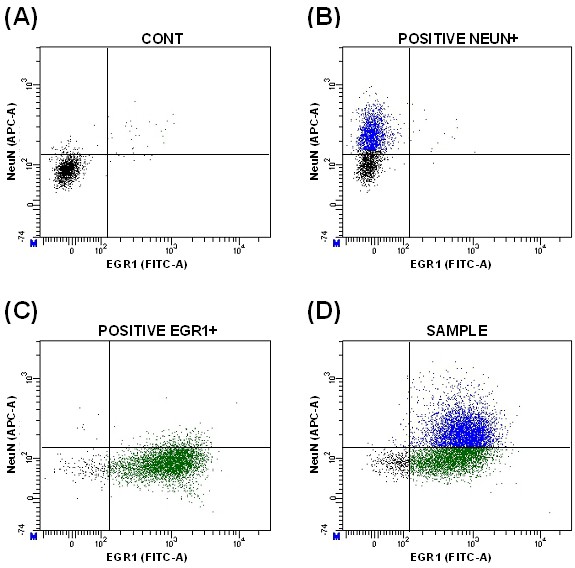
Figure 6. Dot plots representing the fluorescence signals for NeuN and EGR1 in secondary antibody control (A), positive control for NeuN (B), positive control for EGR1 (C), and sample populations (D) - Select positive populations in the individual positive controls (Figure 5C-D and Figure 6B-C). This allows to identify the double-labeled population (Figure 6D).
Note: Sometimes nuclei of interest have specific characteristics of size or complexity that can be used for defining specific nuclei populations. For instance, most of the NeuN positive nuclei from adult mice has higher SSC-A than the negative ones (Figure 7). This allows to improve the correct gating of this population.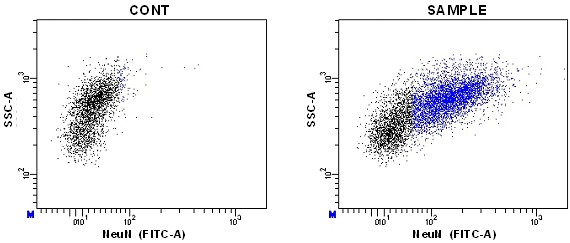
Figure 7. Dot plots of NeuN vs. SSC-A signals - Finally, analyze the DNA content of the population/s of interest in each sample.
Note: This method is useful for both cell cycle analysis in proliferating tissues (Figure 8A-B) and quantification of DNA content in NeuN+ nuclei (Figure 8C-D).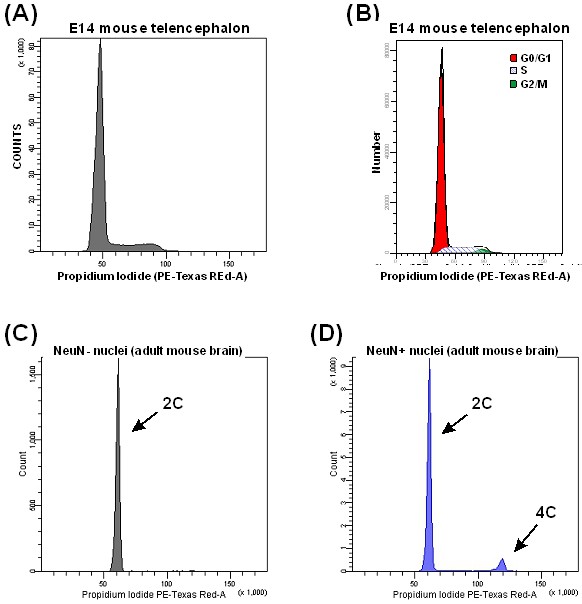
Figure 8. DNA content analysis. Typical histogram of DNA content from a proliferating tissue (A), and analysis of its cell cycle phases quantified with Modfit software (B). Representative histograms showing non-neuronal (C) and neuronal (D) cell nuclei from adult mouse brain. 2C: diploid, 4C: tetraploid.
- Analyze the samples using a flow cytometer.
Notes
- Similar results have been obtained with nervous tissue from other vertebrates, including embryonic and post hatch chicken (Lopez-Sanchez and Frade, 2013) and human (unpublished data).
Acknowledgments
This protocol is adapted from a previous paper by Lopez-Sanchez and Frade (2013). The experimental work was supported by grants from the “Ministerio de Ciencia e Innovación” (BFU2009-07671 and SAF2012-38316) and “Fundación Areces” (CIVP16A1815). Noelia López-Sánchez acknowledges a JAE-Doc contract (JAEDoc026, 2008 call) from the CSIC program “Junta para la Ampliación de Estudios”, co-funded by European Social Fund.
References
- López-Sánchez, N. and Frade, J. M. (2013). Genetic evidence for p75NTR-dependent tetraploidy in cortical projection neurons from adult mice. J Neurosci 33(17): 7488-7500.
Article Information
Copyright
© 2013 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Lopez-Sanchez, N. and Frade, J. M. (2013). Cell Cycle Analysis in the Vertebrate Brain Using Immunolabeled Fresh Cell Nuclei. Bio-protocol 3(22): e973. DOI: 10.21769/BioProtoc.973.
-
López-Sánchez, N. and Frade, J. M. (2013). Genetic evidence for p75NTR-dependent tetraploidy in cortical projection neurons from adult mice. J Neurosci 33(17): 7488-7500.
Category
Neuroscience > Cellular mechanisms
Cell Biology > Cell-based analysis > Flow cytometry
Cell Biology > Organelle isolation > Nuclei
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.